

Supramolecular Assemblies of 3/4-Chlorobenzoic Acid and Amino-Chloropyridine Derivatives: Synthesis, X-ray Diffraction, DFT Calculations, and Biological Screening

 and
and 
Abstract
:1. Introduction
2. Results and Discussion
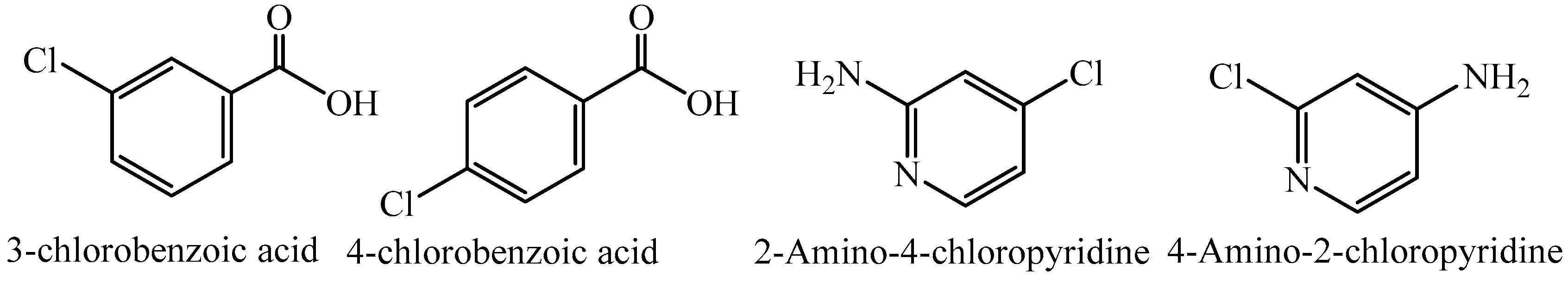
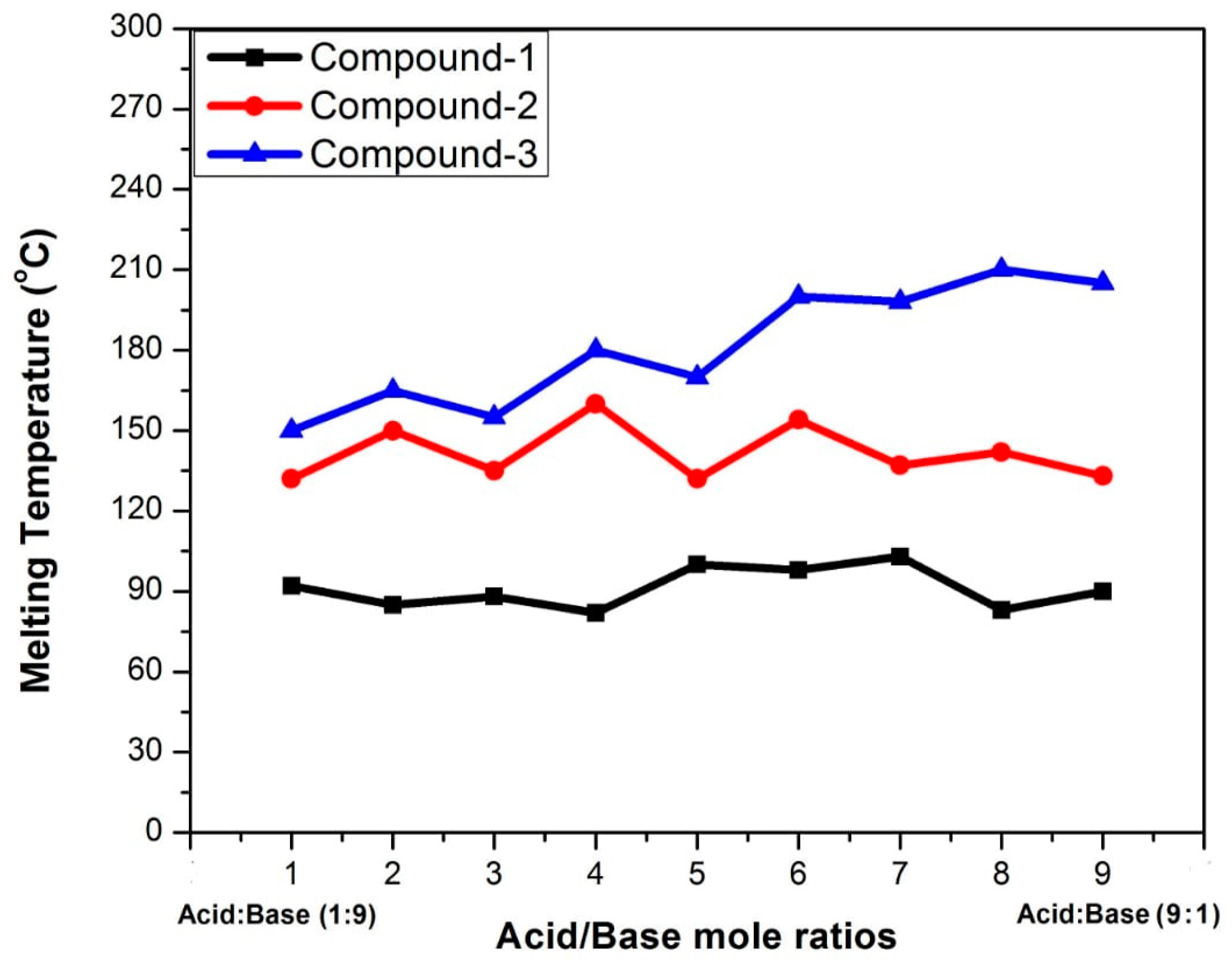
