
Nitrate Reduction Reaction on Zr-Doped TiO2 (101) Surfaces Investigated by First-Principles Calculations


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
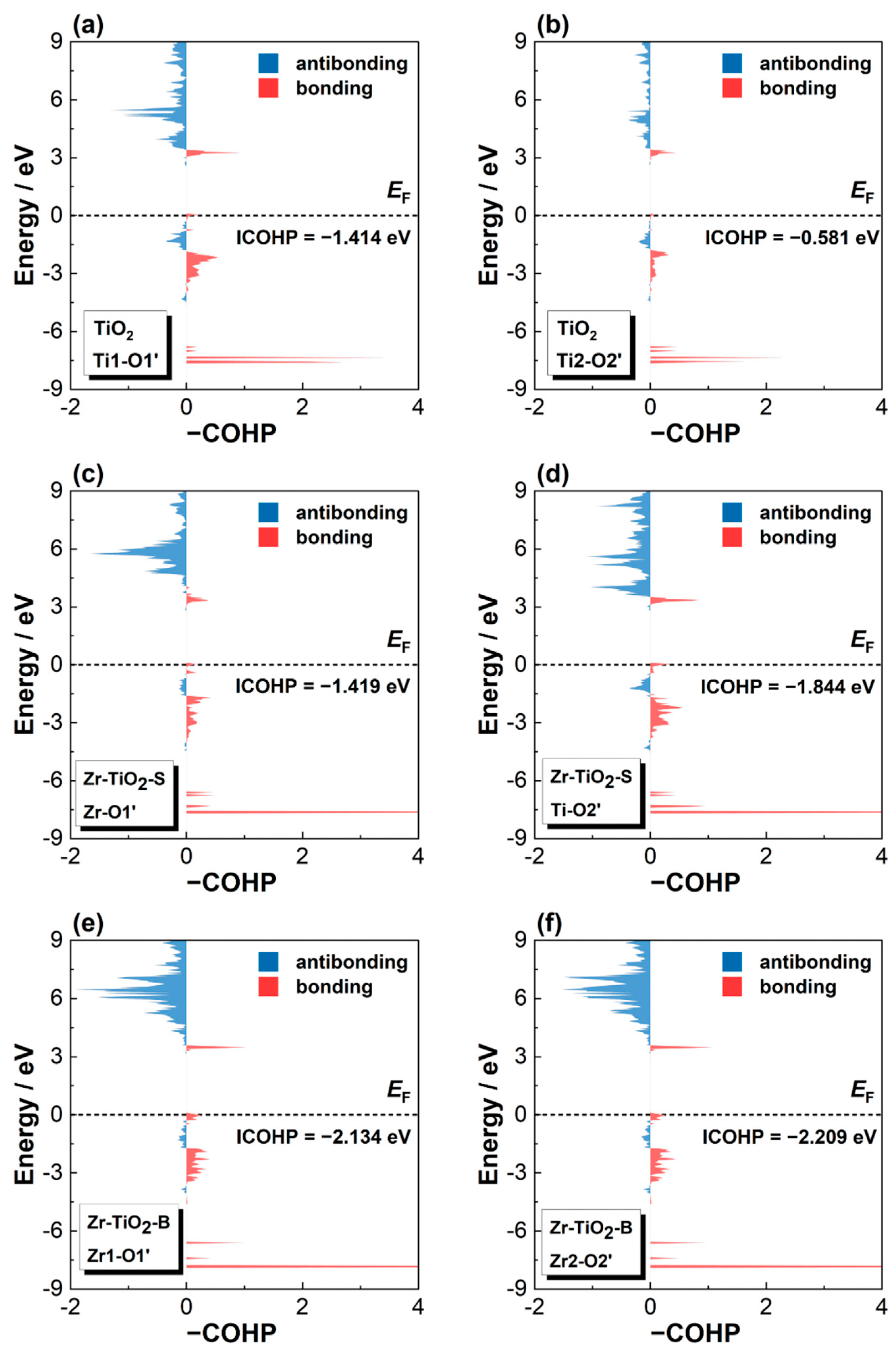
3.1. Electronic Structure
3.2. Adsorption and Charge Transfer of Nitrate
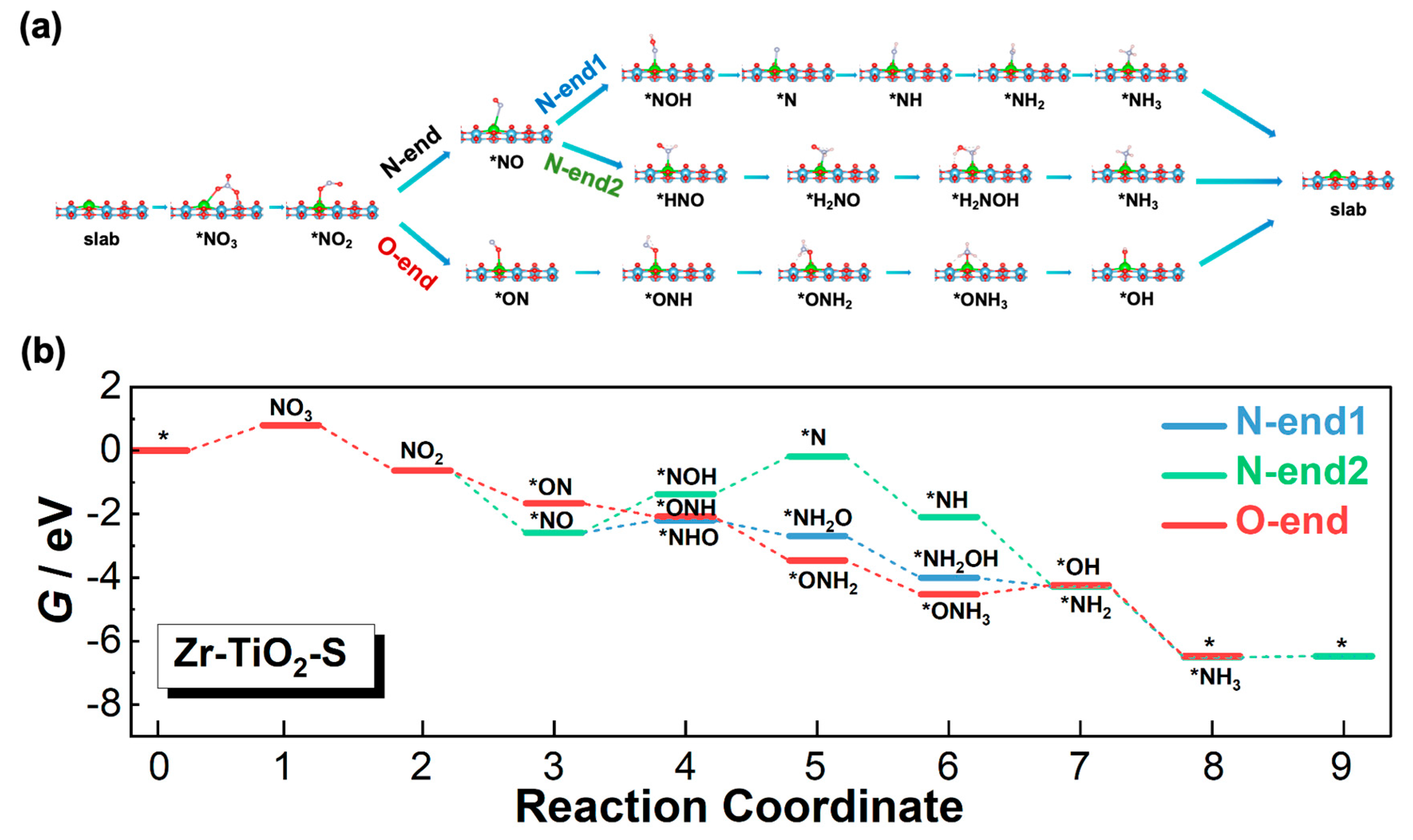
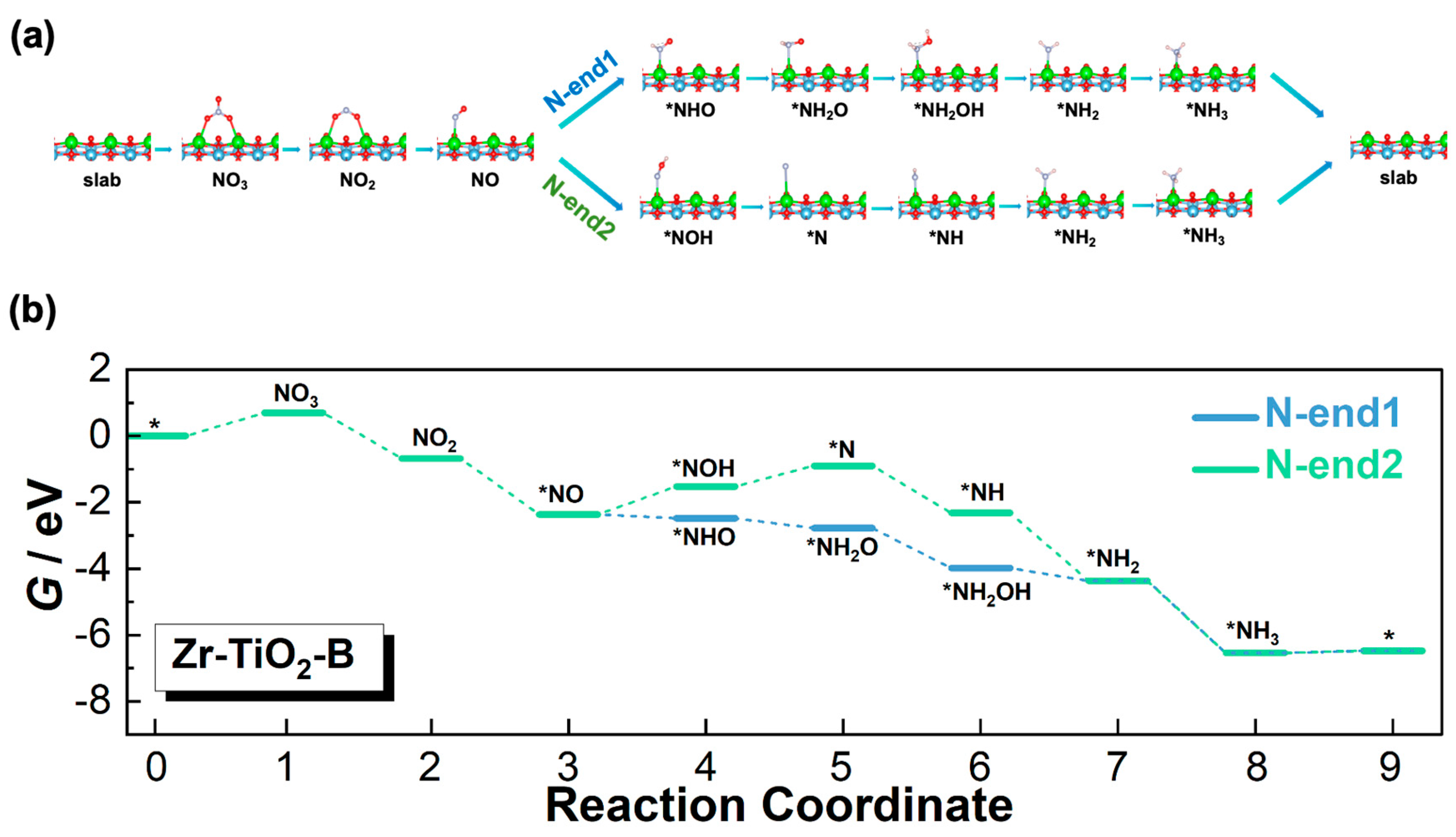
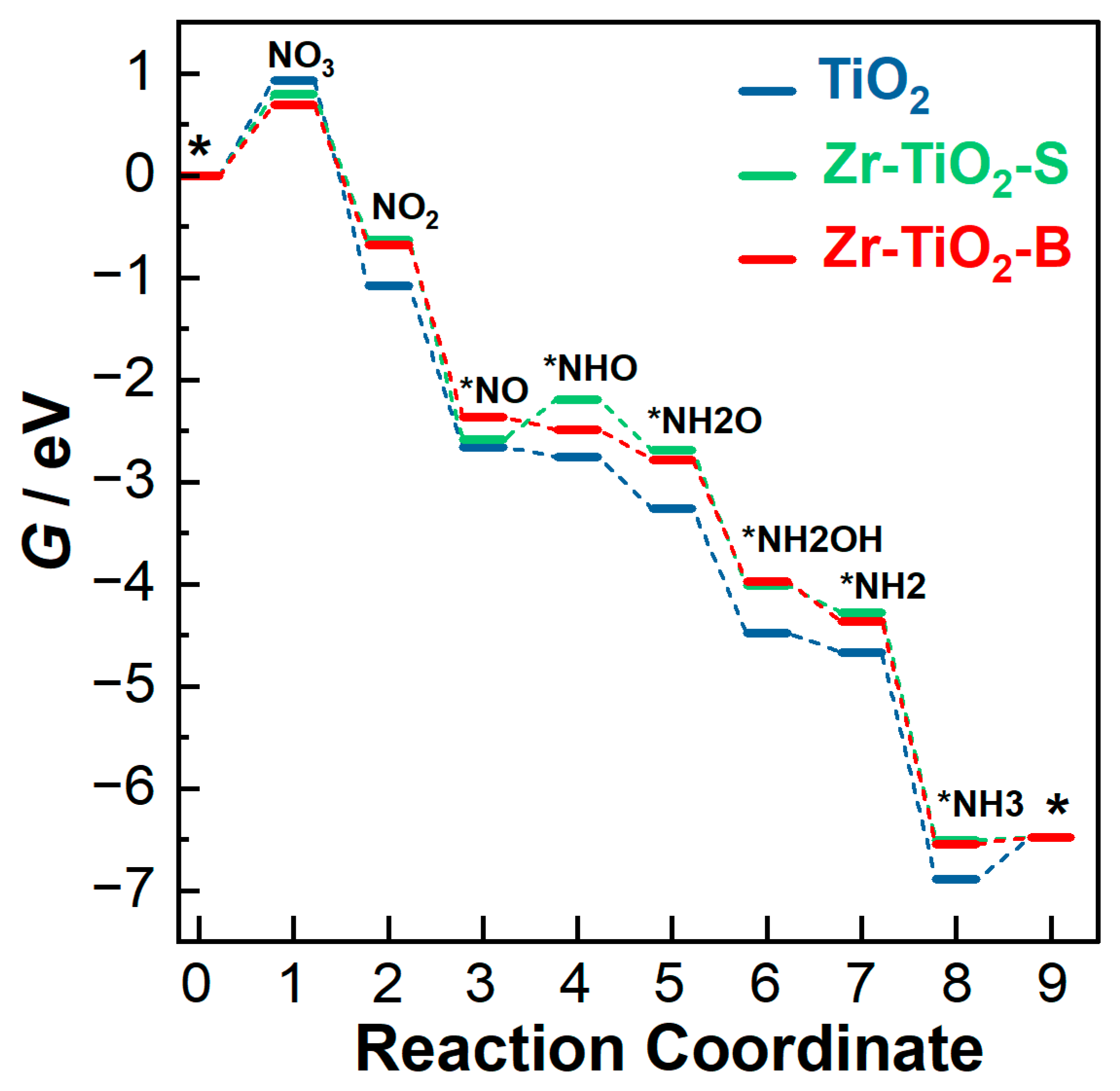
3.3. Reaction Pathways
3.3.1. Undoped TiO2 Model
3.3.2. Zr-TiO2-S Model
3.3.3. Zr-TiO2-B Model
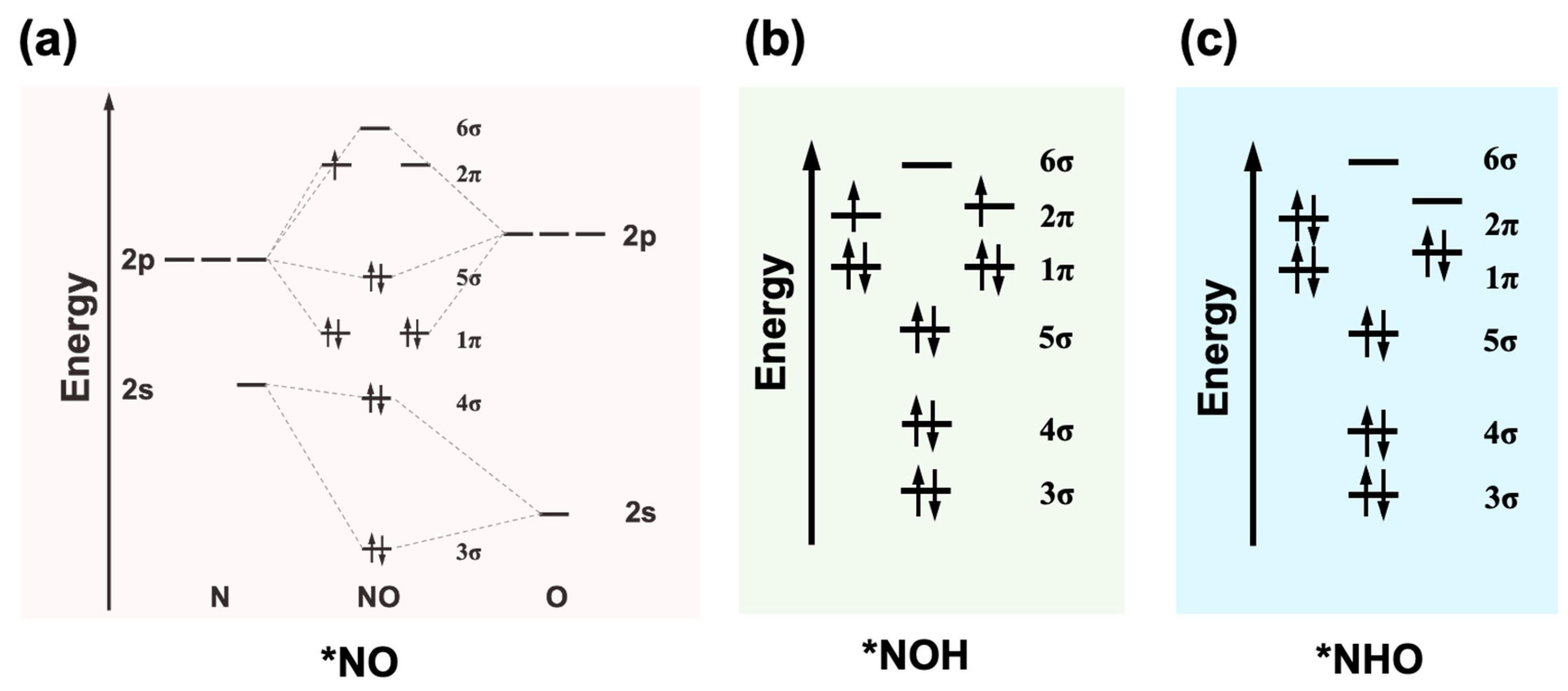
3.4. Adsorption of Intermediates and Hydrogen
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rosca, V.; Duca, M.; de Groot, M.T.; Koper, M.T.M. Nitrogen Cycle Electrocatalysis. Chem. Rev. 2009, 109, 2209–2244. [Google Scholar] [CrossRef] [PubMed]
- Comer, B.M.; Fuentes, P.; Dimkpa, C.O.; Liu, Y.-H.; Fernandez, C.A.; Arora, P.; Realff, M.; Singh, U.; Hatzell, M.C.; Medford, A.J. Prospects and Challenges for Solar Fertilizers. Joule 2019, 3, 1578–1605. [Google Scholar] [CrossRef]
- Martínez, J.; Ortiz, A.; Ortiz, I. State-of-the-art and perspectives of the catalytic and electrocatalytic reduction of aqueous nitrates. Appl. Catal. B Environ. 2017, 207, 42–59. [Google Scholar] [CrossRef]
