Substituent Effects in Weak Charge-Transfer Cocrystals of Benzene Derivatives with Classical TCNQ Acceptors: Experimental and Theoretical Study

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Crystal Growth
2.2. Crystallography
2.3. Quantum Chemical Calculations
3. Results and Discussion
- -
- o-xylene/F1TCNQ (D:A = 2:1) (1);
- -
- o-xylene/F2TCNQ (D:A = 1:1) (2);
- -
- o-xylene/F4TCNQ (D:A = 2:1) (3);
- -
- p-xylene/F1TCNQ (D:A = 1:1) (4).
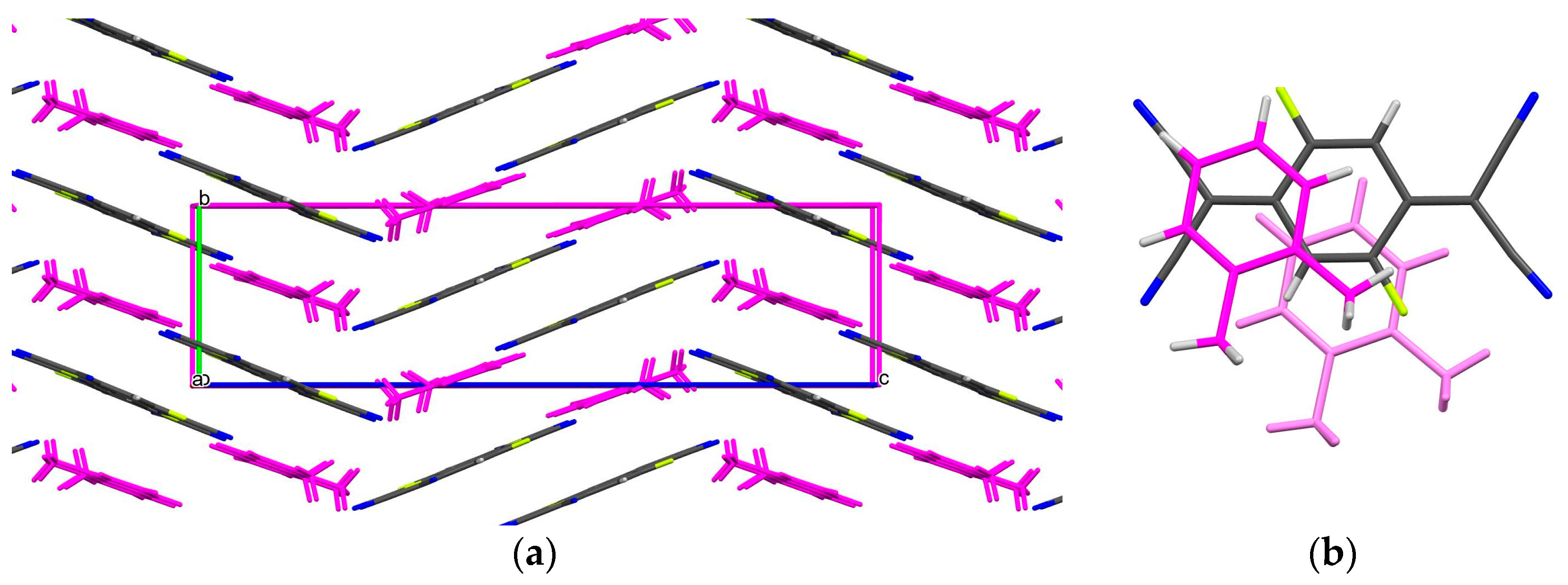
3.1. Crystal Structure of the Cocrystals 1–4 in Comparison with Toluene Cocrystals
- -
- The introduction of the two methyl groups into the benzene molecule significantly changes the relative orientation of the donor and acceptor molecules within the stacks and results in the formation of 2:1 donor–acceptor cocrystals with F1TCNQ and F4TCNQ in comparison to a 1:1 ratio for complexes of toluene.
