
Heat-Mediated Transformation of PMMA-SiO2 Core-Shell Particles into Hollow SiO2 Particles

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Transformation of PMMA-SiO2 Core-Shell Particles under the Action of the Electron Microscope Beam
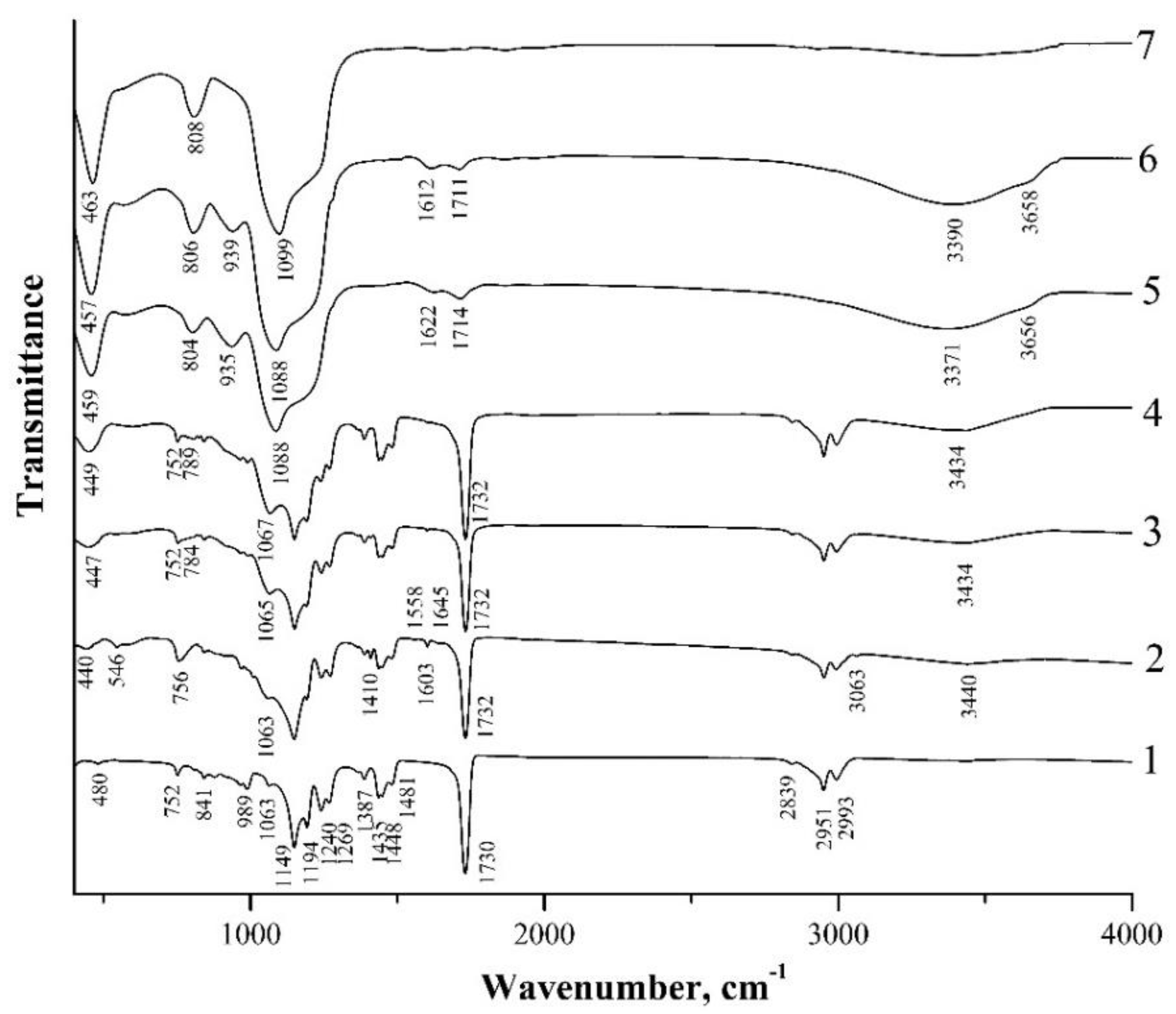
3.2. IR-Transmission Spectra of PMMA-SiO2 Particles
3.3. IR-Transmission Spectra of PMMA-SiO2 Particles at Different Annealing Temperatures
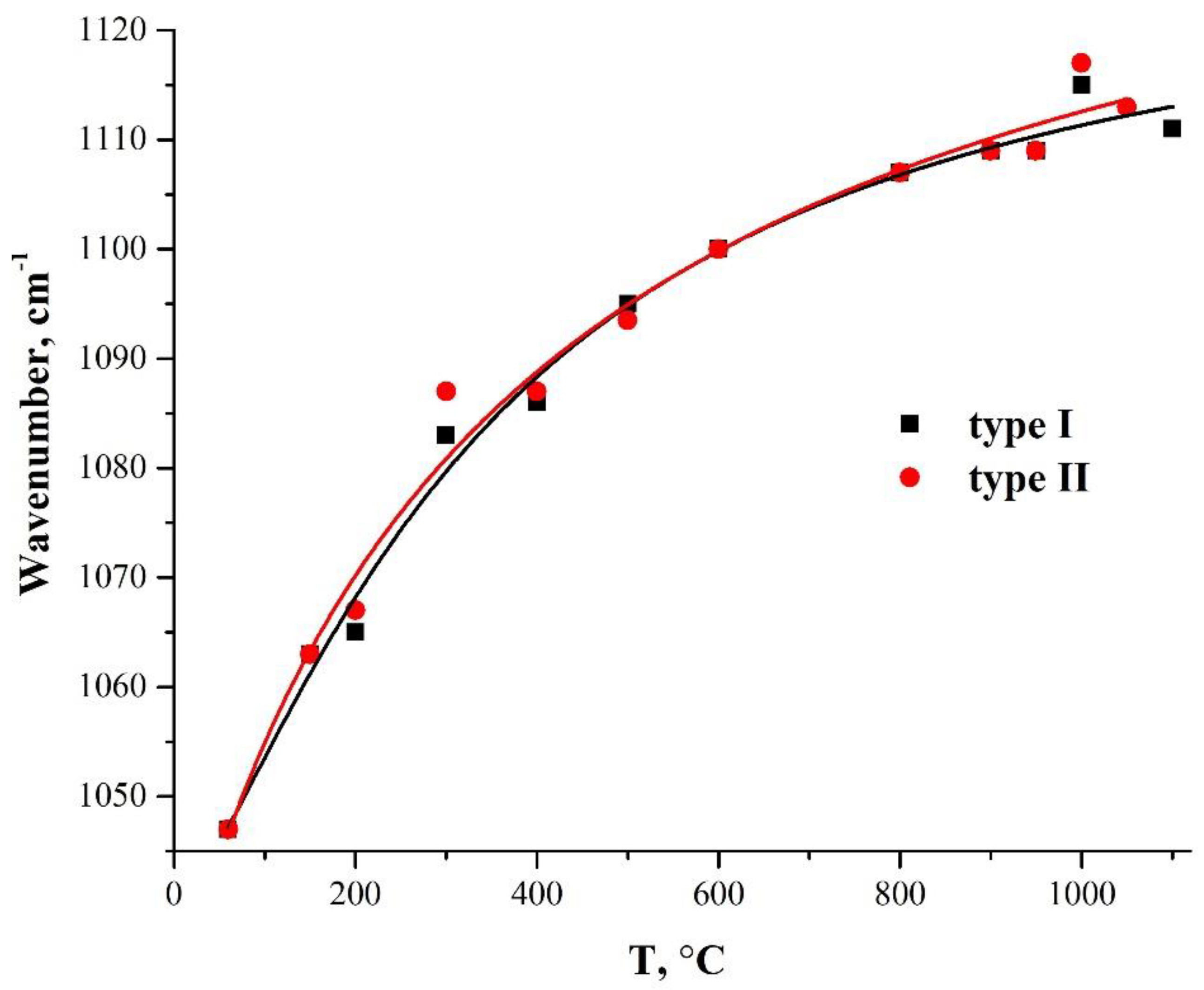
3.4. Variation of the TO3 Mode-Oscillation Frequency with an Annealing Temperature
3.5. Evolution of the Shell Structure during Heat Treatment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Wang, P.; Sun, D.; Li, X.; An, J.; Yu, T.; Yang, E.-H.; Yang, J. Dynamic plastic deformation and failure mechanisms of individual microcapsule and its polymeric composites. J. Mech. Phys. Solids 2020, 139, 103933. [Google Scholar] [CrossRef]
- Eisinaite, V.; Juraite, D.; Schroen, K.; Leskauskaite, D. Food-grade double emulsions as effective fat replacers in meat systems. J. Food Eng. 2017, 213, 54–59. [Google Scholar] [CrossRef]
- Ouarga, A.; Noukrati, H.; Iraola-Arregui, I.; Elaissari, A.; Barroug, A.; Ben Youcef, H. Development of anti-corrosion coating based on phosphorylated ethyl cellulose microcapsules. Prog. Org. Coat. 2020, 148, 105885. [Google Scholar] [CrossRef]
- Huang, L.; Zhou, J.; Chen, Y.; Li, W.; Han, X.; Wang, L. Engineering Microcapsules for Simultaneous Delivery of Combinational Therapeutics. Adv. Mater. Technol. 2020, 5, 2000623. [Google Scholar] [CrossRef]
- Ma, G.; Yue, H. Advances in Uniform Polymer Microspheres and Microcapsules: Preparation and Biomedical Applications. Chin. J. Chem. 2020, 38, 911–923. [Google Scholar] [CrossRef]
- Shang, X.; Zhan, B.; Li, J.; Zhong, R. Novel microcapsules for internal curing of high-performance cementitious system. Sci. Rep. 2020, 10, 8318. [Google Scholar] [CrossRef]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef]
- Hofmeister, I.; Landfester, K.; Taden, A. pH-Sensitive Nanocapsules with Barrier Properties: Fragrance Encapsulation and Controlled Release. Macromolecules 2014, 47, 5768–5773. [Google Scholar] [CrossRef]