- Bae, S.-E.; Stewart, K.L.; Gewirth, A.A. Nitrate Adsorption and Reduction on Cu(100) in Acidic Solution. J. Am. Chem. Soc. 2007, 129, 10171–10180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, Z.; Gao, S.; Sun, X.; Zhu, S. CoS2@TiO2 nanoarray: A heterostructured electrocatalyst for high-efficiency nitrate reduction to ammonia. Chem. Commun. 2022, 58, 12995–12998. [Google Scholar] [CrossRef]
- Xu, T.; Zheng, H.; Zhang, P.; Lin, W.; Sekiguchi, Y. Hydrothermal preparation of nanoporous TiO2 films with exposed {001} facets and superior photocatalytic activity. J. Mater. Chem. A 2015, 3, 19115–19122. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.L.; Reller, A. Photoinduced reactivity of titanium dioxide. Prog. Solid State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Jia, R.; Wang, Y.; Wang, C.; Ling, Y.; Yu, Y.; Zhang, B. Boosting selective nitrate electroreduction to ammonium by constructing oxygen vacancies in TiO2. ACS Catal. 2020, 10, 3533–3540. [Google Scholar] [CrossRef]
- Wei, Z.; Niu, X.; Yin, H.; Yu, S.; Li, J. Synergistic effect of oxygen defects and hetero-phase junctions of TiO2 for selective nitrate electroreduction to ammonia. Appl. Catal. A Gen. 2022, 636, 118596. [Google Scholar] [CrossRef]