- -
- All toluene cocrystals exhibit disorder of the donor molecule, while in xylene cocrystals, two methyl groups fix the position of the donor within the stacks.
- -
- The number of fluorine atoms in the acceptor molecule significantly changes the intermolecular interactions between acceptor molecules; in the case of F1TCNQ cocrystals, fluorine atoms are in most cases disordered over all four possible positions due to their small volume.
3.2. The Analysis of Intermolecular Interactions in the Complexes of Xylenes Using Quantum Chemical Calculations
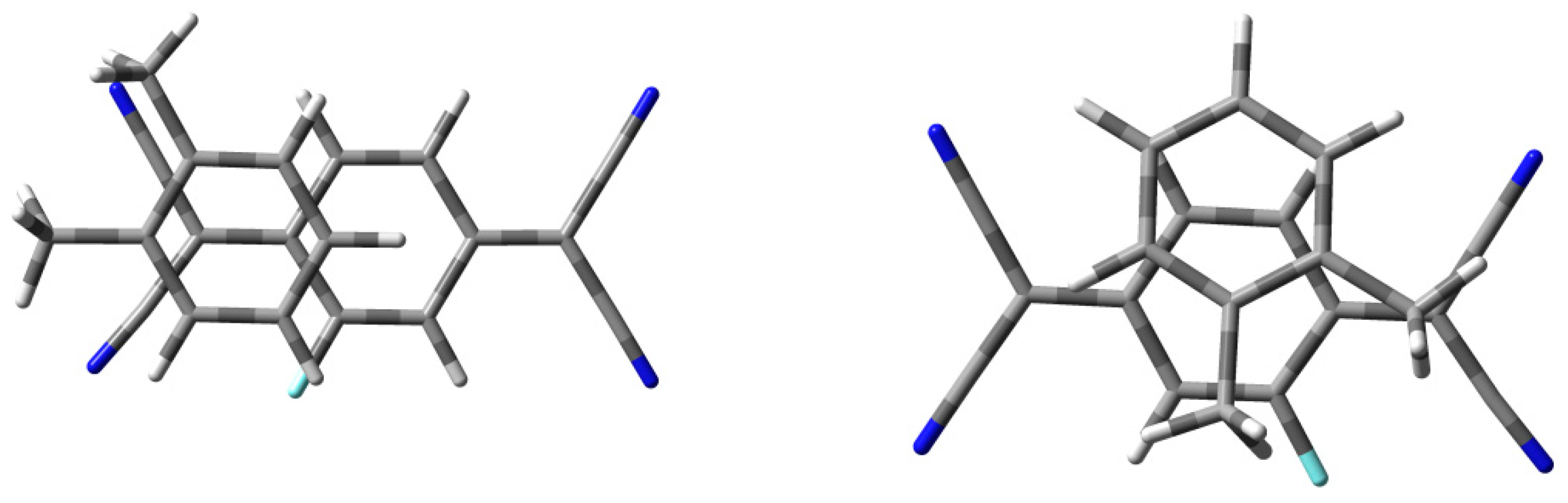
3.2.1. The Analysis of Charge-Transfer D-A Pairs
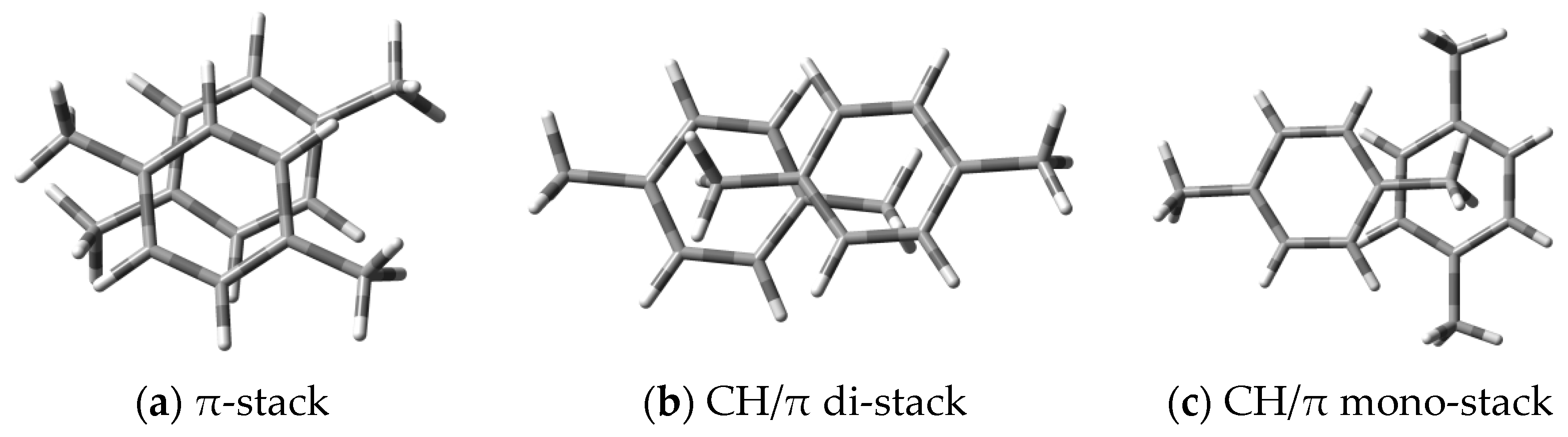
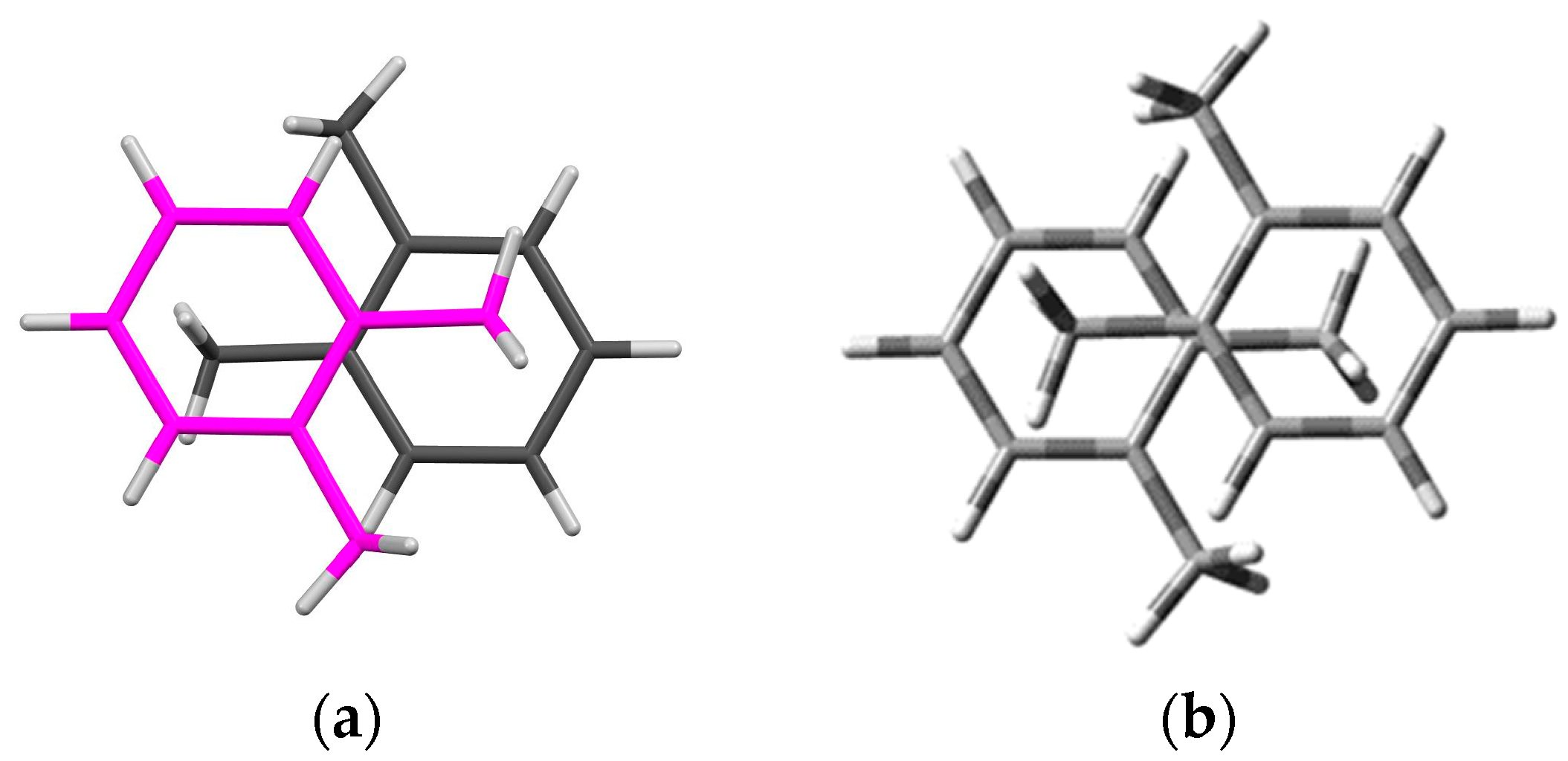
3.2.2. The Analysis of Donor–Donor Interactions
3.2.3. The Analysis of Acceptor–Acceptor Lateral Interactions
3.2.4. The QTAIM Analysis of Intermolecular Interactions in Xylene Cocrystals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dong, H.; Fu, X.; Liu, J.; Wang, Z.; Hu, W. 25th Anniversary article: Key points for high-mobility organic field-effect transistors. Adv. Mater. 2013, 25, 6158–6183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, W.; Dong, H.; Zhang, X.; Li, R.; Hu, W. Organic cocrystals: New strategy for molecular collaborative innovation. Top. Curr. Chem. 2016, 374, 83. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hu, P.; Ye, J.; Zhang, K.K.; Long, Y.; Hu, W.; Kloc, C. Tuning of the degree of charge transfer and the electronic properties in organic binary compounds by crystal engineering: A perspective. J. Mater. Chem. C 2018, 6, 1884–1902. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, X.; Hu, W. Molecular cocrystal odyssey to unconventional electronics and photonics. Sci. Bull. 2021, 66, 512–520. [Google Scholar] [CrossRef]