2.1. Physical Phase Diagram of the Starting Materials of Compounds 1, 2, and 3
2.2. FT-IR Studies of Compounds 1, 2 and 3
2.3. Powder and Single X-Ray Diffraction Studies of Compounds 1–3
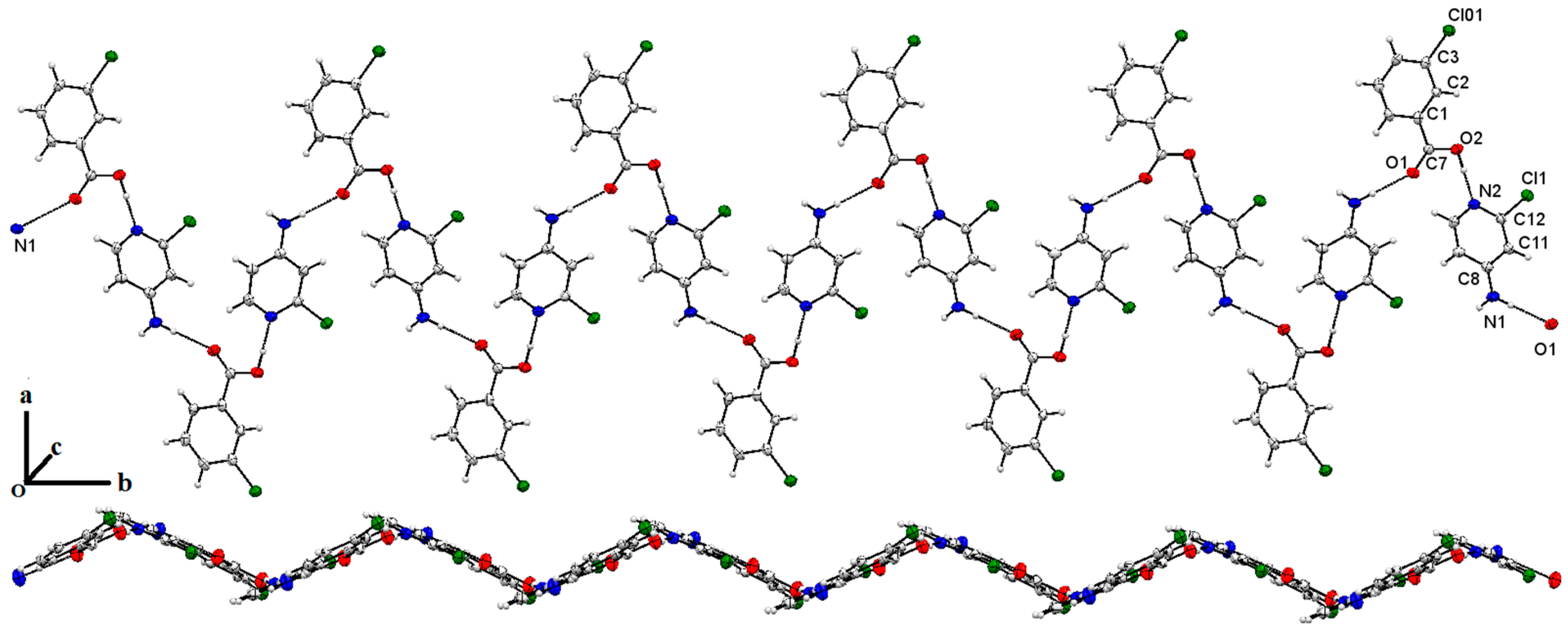
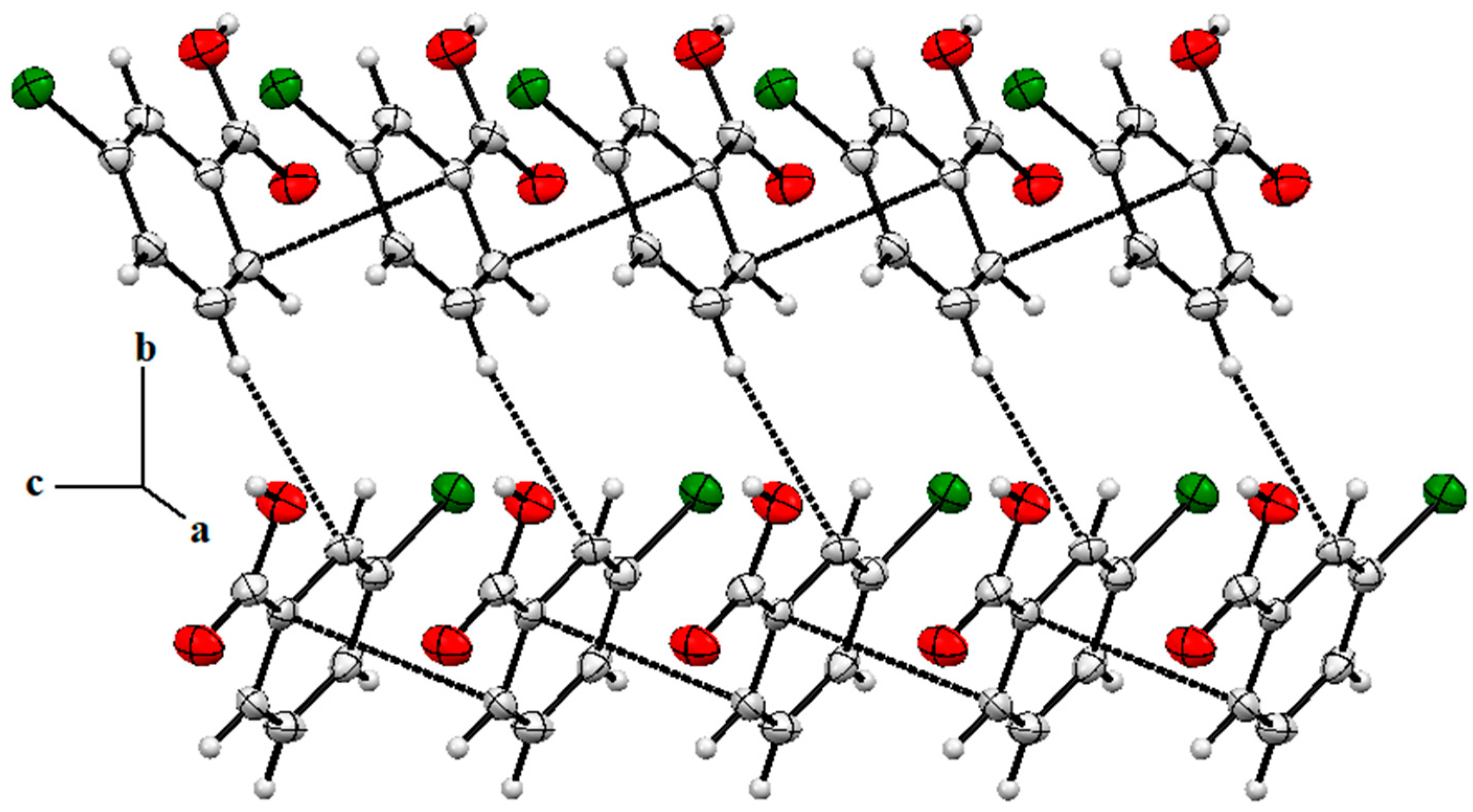
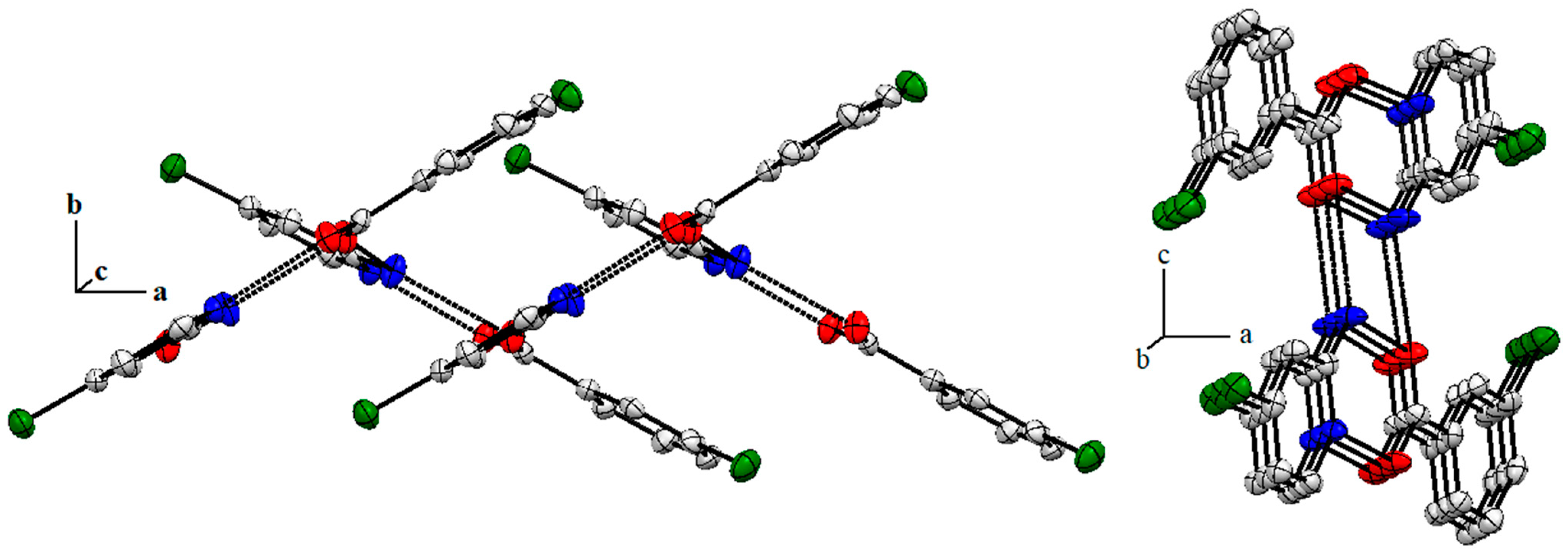
