
How the Hinge Region Affects Interactions between the Catalytic and β-Propeller Domains in Oligopeptidase B
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Production of the Recombinant Protein
2.2. Crystallography, X-ray, and Structural Analysis
2.3. SAXS Experiment
2.4. MD Simulation and Analysis of MD Trajectories
2.5. Comparison of the SAXS Experimental Data with Calculated SAXS Curves
2.6. Data Bank Accession Numbers
3. Results and Discussion
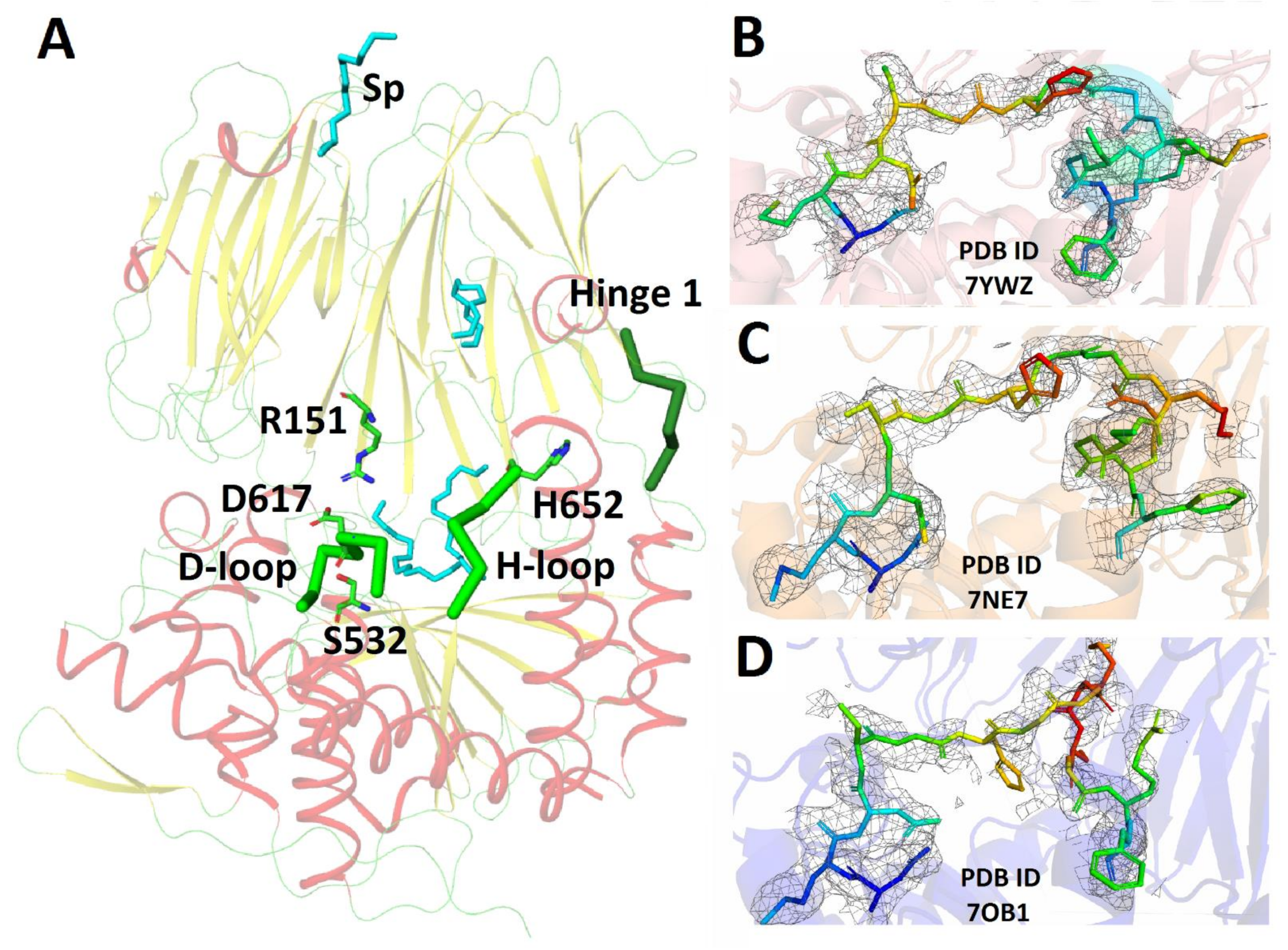
3.1. Crystal Structure of SpOpBmod in the Intermediate Conformation with an Improved Electrone Density in the H-Loop Region
3.2. Generation of the Model of the Open Conformation of SpOpBmod Using MD Simulations Combined with the SAXS Experiment
3.3. Comparison of Interdomain Interactions in Intermediate and Open Conformations of SpOpB with Intact and Modified Hinge Regions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, F.H.; Taylor, R. Librarians, Crystal Structures and Drug Design. Chem. Commun. 2005, 41, 5135–5140. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Maheshwari, S.; McRobb, F.M.; Kinzler, K.W.; Amzel, L.M.; Vogelstein, B.; Gabelli, S.B. Identification of Allosteric Binding Sites for PI3Kα Oncogenic Mutant Specific Inhibitor Design. Bioorganic Med. Chem. 2017, 25, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Agapova, Y.K.; Altukhov, D.A.; Timofeev, V.I.; Stroylov, V.S.; Mityanov, V.S.; Korzhenevskiy, D.A.; Vlaskina, A.V.; Smirnova, E.V.; Bocharov, E.V.; Rakitina, T.V. Structure-Based Inhibitors Targeting the Alpha-Helical Domain of the Spiroplasma Melliferum Histone-like HU Protein. Sci. Rep. 2020, 10, 15128. [Google Scholar] [CrossRef]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Protein Stability, Flexibility and Function. Biochim. Biophys. Acta BBA-Proteins Proteom. 2011, 1814, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Polgár, L. The Prolyl Oligopeptidase Family. Cell. Mol. Life Sci. CMLS 2002, 59, 349–362. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Rea, D.; Fülöp, V. Structure-Function Properties of Prolyl Oligopeptidase Family Enzymes. Cell Biochem. Biophys. 2006, 44, 349–365. [Google Scholar] [CrossRef]
- Fülöp, V.; Böcskei, Z.; Polgár, L. Prolyl Oligopeptidase: An Unusual β-Propeller Domain Regulates Proteolysis. Cell 1998, 94, 161–170. [Google Scholar] [CrossRef]