- Gao, J.; Guo, J.; Chen, Y.; Deng, S.; Lu, Q.; Ren, Y.; Wang, X.; Fan, H.; Teng, F.; He, X.; et al. The competitive role of C–H⋯X (X = F, O) and π–π interactions in contributing to the degree of charge transfer in organic cocrystals: A case study of heteroatom-free donors with p-fluoranil (FA). CrystEngComm. 2022, 24, 6429–6438. [Google Scholar] [CrossRef]
- Li, F.; Zheng, L.; Sun, Y.; Li, S.; Sun, L.; Yang, F.; Dong, W.; Zhang, X.; Hu, W. Cocrystal engineering: Towards high-performance near-infrared organic phototransistors based on donor-acceptor charge transfer cocrystals. Sci. China Chem. 2023, 66, 266–272. [Google Scholar] [CrossRef]
- Goetz, K.P.; Vermeulen, D.; Payne, M.E.; Kloc, C.; McNeil, L.E.; Jurchescu, O.D. Charge-transfer complexes: New perspectives on an old class of compounds. J. Mater. Chem. C 2014, 2, 3065–3076. [Google Scholar] [CrossRef]
- Sultan, M.; Wu, J.; UI Haq, I.; Imran, M.; Yang, L.; Wu, J.; Lu, J.; Chen, L. Recent progress on synthesis, characterization, and performance of energetic cocrystals: A review. Molecules 2022, 27, 4775. [Google Scholar] [CrossRef]
- Ekim, S.D.; Kaya, G.E.; Daştemir, M.; Yildirim, E.; Baytekin, H.T.; Baytekin, B. Organic charge transfer cocrystals as additives for dissipation of contact charges on polymers. ACS Appl. Mater. Interfaces 2022, 14, 56018–56026. [Google Scholar] [CrossRef]
- Zhu, T.; Van Voorhis, T.; de Silva, P. Charge Transfer in Molecular Materials; Andreoni, W., Yip, S., Eds.; Handbook of Materials Modeling; Springer: Berlin/Heidelberg, Germany, 2020; pp. 227–257. [Google Scholar] [CrossRef]
- Buurma, A.J.C.; Jurchescu, O.D.; Shokaryev, I.; Baas, J.; Meetsma, A.; de Wijs, G.A.; de Groot, R.A.; Palstra, T.T.M. Crystal growth, structure, and electronic band structure of tetracene-TCNQ. J. Phys. Chem. C 2007, 111, 3486–3489. [Google Scholar] [CrossRef]
- Hu, P.; Li, H.; Li, Y.; Jiang, H.; Kloc, C. Single-crystal growth, structures, charge transfer and transport properties of anthracene-F4TCNQ and tetracene-F4TCNQ charge-transfer compounds. CrystEngComm 2017, 19, 618–624. [Google Scholar] [CrossRef]