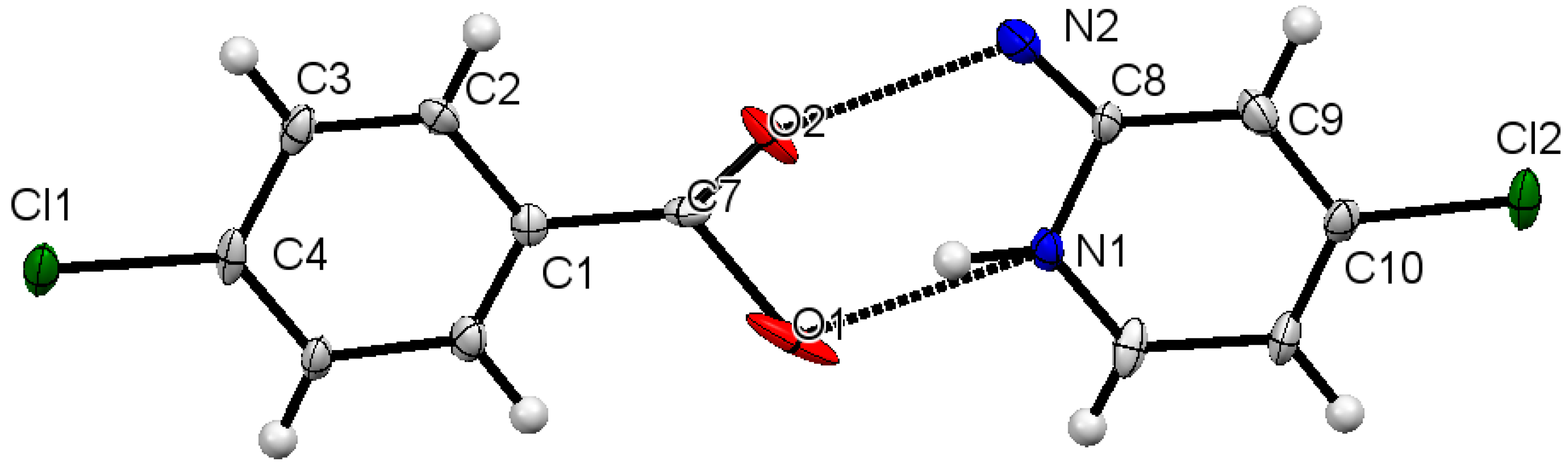
2.4. Single X-Ray Structural Description of Cocrystal of 3-Chlorobenzoic Acid:4-Amino-2-Chloropyridine, Compound 1