- Shan, L.; Mathews, I.I.; Khosla, C. Structural and Mechanistic Analysis of Two Prolyl Endopeptidases: Role of Interdomain Dynamics in Catalysis and Specificity. Proc. Natl. Acad. Sci. USA 2005, 102, 3599–3604. [Google Scholar] [CrossRef]
- Li, M.; Chen, C.; Davies, D.; Chiu, T. Induced-Fit Mechanism for Prolyl Endopeptidase. J. Biol. Chem. 2010, 285, 21487–21495. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Rea, D.; Morty, R.E.; Fülöp, V. Crystal Structures of Trypanosoma Brucei Oligopeptidase B Broaden the Paradigm of Catalytic Regulation in Prolyl Oligopeptidase Family Enzymes. PLoS ONE 2013, 8, e79349. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, D.E.; Timofeev, V.I.; Britikov, V.V.; Britikova, E.V.; Kleymenov, S.Y.; Vlaskina, A.V.; Kuranova, I.P.; Mikhailova, A.G.; Rakitina, T.V. First Crystal Structure of Bacterial Oligopeptidase B in an Intermediate State: The Roles of the Hinge Region Modification and Spermine. Biology 2021, 10, 1021. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Paterson, N.G.; Bland, N.D.; Isaacs, N.W.; Mottram, J.C. Crystal Structure of Leishmania Major Oligopeptidase B Gives Insight into the Enzymatic Properties of a Trypanosomatid Virulence Factor*. J. Biol. Chem. 2010, 285, 39249–39259. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, D.E.; Karlinsky, D.M.; Gordeeva, V.D.; Arapidi, G.P.; Britikova, E.V.; Britikov, V.V.; Nikolaeva, A.Y.; Boyko, K.M.; Timofeev, V.I.; Kuranova, I.P.; et al. Crystal Structure of Inhibitor-Bound Bacterial Oligopeptidase B in the Closed State: Similarity and Difference between Protozoan and Bacterial Enzymes. Int. J. Mol. Sci. 2023, 24, 2286. [Google Scholar] [CrossRef]
- Haffner, C.D.; Diaz, C.J.; Miller, A.B.; Reid, R.A.; Madauss, K.P.; Hassell, A.; Hanlon, M.H.; Porter, D.J.T.; Becherer, J.D.; Carter, L.H. Pyrrolidinyl Pyridone and Pyrazinone Analogues as Potent Inhibitors of Prolyl Oligopeptidase (POP). Bioorganic Med. Chem. Lett. 2008, 18, 4360–4363. [Google Scholar] [CrossRef]
- Kaushik, S.; Etchebest, C.; Sowdhamini, R. Decoding the Structural Events in Substrate-Gating Mechanism of Eukaryotic Prolyl Oligopeptidase Using Normal Mode Analysis and Molecular Dynamics Simulations. Proteins Struct. Funct. Bioinform. 2014, 82, 1428–1443. [Google Scholar] [CrossRef]