- Wang, C.; Ge, X.; Fan, H.; Yang, F.; Hu, Q.; Li, J. Simultaneous tuning of particle size and phase composition of TiO2- nanoparticles by a simple liquid immiscibility strategy. J. Mater. Sci. Technol. 2023, 145, 1–6. [Google Scholar] [CrossRef]
- Chen, J.; He, X.; Zhao, D.; Li, J.; Sun, S.; Luo, Y.; Zheng, D.; Li, T.; Liu, Q.; Xie, L.; et al. Greatly enhanced electrochemical nitrate-to-ammonia conversion over an Fe-doped TiO2 nanoribbon array. Green Chem. 2022, 24, 7913–7917. [Google Scholar] [CrossRef]
- Zhao, D.; Ma, C.; Li, J.; Li, R.; Fan, X.; Zhang, L.; Dong, K.; Luo, Y.; Zheng, D.; Sun, S.; et al. Direct eight-electron NO3−-to-NH3 conversion: Using a Co-doped TiO2 nanoribbon array as a high-efficiency electrocatalyst. Inorg. Chem. Front. 2022, 9, 6412–6417. [Google Scholar] [CrossRef]
- Hu, T.; Wang, C.; Wang, M.; Li, C.M.; Guo, C. Theoretical insights into superior nitrate reduction to ammonia performance of copper catalysts. ACS Catal. 2021, 11, 14417–14427. [Google Scholar] [CrossRef]
- Gao, B.; Lim, T.M.; Subagio, P.; Lim, T. Zr-doped TiO2 for enhanced photocatalytic degradation of bisphenol A. Appl. Catal. A Gen. 2010, 375, 107–115. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 31, 751–767. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, D.; Cao, D.; Zeng, X.C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weirich, T.E.; Winterer, M.; Seifried, S.; Mayer, J. Sturcture of nanocrystalline anatase solved and refined from electron powder data. Acta Crystallogr. Sect. A 2002, 58, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhou, C.; Ma, Z.; Ren, Z.; Fan, H.; Yang, X. Elementary photocatalytic chemistry on TiO2 surfaces. Chem. Soc. Rev. 2016, 45, 3701–3730. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, W.; Bi, Z.; Yuan, X.; Zhang, J.; Wu, Y. First-principles study of methanol adsorption and dissociation reactivity on the anatase TiO2 (101) surface: The effect of Co doping and oxygen vacancy. Catal. Lett. 2023, 153, 104–113. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 90th ed.; (CD-ROM Version 2010); CRC Press/Taylor and Francis: Boca Raton, FL, USA, 2010. [Google Scholar]
- Chase, M.W. NIST-JANAF Thermochemical Tables; 2 Volume-Set, Journal of Physical and Chemical Reference Data Monographs; American Institute of Physics: Melville, NY, USA, 1998. [Google Scholar]
- Guo, Y.; Cai, X.; Shen, S.; Wang, G.; Zhang, J. Computational prediction and experimental evaluation of nitrate reduction to ammonia on rhodium. J. Catal. 2021, 402, 1–9. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Huang, M.; Henry, J.B.; Koper, M.T.M.; Bandarenka, A.S. Theoretical design and experimental implementation of Ag/Au electrodes for the electrochemical reduction of nitrate. Phys. Chem. Chem. Phys. 2013, 15, 3196–3202. [Google Scholar] [CrossRef]
- Chen, W.; Yang, X.; Chen, Z.; Ou, Z.; Hu, J.; Xu, Y.; Li, Y.; Ren, X.; Ye, S.; Qiu, J.; et al. Emerging Applications, developments, prospects, and challenges of electrochemical nitrate-to-ammonia conversion. Adv. Funct. Mater. 2023, 33, 2300512. [Google Scholar] [CrossRef]
- Qin, J.; Liu, N.; Wei, Y.; Lu, Y.; Huang, Y.; Zhao, Q.; Ye, Z. The mechanism of efficient photoreduction nitrate over anatase TiO2 in simulated sunlight. Chemosphere 2022, 307, 135921. [Google Scholar] [CrossRef]
- Wang, K.; Gong, Z.; Luo, H.; Jin, K.; Hua, Y.; Wu, W. DRIFT study of the adsorption of NH3 and NOx over rare earth concentrate enriched from Bayan Obo Tailings. Combust. Sci. Technol. 2018, 190, 770–783. [Google Scholar] [CrossRef]