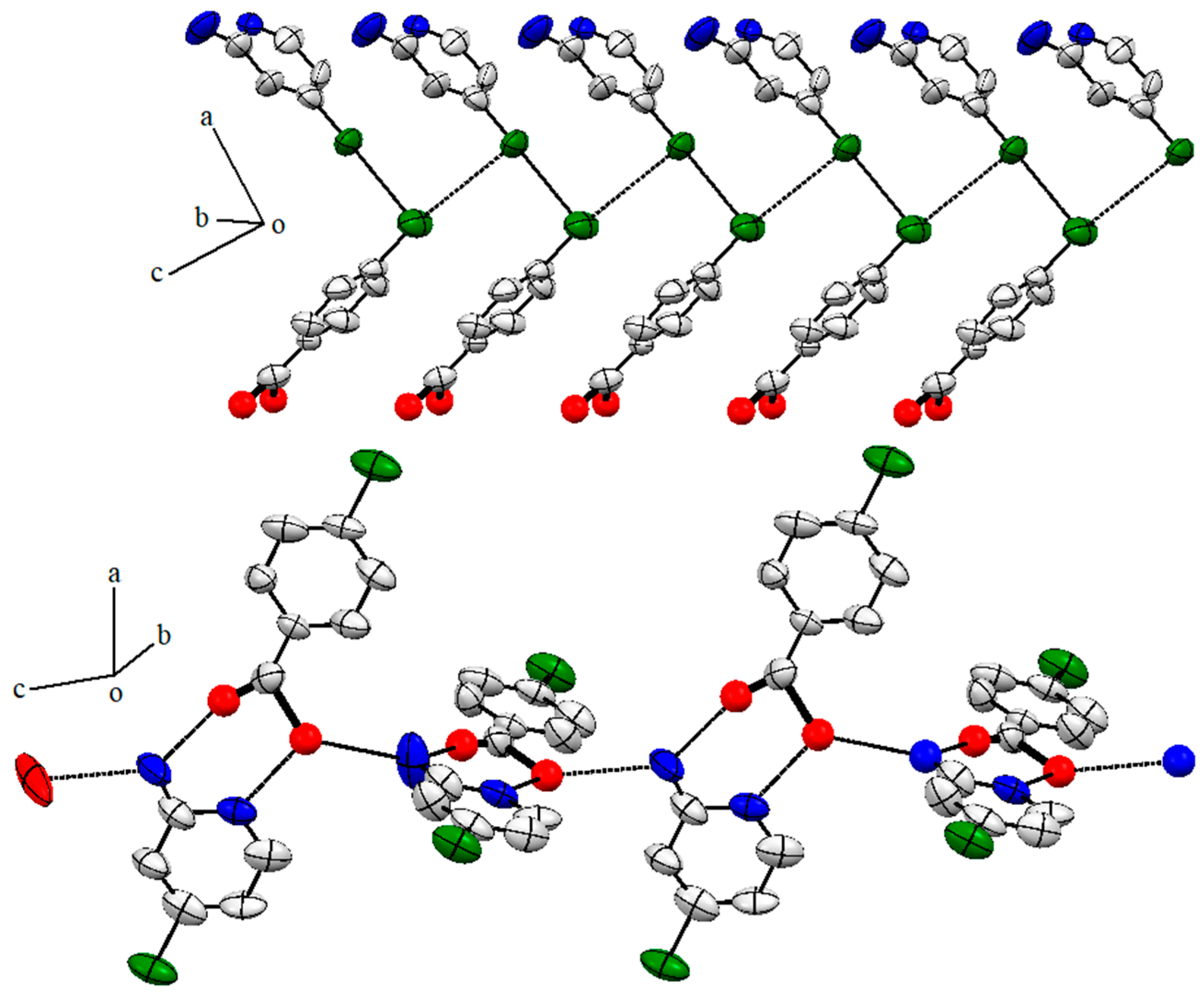
2.5. Single X-ray of Compound 2 (2-Amino-4-Chloropyridinium 3-Chlorobenzoate)
2.6. Structure Description of Compound 3 (2-Amino-4-Chloropyridinium 4-Chlorobenzoate)
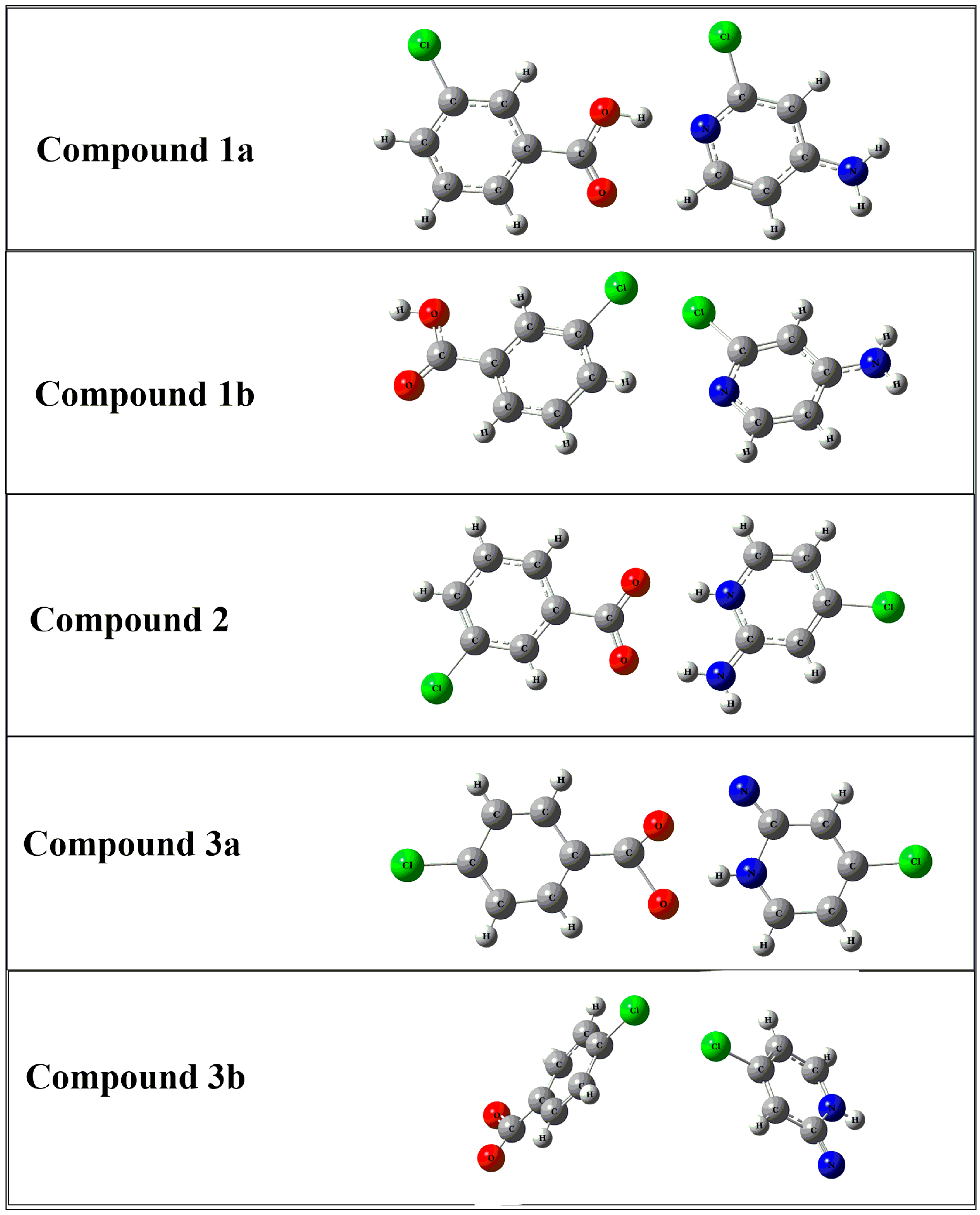
3. Supramolecular Interactions via DFT Calculations
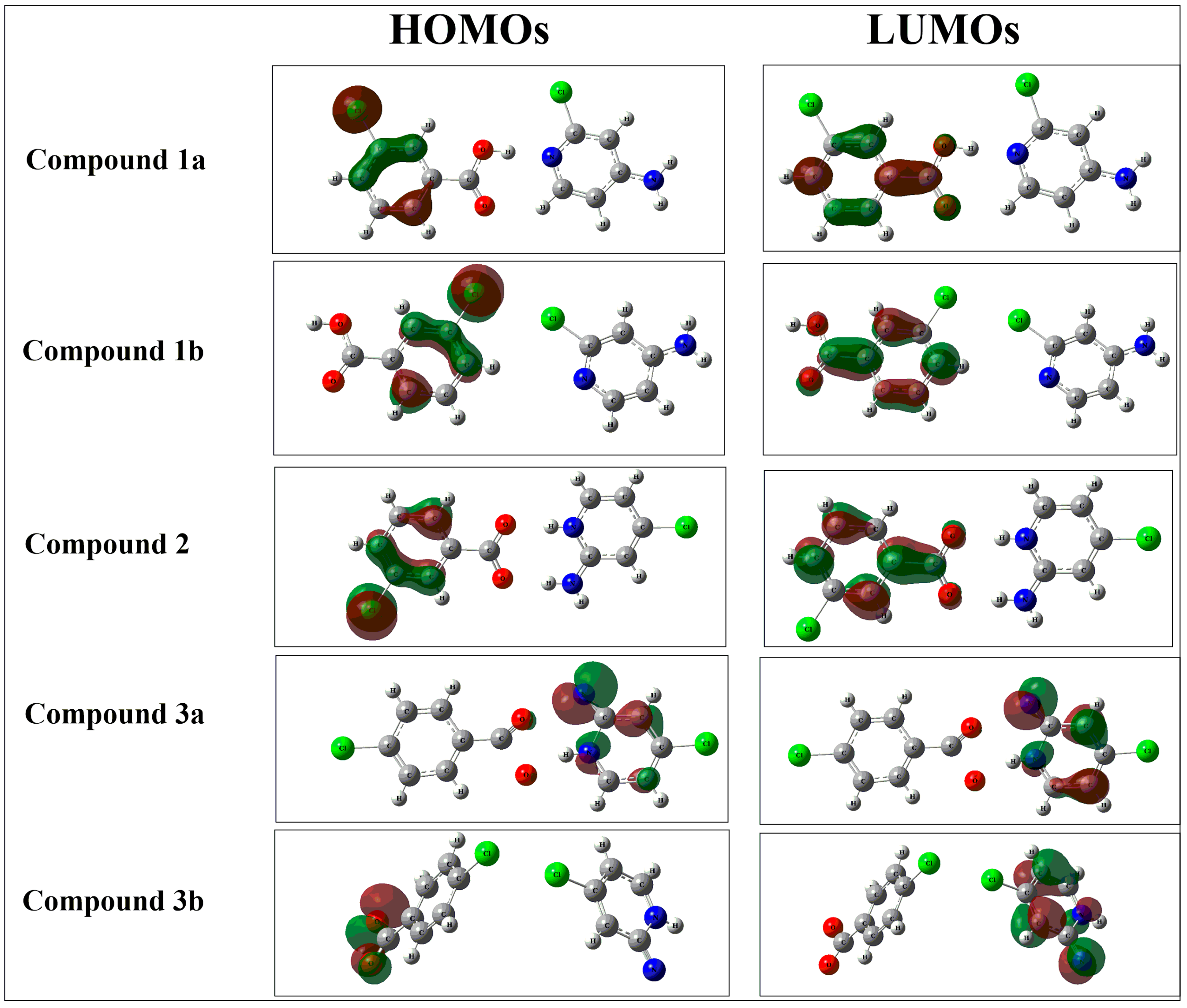