- Gao, T.; Sandberg, L.I.C.; Jelle, B.P.; Gustavsen, A. Fuelling the Future: Advances in Science and Technologies for Energy Generation, Transmission and Storage; Mendez-Vilas, A.A., Ed.; Brown Walker Press: Boca Raton, FL, USA, 2012; pp. 535–539. [Google Scholar]
- Gao, T.; Jelle, B.P.; Sandberg, L.I.C.; Gustavsen, A. Monodisperse Hollow Silica Nanospheres for Nano Insulation Materials: Synthesis, Characterization, and Life Cycle Assessment. ACS Appl. Mater. Interfaces 2013, 5, 761–767. [Google Scholar] [CrossRef]
- Ruckdeschel, P.; Philipp, A.; Retsch, M. Understanding Thermal Insulation in Porous, Particulate Materials. Adv. Funct. Mater. 2017, 27, 1702256–1702266. [Google Scholar] [CrossRef]
- Barthelemy, H.; Weber, M.; Barbier, F. Hydrogen storage: Recent improvements and industrial perspectives. Int. J. Hydrog. Energy 2017, 42, 7254–7262. [Google Scholar] [CrossRef]
- Zhu, A.; Shi, Z.; Cai, A.; Zhao, F.; Liao, T. Synthesis of core–shell PMMA–SiO2 nanoparticles with suspension–dispersion–polymerization in an aqueous system and its effect on mechanical properties of PVC composites. Polym. Test. 2008, 27, 540–547. [Google Scholar] [CrossRef]
- Choi, W.S.; Koo, H.Y.; Kim, D.-Y. Facile Fabrication of Core-in-Shell Particles by the Slow Removal of the Core and Its Use in the Encapsulation of Metal Nanoparticles. Langmuir 2008, 24, 4633–4636. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Ding, Z.; Fu, A.; Wang, H.; Guo, P.; Yu, J.; Wang, C.; Zhao, X.S. Preparation of Porous Hollow SiO2 Spheres by a Modified Stöber Process Using MF Microspheres as Templates. J. Clust. Sci. 2012, 23, 273–285. [Google Scholar] [CrossRef]
- Huang, Z.F.; Qu, X.Y.; Chen, Z. Nano-SiO2/PMMA-PU composite particles with core-shell structure via emulsion polymerization and their application in epoxy resin. J. Appl. Polym. Sci. 2015, 132, 41919. [Google Scholar] [CrossRef]
- Mostafa, H.Y.; Hussain, A.I.; El-Masry, A.M.; Maher, A. Novel Core–Shell of Polymethyl Methacrylate/butyl Acrylate/vinyl Silica Nanocomposite Particles through Seed Emulsion Polymerization. Polym. Technol. Eng. 2017, 56, 411–420. [Google Scholar] [CrossRef]
- Yamada, Y.; Mizutani, M.; Nakamura, T.; Yano, K. Mesoporous Microcapsules with Decorated Inner Surface: Fabrication and Photocatalytic Activity. Chem. Mater. 2010, 22, 1695–1703. [Google Scholar] [CrossRef]