- Kichik, N.; Tarrago, T.; Claasen, B.; Gairi, M.; Millet, O.; Giralt, E. 15N Relaxation NMR Studies of Prolyl Oligopeptidase, an 80 KDa Enzyme, Reveal a Pre-Existing Equilibrium between Different Conformational States. Chembiochem Eur. J. Chem. Biol. 2011, 12, 2737–2739. [Google Scholar] [CrossRef]
- Szeltner, Z.; Rea, D.; Juhász, T.; Renner, V.; Fülöp, V.; Polgár, L. Concerted Structural Changes in the Peptidase and the Propeller Domains of Prolyl Oligopeptidase Are Required for Substrate Binding. J. Mol. Biol. 2004, 340, 627–637. [Google Scholar] [CrossRef]
- Ellis-Guardiola, K.; Rui, H.; Beckner, R.L.; Srivastava, P.; Sukumar, N.; Roux, B.; Lewis, J.C. Crystal Structure and Conformational Dynamics of Pyrococcus Furiosus Prolyl Oligopeptidase. Biochemistry 2019, 58, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, V.I.; Petrenko, D.E.; Agapova, Y.K.; Vlaskina, A.V.; Karlinsky, D.M.; Mikhailova, A.G.; Kuranova, I.P.; Rakitina, T.V. The Crystal Structure of Nα-p-Tosyl-Lysyl Chloromethylketone-Bound Oligopeptidase B from Serratia Proteamaculans Revealed a New Type of Inhibitor Binding. Crystals 2021, 11, 1438. [Google Scholar] [CrossRef]
- Britikov, V.V.; Timofeev, V.I.; Petrenko, D.E.; Britikova, E.V.; Nikolaeva, A.Y.; Vlaskina, A.V.; Boyko, K.M.; Mikhailova, A.G.; Rakitina, T.V. Elucidation of the Conformational Transition of Oligopeptidase B by an Integrative Approach Based on the Combination of X-Ray, SAXS, and Essential Dynamics Sampling Simulation. Crystals 2022, 12, 712. [Google Scholar] [CrossRef]
- Petrenko, D.E.; Nikolaeva, A.Y.; Lazarenko, V.A.; Dorovatovskii, P.V.; Timofeev, V.I.; Vlaskina, A.V.; Korzhenevskiy, D.A.; Mikhailova, A.G.; Rakitina, T.V. Screening of Conditions That Facilitate Crystallization of Oligopeptidase B from Serratia Proteamaculans by Differential Scanning Fluorimetry. Crystallogr. Rep. 2020, 65, 264–268. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A New Graphical Interface for Diffraction-Image Processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. BALBES: A Molecular-Replacement Pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computational Project, Number 4 The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [CrossRef] [PubMed]