- Kataeva, O.; Nohr, M.; Ivshin, K.; Hampel, S.; Büchner, B.; Knupfer, M. Understanding intermolecular interactions in a tetracene-F4TCNQ cocrystal via its electron density distribution and topology. Cryst. Growth Des. 2021, 21, 471–481. [Google Scholar] [CrossRef]
- Liu, X.-X.; Shi, P.; Dai, X.-L.; Huang, Y.-L.; Lu, T.-B.; Chen, J.-M. Photophysics of charge transfer cocrystals composed of fluorene and its heterocyclic analogues as donors and TCNQ as an acceptor. CrystEngComm. 2022, 24, 8449–8456. [Google Scholar] [CrossRef]
- Fijahi, L.; Salzillo, T.; Tamayo, A.; Bardini, M.; Ruzié, C.; Quarti, C.; Beljonne, D.; d’Agostino, S.; Geerts, Y.H.; Mas-Torrent, M. Charge transfer complexes of a benzothienobenzothiophene derivative and their implementation as active layer in solution-processed thin film organic field-effect transistors. J. Mater. Chem. C 2022, 10, 7319–7328. [Google Scholar] [CrossRef]
- Kataeva, O.; Ivshin, K.; Metlushka, K.; Nikitina, K.; Khrizanforova, V.; Budnikova, Y.; Fayzullin, R.R.; Latypov, S.; Schiemenz, S.; Bretschneider, M.; et al. New charge transfer cocrystals of F2TCNQ with polycyclic aromatic hydrocarbons: Acceptor–acceptor interactions and their contribution to supramolecular arrangement and charge transfer. Cryst. Growth Des. 2022, 22, 751–762. [Google Scholar] [CrossRef]
- Morherr, A.; Witt, S.; Chernenkaya, A.; Bäcker, J.-P.; Schönhense, G.; Bolte, M.; Krellner, C. Crystal growth of new charge-transfer salts based on π-conjugated donor molecules. Phys. B Condens. 2016, 496, 98–105. [Google Scholar] [CrossRef]