- Cao, S.; Zhao, Z.; Jin, X.; Sheng, W.; Li, S.; Ge, Y.; Dong, M.; Wu, W.; Fang, L. Unique double-shelled hollow silica microspheres: Template-guided self-assembly, tunable pore size, high thermal stability, and their application in removal of neutral red. J. Mater. Chem. 2011, 21, 19124–19131. [Google Scholar] [CrossRef]
- Ernawati, L.; Ogi, T.; Balgis, R.; Okuyama, K.; Stucki, M.; Hess, S.C.; Stark, W.J. Hollow Silica as an Optically Transparent and Thermally Insulating Polymer Additive. Langmuir 2016, 32, 338–345. [Google Scholar] [CrossRef]
- Tissot, I.; Reymond, J.P.; Lefebvre, F.; Bourgeat-Lami, E. SiOH-Functionalized Polystyrene Latexes. A Step toward the Synthesis of Hollow Silica Nanoparticles. Chem. Mater. 2002, 14, 1325–1331. [Google Scholar] [CrossRef]
- Chen, M.; Wu, L.; Zhou, S.; You, B. Synthesis of Raspberry-like PMMA/SiO2 Nanocomposite Particles via a Surfactant-Free Method. Macromolecules 2004, 37, 9613–9619. [Google Scholar] [CrossRef]
- Cheng, X.; Chen, M.; Zhou, S.; Wu, L. Preparation of SiO2/PMMA composite particles via conventional emulsion polymerization. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 3807–3816. [Google Scholar] [CrossRef]
- Jiu, H.; Jia, W.; Zhang, L.; Huang, C.; Jiao, H.; Chang, J. Synthesis, luminescent and drug-release properties of SiO2@Y2O3:Eu hollow mesoporous microspheres. J. Porous Mater. 2015, 22, 1511–1518. [Google Scholar] [CrossRef]
- Kato, N.; Ishii, T.; Koumoto, S. Synthesis of Monodisperse Mesoporous Silica Hollow Microcapsules and Their Release of Loaded Materials. Langmuir 2010, 26, 14334–14344. [Google Scholar] [CrossRef] [PubMed]
- Nandiyanto, A.B.D.; Akane, Y.; Ogi, T.; Okuyama, K. Mesopore-Free Hollow Silica Particles with Controllable Diameter and Shell Thickness via Additive-Free Synthesis. Langmuir 2012, 28, 8616–8624. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Fang, L.; Zhao, Z.; Ge, Y.; Piletsky, S.; Turner, A.P.F. Hierachically Structured Hollow Silica Spheres for High Efficiency Immobilization of Enzymes. Adv. Funct. Mater. 2013, 23, 2162–2167. [Google Scholar] [CrossRef]