- Diederichs, K.; Karplus, P.A. Improved R-Factors for Diffraction Data Analysis in Macromolecular Crystallography. Nat. Struct. Biol. 1997, 4, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded Functionality and New Tools for Small-Angle Scattering Data Analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Hammersley, A.P.; Svensson, S.O.; Hanfland, M.; Fitch, A.N.; Hausermann, D. Two-Dimensional Detector Software: From Real Detector to Idealised Image or Two-Theta Scan. High Press. Res. 1996, 14, 235–248. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain Fluctuations and Elastic Constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The Effect of Long-Range Electrostatic Interactions in Simulations of Macromolecular Crystals: A Comparison of the Ewald and Truncated List Methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A Comprehensive Data Analysis Suite for Small-Angle Scattering from Macromolecular Solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I.; Feigin, L.A. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Taylor, G.W., Ed.; Plenum Press: New York, NY, USA, 1987; ISBN 978-0-306-42629-2. [Google Scholar]
- Mikhailova, A.G.; Rakitina, T.V.; Timofeev, V.I.; Karlinsky, D.M.; Korzhenevskiy, D.A.; Agapova, Y.K.; Vlaskina, A.V.; Ovchinnikova, M.V.; Gorlenko, V.A.; Rumsh, L.D. Activity Modulation of the Oligopeptidase B from Serratia Proteamaculans by Site-Directed Mutagenesis of Amino Acid Residues Surrounding Catalytic Triad Histidine. Biochimie 2017, 139, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, D.; Mikhailova, A.; Timofeev, V.; Agapova, Y.; Karlinsky, D.; Komolov, A.; Korzhenevskiy, D.; Vlaskina, A.; Rumsh, L.; Rakitina, T. Molecular Dynamics Complemented by Site-Directed Mutagenesis Reveals Significant Difference between the Interdomain Salt Bridge Networks Stabilizing Oligopeptidases B from Bacteria and Protozoa in Their Active Conformations. J. Biomol. Struct. Dyn. 2019, 38, 4868–4882. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Larsen, A.H.; Wang, Y.; Bottaro, S.; Grudinin, S.; Arleth, L.; Lindorff-Larsen, K. Combining Molecular Dynamics Simulations with Small-Angle X-Ray and Neutron Scattering Data to Study Multi-Domain Proteins in Solution. PLoS Comput. Biol. 2020, 16, e1007870. [Google Scholar] [CrossRef]