- Mahns, B.; Kataeva, O.; Islamov, D.; Hampel, S.; Steckel, F.; Hess, C.; Knupfer, M.; Büchner, B.; Himcinschi, C.; Hahn, T.; et al. Crystal growth, structure, and transport properties of the charge-transfer salt picene/2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane. Cryst. Growth Des. 2014, 14, 1338–1346. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Yasutake, M.; Sakamoto, Y.; Onaka, S.; Sako, K.; Tatemitsu, H.; Shinmyozu, T. Crystal structural properties of a pinwheel compound: [36](1,2,3,4,5,6)cyclophane. Tetrahedron Lett. 2000, 41, 7933–7938. [Google Scholar] [CrossRef]
- Shirai, M.; Hasegawa, M.; Sato, H.; Mazaki, Y. Molecular and electronic structure of distannine-fused tetrathiafulvalene dimer and its cationic species. Chem. Lett. 2014, 43, 592–594. [Google Scholar] [CrossRef]
- Sun, H.; Wang, M.; Wei, X.; Zhang, R.; Wang, S.; Khan, A.; Usman, R.; Feng, Q.; Du, M.; Yu, F.; et al. Understanding charge-transfer interaction mode in cocrystals and solvates of 1-phenyl-3-(pyren-1-yl)prop-2-en-1-one and TCNQ. Cryst. Growth Des. 2015, 15, 4032–4038. [Google Scholar] [CrossRef]
- Johnson, N.T.; Probert, M.R.; Waddell, P.G. Structural investigations into a new polymorph of F4TCNQ: Towards enhanced semiconductor properties. Acta Crystallogr. C 2021, 77, 426–434. [Google Scholar] [CrossRef]
- Ivshin, K.A.; Fedonin, A.P.; Zinnatullin, R.G.; Metlushka, K.E.; Latypov, S.K.; Kataeva, O.N. Non-covalent interactions in weak donor-acceptor systems based on toluene and tetracyanoquinodimethane derivatives. Russ. J. Gen. Chem. 2022, 92, 2561–2568. [Google Scholar] [CrossRef]
- Ivshin, K.A.; Metlushka, K.; Fedonin, A.; Latypov, S.K.; Khrizanforova, V.V.; Budnikova, Y.G.; Vandyukov, A.E.; Kiiamov, A.G.; Laskin, A.; Avdoshenko, S.M.; et al. Substituent controllable assembly of anthracene donors and TCNQ acceptors in charge transfer cocrystals. Cryst. Growth Des. 2023, 23, 954–964. [Google Scholar] [CrossRef]