- Wang, J.; Pan, M.; Yuan, J.; Lin, Q.; Zhang, X.; Liu, G.; Zhu, L. Hollow mesoporous silica with a hierarchical shell from in situ synergistic soft–hard double templates. Nanoscale 2020, 12, 10863–10871. [Google Scholar] [CrossRef]
- Masalov, V.M.; Sukhinina, N.S.; Khodos, I.I.; Zverkova, I.I.; Zhokhov, A.A.; Emelchenko, G.A. Effect of Heat Treatment on the Physical Properties and Morphology of Hollow Submicron SiO2 Particles. J. Surf. Investig. 2021, 15, 1174–1180. [Google Scholar] [CrossRef]
- Nemtsev, I.V.; Shabanov, A.V.; Shabanova, O.V. Electron Microscopy Investigation of Polymethylmethacrylate Spherical Particles & Artificial Opals Based on it. Sib. J. Sci. Technol. 2012, 1, 126–129. [Google Scholar]
- Soucek, M.D.; He, J. UV-Curable Vinyl Functionalized Siloxane Colloids. Radtech Rep. 2007, 21, 10–16. [Google Scholar]
- Gunji, T.; Kawaguchi, Y.; Okonogi, H.; Sakan, T.; Arimitsu, K.; Abe, Y. Preparation and Properties of Organic-Inorganic Hybrid Gel Films Based on Polyvinylpolysilsesquioxane Synthesized from Trimethoxy(vinyl)silane. J. Sol-Gel Sci. Tech. 2005, 33, 9–13. [Google Scholar] [CrossRef]
- Tommasini, F.J.; Ferreira, L.; Tienne, L.G.P.; de Oliveira Aguiar, V.; Prado da Silva, M.H.; da Mota Rocha, L.F.; Vieira Marques, M.D.F. Poly (Methyl Methacrylate)-SiC Nanocomposites Prepared Through in situ Polymerization. Mater. Res. 2018, 21, e20180086. [Google Scholar] [CrossRef]
- Tham, D.Q.; Hoang, T.; Giang, N.V.; Dung, N.T.K.; Chung, I. Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites. Green Process. Synth. 2018, 7, 391–398. [Google Scholar] [CrossRef]
- Duan, G.; Zhang, C.; Li, A.; Yang, X.; Lu, L.; Wang, X. Preparation and Characterization of Mesoporous Zirconia Made by Using a Poly (methyl methacrylate) Template. Nanoscale Res. Lett. 2008, 3, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Mas Haris, M.R.H.; Kathiresan, S.; Mohan, S. FT-IR and FT-Raman Spectra and Normal Coordinate Analysis of Poly methyl methacrylate. Der Pharma Chem. 2010, 2, 316–323. [Google Scholar]
- Ou, D.L.; Seddon, A. Near- and mid-infrared spectroscopy of sol–gel derived ormosils: Vinyl and phenyl silicates. J. Non-Cryst. Solids 1997, 210, 187–203. [Google Scholar] [CrossRef]