- He, W.; Henning-Knechtel, A.; Kirmizialtin, S. Visualizing RNA Structures by SAXS-Driven MD Simulations. Front. Bioinform. 2022, 2, 781949. [Google Scholar] [CrossRef]
- Bengtsen, T.; Holm, V.L.; Kjølbye, L.R.; Midtgaard, S.R.; Johansen, N.T.; Tesei, G.; Bottaro, S.; Schiøtt, B.; Arleth, L.; Lindorff-Larsen, K. Structure and Dynamics of a Nanodisc by Integrating NMR, SAXS and SANS Experiments with Molecular Dynamics Simulations. eLife 2020, 9, e56518. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID Protein | 7YWZ SpOpBmod |
|---|---|
| Data Collection | |
| Diffraction source | SPring-8 |
| Wavelength (Å) | 0.8 |
| Temperature (K) | 100 |
| Detector | DECTRIS EIGER X 16 M |
| Space group | P212121 |
| a, b, c (Å) | 72.89, 100.44, 108.55 |
| α, β, γ (°) | 90.0 |
| Unique reflections | 76,338 |
| Resolution range (Å) | 29.51–1.75 (1.80–1.75) |
| Completeness (%) | 99.50 (99.93) |
| Average redundancy | 4.96 (4.87) |
| 〈I/σ(I)〉 | 7.4706 (2.53) |
| Rmrgd-F * (%) | 6.6 (29) |
| Willson B | 18.3 |
| Refinement | |
| Rfact (%) | 17.3 |
| Rfree (%) | 20.2 |
| Rfree set size (%) | 5 |
| RMSD of bonds (Å) | 0.011 |
| RMSD of angles (°) | 1.73 |
| Ramachandran plot | |
| Most favored (%) | 99.6 |
| Allowed (%) | 0.4 |
| No. atoms | |
| Protein | 5545 |
| Water | 562 |
| Ligands | 62 |
| B-factor (Å2) | |
| Average | 22.0 |
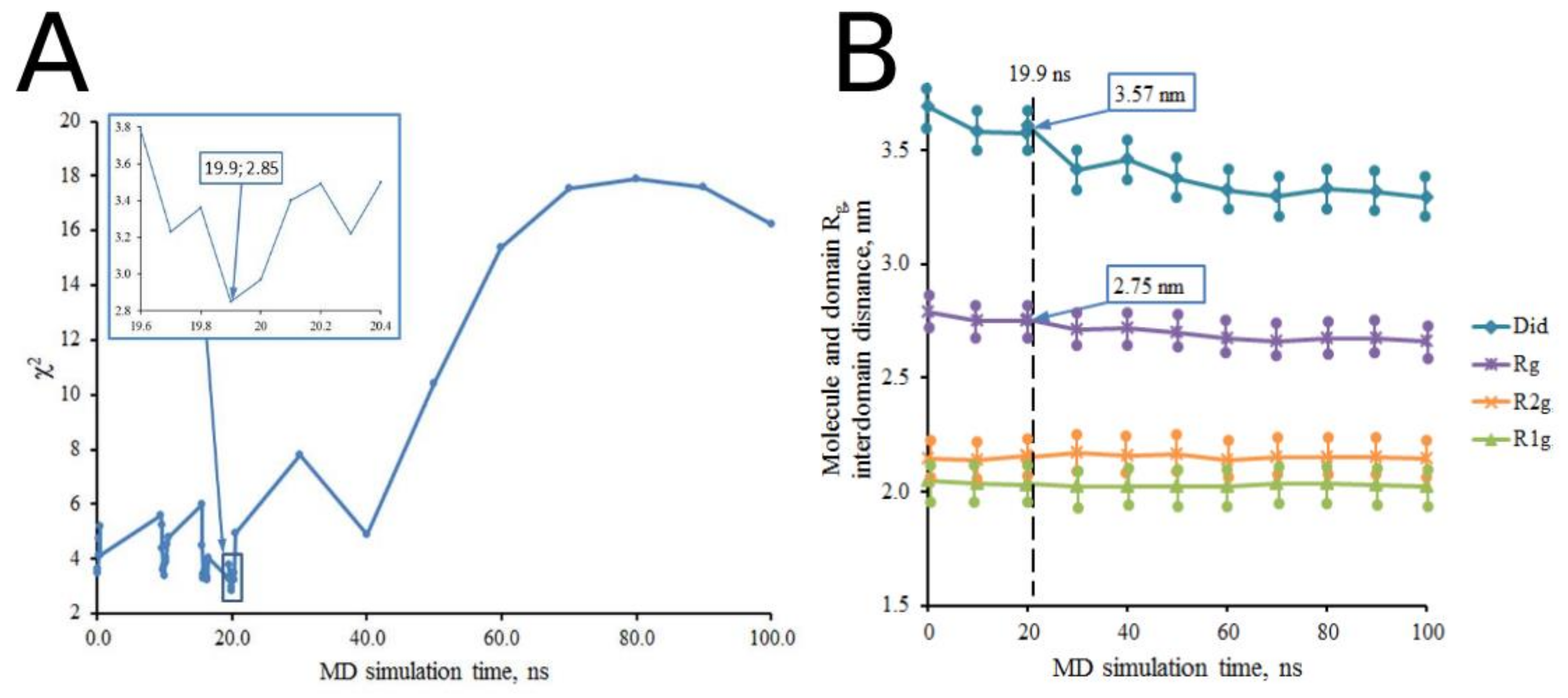
| Sample Description | χ2 | Rg, nm (ΔRg = ±0.03 nm) | Dmax, nm (ΔDmax = ±0.1 nm) | Did, nm (ΔDid = ±0.01 nm) | R1g and R2g, nm (ΔRg = ±0.03 nm) | ||
|---|---|---|---|---|---|---|---|
| R1g | R2g | ||||||
| Data from experimental SAXS curve | |||||||
| Solution | 2.76 | 8.7 | |||||
| Data from model SAXS curves calculated for starting structures | |||||||
| Open conformation | 3.36 | 2.78 | 8.7 | 3.69 | 2.04 | 2.14 | |
| Intermediate conformation | 18.12 | 2.64 | 7.9 | 3.23 | 2.01 | 2.12 | |
| Closed conformation | 31.62 | 2.57 | 7.9 | 3.05 | 2.00 | 2.11 | |
| Data from model SAXS curves calculated for structures derived from MD experiments | |||||||
| Open | t = 0 ns | 3.49 | 2.79 | 8.7 | 3.69 | 2.05 | 2.14 |
| Main cluster * | 17.20 | 2.68 | 8.2 | 3.32 | 2.03 | 2.16 | |
| t = 10 ns | 3.37 | 2.75 | 8.2 | 3.58 | 2.04 | 2.14 | |