- Chen, G.-F.; Yuan, Y.; Jiang, H.; Ren, S.-Y.; Ding, L.-X.; Ma, L.; Wu, T.; Lu, J.; Wang, H. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 2020, 5, 605–613. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Geng, M.; Zhu, J.; Sun, H.; Jiang, B. Defect-engineered TiO2 nanotube cathode for nitrate reduction to ammonia and upcycling into (NH4)2SO4 in the paired electrolysis system. Appl. Catal. B Environ. 2023, 330, 122658. [Google Scholar] [CrossRef]
- Hoffman, R. Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures; VCH Publishers, Inc.: New York, NY, USA, 1988. [Google Scholar]
- Hughbanks, T.; Hoffmann, R. Chains of trans-edge-sharing molybdenum octahedra: Metal-metal bonding in extended systems. J. Am. Chem. Soc. 1983, 105, 3528–3537. [Google Scholar] [CrossRef]
- Luo, Y.R. Compressive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Sun, Y.; Ren, X.; Sun, S.; Liu, Z.; Xi, S.; Xu, Z.J. Engineering high-spin state cobalt cations in spinel zinc cobalt oxide for spin channel propagation and active site enhancement in water oxidation. Angew. Chem. Int. Ed. 2021, 60, 14536–14544. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Active Site | M-O Bond Length/Å | QATIM Atomic Basin Charge | |||
|---|---|---|---|---|---|---|
| Before | After | Before | After | Variation | ||