- APEX3 Crystallography Software Suite; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SAINT Crystallography Software Suite; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.C.M.; van Lenthe, J.H. State of the art in counterpoise theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Ehrlich, S.; Moellmann, J.; Grimme, S. Dispersion-corrected density functional theory for aromatic interactions in complex systems. Acc. Chem. Res. 2013, 46, 916–926. [Google Scholar] [CrossRef]
- Langejuergen, J.; Allers, M.; Oermann, J.; Kirk, A.; Zimmermann, S. Quantitative Detection of Benzene in Toluene- and Xylene-Rich Atmospheres Using High-Kinetic-Energy Ion Mobility Spectrometry (IMS). Analyt. Chem. 2014, 86, 11841–11846. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Pitoňák, M.; Jurečka, P.; Hobza, P. Stabilization and structure calculations for noncovalent interactions in extended molecular systems based on wave function and density functional theories. Chem. Rev. 2010, 110, 5023–5063. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimer | PBE0 | wB97XD | MP2 | |||
|---|---|---|---|---|---|---|
| d (Å) | E (kcal/mol) | d (Å) | E (kcal/mol) | d (Å) | E (kcal/mol) | |
| o-xylene/TCNQ * | 3.794 | 2.98 | 3.320 | 11.20 | 3.211 | 9.27 |
| o-xylene/F1TCNQ | 3.681 | 3.47 | 3.332 | 12.08 | 3.178 | 10.23 |
| o-xylene/F2TCNQ | 3.633 | 3.78 | 3.265 | 12.53 | 3.154 | 10.81 |
| o-xylene/F4TCNQ | 3.522 | 4.61 | 3.242 | 13.80 | 3.155 | 12.52 |
| m-xylene/TCNQ | 3.709 | 2.86 | 3.322 | 12.09 | ||
| m-xylene/F1TCNQ | 3.635 | 3.18 | 3.289 | 12.45 | 3.184 | 10.24 |
| m-xylene/F2TCNQ | 3.618 | 3.45 | 3.284 | 12.83 | ||
| m-xylene/F4TCNQ | 3.562 | 4.49 | 3.252 | 14.19 | ||
| p-xylene/TCNQ | 3.797 | 2.80 | 3.339 | 12.43 | 3.233 | 9.82 |
| p-xylene/F1TCNQ | 3.685 | 3.37 | 3.316 | 13.32 | 3.198 | 10.78 |
| p-xylene/F2TCNQ | 3.668 | 3.55 | 3.313 | 13.56 | 3.186 | 11.19 |
| p-xylene/F4TCNQ | 3.556 | 4.43 | 3.285 | 14.84 | 3.143 | 12.86 |
| o-xylene/o-xylene ** | 3.919 | 1.52 | 3.471 | 8.23 | 3.413 | 4.83 |
| m-xylene/m-xylene | 3.929 | 1.19 | 3.295 | 6.89 | 3.314 | 3.91 |