| t = 20 ns | 2.97 | 2.76 | 8.3 | 3.61 | 2.03 | 2.15 | |
| t = 30 ns | 7.82 | 2.71 | 8.4 | 3.42 | 2.03 | 2.17 | |
| t = 40 ns | 4.87 | 2.72 | 8.2 | 3.46 | 2.03 | 2.16 | |
| t = 50 ns | 10.42 | 2.70 | 8.4 | 3.38 | 2.02 | 2.17 | |
| t = 60 ns | 15.38 | 2.67 | 8.5 | 3.32 | 2.03 | 2.14 | |
| t = 70 ns | 17.56 | 2.66 | 8.1 | 3.30 | 2.04 | 2.15 | |
| t = 80 ns | 17.88 | 2.67 | 8.3 | 3.33 | 2.04 | 2.15 | |
| t = 90 ns | 17.60 | 2.67 | 8.1 | 3.32 | 2.03 | 2.15 | |
| t = 100 ns | 16.22 | 2.66 | 8.4 | 3.29 | 2.02 | 2.15 | |
| Best structure (SpOpBmodOpen) | |||||||
| t = 19.9 ns | 2.85 | 2.75 | 8.4 | 3.57 | 2.03 | 2.16 | |
| Intermediate | t = 0 ns | 18.12 | 2.64 | 7.9 | 3.23 | 2.01 | 2.12 |
| Main cluster * | 19.23 | 2.67 | 8.3 | 3.28 | 2.04 | 2.14 | |
| Closed | t = 0 ns | 31.62 | 2.57 | 7.9 | 3.05 | 2.00 | 2.11 |
| Main cluster * | 28.96 | 2.61 | 8.0 | 3.15 | 2.03 | 2.14 | |
| Crystal Structures Domains | SpOpBmod (7YWZ) | SpOpB-S532A (7ZJZ) | SpOpBmod-TCK (7NE7) | ||||
|---|---|---|---|---|---|---|---|
| Propeller | Catalytic | Atom 1 prop. | Atom 2 cat. | Atom 1 prop. | Atom 2 cat. | Atom 1 prop. | Atom 2 cat. |
| Hinge1 | α2 | E71N | V68O | I71N | V68O | E71N | V68O |
| β5/β6, Blade 1 | H-loop (α12) | E92O | (K655NZ) | ||||
| P93O | (K655NZ) | ||||||
| N95O | (K655NZ) | ||||||
| E96OE2 | H652ND1 | E96OE2 | H652NE2 | ||||
| E96OE2 | (K655N) | E96OE2 | (K655N) | ||||
| E96O | (K655N) | E96O | (K655N) | ||||
| E96OE2 | (S656N) | E96OE2 | (S656N) | ||||
| E96OE2 | (S656OG) | E96O | S656OG | E96OE1 | (S656OG) | ||
| E96O | (R658NH2) | ||||||
| Y97OH | S656N | ||||||
| Blade 1/ Blade 2 | A121O | K655NZ | |||||
| R124O | K655NZ | ||||||
| β9/β10, Blade 2 | S149OG | S650O | S149OG | S650O | |||
| S149OG | G651O | ||||||
| D-loop | R151NH1 | D617OD2 | R151NH2 | D617OD1 | |||
| β17/β18, Blade 4 | α8/α9 | T244OG1 | D578OD2 | T244OG1 | D578OD2 | ||
| Blade 4/ Blade 5 | K269NZ | D578OD½ | K269NZ | D578OD½ | |||
| β21/β22, Blade 5 | α5 | K291NZ | E494OE½ | K291NZ | E494OE½ | K291NZ | E494OE1 |
| N292OD1 | Q490N | N292OD1 | Q490N | N292OD1 | Q490N | ||
| β24, Blade 6 | M317SD | Q490N | M317SD | Q490N | M317SD | Q490N | |
| β25/β26, Blade 6 | β34/α4 | R333NH½ | D460OD½ | R333NH½ | D460OD½ | R333NH½ | D460OD½ |