| TiO2 | Ti1 | 2.66 | 2.32 | 7.724 | 7.758 | 0.034 |
| Ti2 | 2.85 | 2.73 | 7.724 | 7.741 | 0.017 | |
| Zr-TiO2-S | Zr | 2.25 | 2.62 | 9.058 | 9.090 | 0.032 |
| Ti | 2.26 | 2.21 | 7.744 | 7.735 | −0.009 | |
| Zr-TiO2-B | Zr1 | 1.51 | 2.39 | 9.100 | 9.166 | 0.066 |
| Zr2 | 1.68 | 2.41 | 9.102 | 9.164 | 0.062 | |
| Model | Adsorption Gibbs Free Energy/eV | ||
|---|---|---|---|
| NO2 | NO | H | |
| TiO2 | 0.595 | 0.331 | 0.603 |
| Zr-TiO2-S | 1.043 | 0.405 | 0.352 |
| Zr-TiO2-B | 0.998 | 0.627 | 0.300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Yang, F.; Hu, Q.; Yin, J.; Zhang, J. Nitrate Reduction Reaction on Zr-Doped TiO2 (101) Surfaces Investigated by First-Principles Calculations. Crystals 2023, 13, 1640. https://doi.org/10.3390/cryst13121640
He X, Yang F, Hu Q, Yin J, Zhang J. Nitrate Reduction Reaction on Zr-Doped TiO2 (101) Surfaces Investigated by First-Principles Calculations. Crystals. 2023; 13(12):1640. https://doi.org/10.3390/cryst13121640
Chicago/Turabian StyleHe, Xiyu, Fan Yang, Qiaodan Hu, Jiewei Yin, and Junliang Zhang. 2023. "Nitrate Reduction Reaction on Zr-Doped TiO2 (101) Surfaces Investigated by First-Principles Calculations" Crystals 13, no. 12: 1640. https://doi.org/10.3390/cryst13121640