- Chen, S.; Osaka, A.; Hayakawa, S.; Shirosaki, Y.; Tsuru, K. Morphology and structure of organosilica hybrid particles derived from tetramethoxysilane and vinyltrimethoxysilane via a catalyst-free sol–gel route. J. Mater. Chem. 2010, 20, 7337–7339. [Google Scholar] [CrossRef]
- Yoshino, H.; Kamiya, K.; Nasu, H. IR study on the structural evolution of sol-gel derived SiO2 gels in the early stage of conversion to glasses. J. Non-Cryst. Solids 1990, 126, 68–78. [Google Scholar] [CrossRef]
- Innocenzi, P. Infrared spectroscopy of sol–gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Vlasova, N.N.; Golovkova, L.P. The adsorption of amino acids on the surface of highly dispersed silica. Colloid J. 2004, 66, 657–662. [Google Scholar] [CrossRef]
- Primeau, N.; Vautey, C.; Langlet, M. The effect of thermal annealing on aerosol-gel deposited SiO2 films: A FTIR deconvolution study. Thin Solid Film. 1997, 310, 47–56. [Google Scholar] [CrossRef]
- Kirk, C.T. Quantitative analysis of the effect of disorder-induced mode coupling on infrared absorption in silica. Phys. Rev. B 1988, 38, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Parrill, T. Heat treatment of spun-on acid-catalyzed sol-gel silica films. J. Mater. Res. 1994, 9, 723–730. [Google Scholar] [CrossRef]
- Parrill, T. Transmission infrared study of acid-catalyzed sol-gel silica coatings during room ambient drying. J. Mater. Res. 1992, 7, 2230–2239. [Google Scholar] [CrossRef]
- Almeida, R.M.; Vasconcelos, H.C.; Ilharco, L.M. Relationship between infrared absorption and porosity in silica-based sol-gel films. SPIE 1994, 2288, 678–687. [Google Scholar] [CrossRef]
- Hussain, R.; Mohammad, D. X-ray Diffraction Study of the Changes Induced During the Thermal Degradation of Poly (Methyl Methacrylate) and Poly (Methacryloyl Chloride). Turk. J. Chem. 2004, 28, 725–729. [Google Scholar]
- Shobhana, E. X-ray Diffraction and UV-Visible Studies of PMMA Thin Films. Int. J. Mod. Eng. Res. 2012, 2, 1092–1095. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Samples | T = 150 °C | T = 200 °C | T = 300 °C | T = 400 °C | T = 600 °C | Particle Type | Assignment | Ref. |
|---|---|---|---|---|---|---|---|---|
| 444 | 453 | 455 | 455 | 459 | 467 | I | δ(O–Si–O) | [37] |