| β32 | G336O | R418NH1 | G336O | R418NH1 | G336O | R418NH1 | |
| Blade 6/ Blade 7 | β34/α4 | T361N/OG1 | P461O | T361N/OG1 | P461O | T361N/OG1 | P461O |
| β29/β30, Blade7 | S380OG | F463N | S380OG | F463N | S380OG | F463N | |
| β33 | M382SD | L433N | M382SD | L433N | M382SD | L433N | |
| Hinge2 | α2 | K407N | R70O | K407N | R70O | K407NE | R70O |
| η6 | T410O | N413N | T410O | N413N | T410O | N413N | |
| T410OG1 | N413ND2 | ||||||
| Models Domains | SpOpBmodOpen | SpOpBopen | |||
|---|---|---|---|---|---|
| Propeller | Catalytic | Atom 1 prop. | Atom 2 cat. | Atom 1 prop. | Atom 2 cat. |
| β21/β22, Blade 5 | α5 (β35/α5) | K291NZ N292ND2 | E494OE2 (L488O) | K291NZ K291O | E494OE1 Q490NE2 |
| N292OD1 | Q490N | N292OD1 | Q490N | ||
| N292OD1 | L491N | N292OD1 | L491N | ||
| β25, Blade 6 | β34/α4 (β35/α5) | R333NH½ R333NH1 | D460OD½ (E487O) | R333NH½ R333NH2 R333NH1 | D460OD½ S458OG (E487O) |
| β32 | E335O | R418NH½ | |||
| Blade 6/ | T359OG1 | S416OG | D357OD1 | S416OG | |
| Blade 7 | β34/α4 | T361N/OG1 | P461O | T361N/OG1 | P461O |
| β29/β30, Blade7 | S380O/OG M382SD M382SD | F463N A462N R467NH1 | S380OG | F463N | |
| Hinge2 | α2 N-loop | K407N K407O N408O/OD1 | R70O R70NE R70NH1 | K407N K407O N408O | R70O R70NE R70NH1 |
| η6 | T410O | N413N | |||
| N-loop | T410OG1 | N412N | T410OG1 | N412N | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timofeev, V.I.; Gaponov, Y.A.; Petrenko, D.E.; Peters, G.S.; Agapova, Y.K.; Nikolaeva, A.Y.; Mikhailova, A.G.; Rakitina, T.V. How the Hinge Region Affects Interactions between the Catalytic and β-Propeller Domains in Oligopeptidase B. Crystals 2023, 13, 1642. https://doi.org/10.3390/cryst13121642
Timofeev VI, Gaponov YA, Petrenko DE, Peters GS, Agapova YK, Nikolaeva AY, Mikhailova AG, Rakitina TV. How the Hinge Region Affects Interactions between the Catalytic and β-Propeller Domains in Oligopeptidase B. Crystals. 2023; 13(12):1642. https://doi.org/10.3390/cryst13121642
Chicago/Turabian StyleTimofeev, Vladimir I., Yury A. Gaponov, Dmitry E. Petrenko, Georgy S. Peters, Yulia K. Agapova, Alena Y. Nikolaeva, Anna G. Mikhailova, and Tatiana V. Rakitina. 2023. "How the Hinge Region Affects Interactions between the Catalytic and β-Propeller Domains in Oligopeptidase B" Crystals 13, no. 12: 1642. https://doi.org/10.3390/cryst13121642