| p-xylene/p-xylene | 3.945 | 1.20 | 3.481 | 6.34 | 3.456 | 3.72 |
| F1TCNQ/F1TCNQ | 2.406 *** | 5.62 | 2.447 | 7.33 | 2.495 | 6.50 |
| Cocrystal | Contact | d (Å) | PBE0/6-31+G(d) | wB97XD/6-31+G(d) | ||
|---|---|---|---|---|---|---|
| E (kcal/mol) | ρ (e) | E (kcal/mol) | ρ (e) | |||
| 1 | D-A | 3.250 (4) | 3.33 | 0.053 | 3.33 | 0.042 |
| D-D | 3.566 (4) | 3.33 | 3.33 | |||
| D-D | 3.734 (4) | 2.67 | 3.29 | |||
| 2 | * D-A | 3.402 (3) | 2.73 | 0.035 | 2.76 | 0.032 |
| ** D-A | 3.309 (2) | 2.70 | 2.73 | 0.041 | ||
| 3 | D-A | 3.348 (3) | 2.98 | 0.085 | 3.01 | 0.063 |
| D-D | 3.672 (4) | 2.98 | 2.98 | |||
| 4 | D-A | 3.342 (4) | 4.05 | 0.039 | 4.05 | 0.031 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latypov, S.; Fedonin, A.; Ivshin, K.; Zinnatullin, R.; Metlushka, K.; Kataeva, O. Substituent Effects in Weak Charge-Transfer Cocrystals of Benzene Derivatives with Classical TCNQ Acceptors: Experimental and Theoretical Study. Crystals 2023, 13, 1515. https://doi.org/10.3390/cryst13101515
Latypov S, Fedonin A, Ivshin K, Zinnatullin R, Metlushka K, Kataeva O. Substituent Effects in Weak Charge-Transfer Cocrystals of Benzene Derivatives with Classical TCNQ Acceptors: Experimental and Theoretical Study. Crystals. 2023; 13(10):1515. https://doi.org/10.3390/cryst13101515
Chicago/Turabian StyleLatypov, Shamil, Anton Fedonin, Kamil Ivshin, Ruzal Zinnatullin, Kirill Metlushka, and Olga Kataeva. 2023. "Substituent Effects in Weak Charge-Transfer Cocrystals of Benzene Derivatives with Classical TCNQ Acceptors: Experimental and Theoretical Study" Crystals 13, no. 10: 1515. https://doi.org/10.3390/cryst13101515
APA StyleLatypov, S., Fedonin, A., Ivshin, K., Zinnatullin, R., Metlushka, K., & Kataeva, O. (2023). Substituent Effects in Weak Charge-Transfer Cocrystals of Benzene Derivatives with Classical TCNQ Acceptors: Experimental and Theoretical Study. Crystals, 13(10), 1515. https://doi.org/10.3390/cryst13101515