| 440 | 447 | 449 | 459 | 457 | 463 | II | ||
| 544 | − | − | − | − | − | I | C=CH2 twist | [37] |
| 546 | − | − | − | − | − | II | ||
| 756 | 791 | 802 | 802 | 802 | 808 | I | νs(Si–O–Si) | [37,40,42] |
| 756 | 784 | 789 | 804 | 806 | 808 | II | ||
| − | − | − | 939 | 937 | − | I | νas(Si–OH) | [37,44,45] |
| − | − | − | 935 | 939 | − | II | ||
| 1047 | 1065 | 1068 | 1084 | 1088 | 1101 | I | νas(Si–O–Si) | [37,40,42] |
| 1063 | 1065 | 1067 | 1088 | 1088 | 1099 | II | ||
| 1410 | 1410 | − | − | − | − | I | δ(CH2) in-plane | [37,38] |
| 1410 | 1410 | − | − | − | − | II | ||
| 1603 | 1603 | − | − | − | − | I | νs(C=C) | [37,38] |
| 1603 | 1603 | − | − | − | − | II | ||
| 1614 | 1630 | I | δ(H–O–H) | [40] | ||||
| 1622 | 1612 | II | ||||||
| 3062 | − | − | − | − | − | I | νas(CH2) | [36,37] |
| 3063 | − | − | − | − | − | II | ||
| 3436 | 3425 | 3371 | 3360 | 3400 | − | I | ν(H–O–H) | [44,45] |
| 3440 | 3434 | 3434 | 3434 | 3371 | − | II | ||
| − | − | − | 3650 | 3650 | − | I | ν(SiO–H) | [44,45] |
| − | − | − | 3656 | 3658 | − | II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhinina, N.S.; Masalov, V.M.; Fursova, T.N.; Khodos, I.I.; Zverkova, I.I.; Zhokhov, A.A.; Emelchenko, G.A. Heat-Mediated Transformation of PMMA-SiO2 Core-Shell Particles into Hollow SiO2 Particles. Crystals 2022, 12, 883. https://doi.org/10.3390/cryst12070883
Sukhinina NS, Masalov VM, Fursova TN, Khodos II, Zverkova II, Zhokhov AA, Emelchenko GA. Heat-Mediated Transformation of PMMA-SiO2 Core-Shell Particles into Hollow SiO2 Particles. Crystals. 2022; 12(7):883. https://doi.org/10.3390/cryst12070883
Chicago/Turabian StyleSukhinina, Nadezhda S., Vladimir M. Masalov, Tatiana N. Fursova, Igor I. Khodos, Irina I. Zverkova, Andrey A. Zhokhov, and Gennadi A. Emelchenko. 2022. "Heat-Mediated Transformation of PMMA-SiO2 Core-Shell Particles into Hollow SiO2 Particles" Crystals 12, no. 7: 883. https://doi.org/10.3390/cryst12070883
APA StyleSukhinina, N. S., Masalov, V. M., Fursova, T. N., Khodos, I. I., Zverkova, I. I., Zhokhov, A. A., & Emelchenko, G. A. (2022). Heat-Mediated Transformation of PMMA-SiO2 Core-Shell Particles into Hollow SiO2 Particles. Crystals, 12(7), 883. https://doi.org/10.3390/cryst12070883