3.1. Frontier Molecular Orbital (MO) Analysis
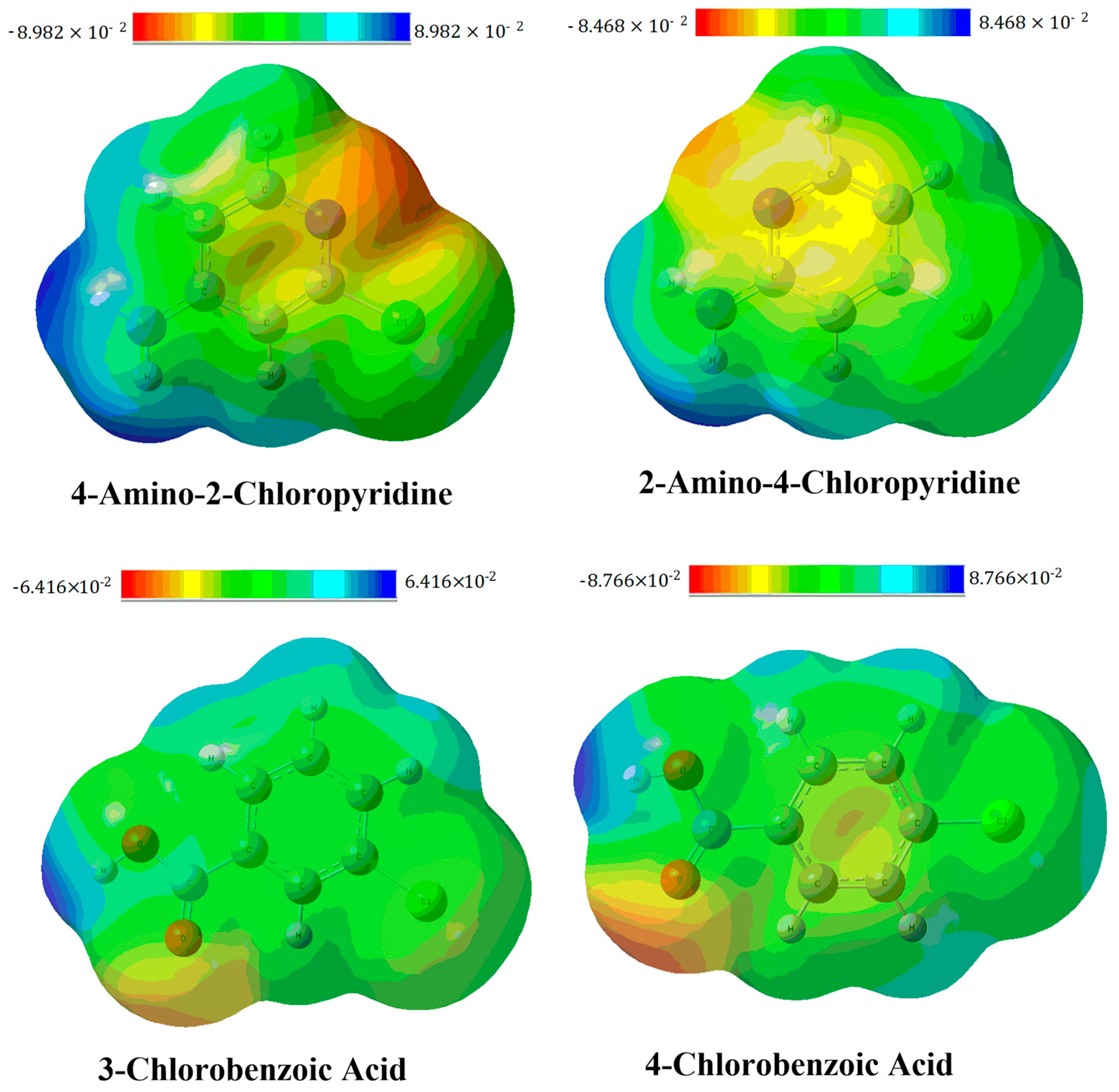
3.2. Molecular Electrostatic Potential (MEP) Analysis
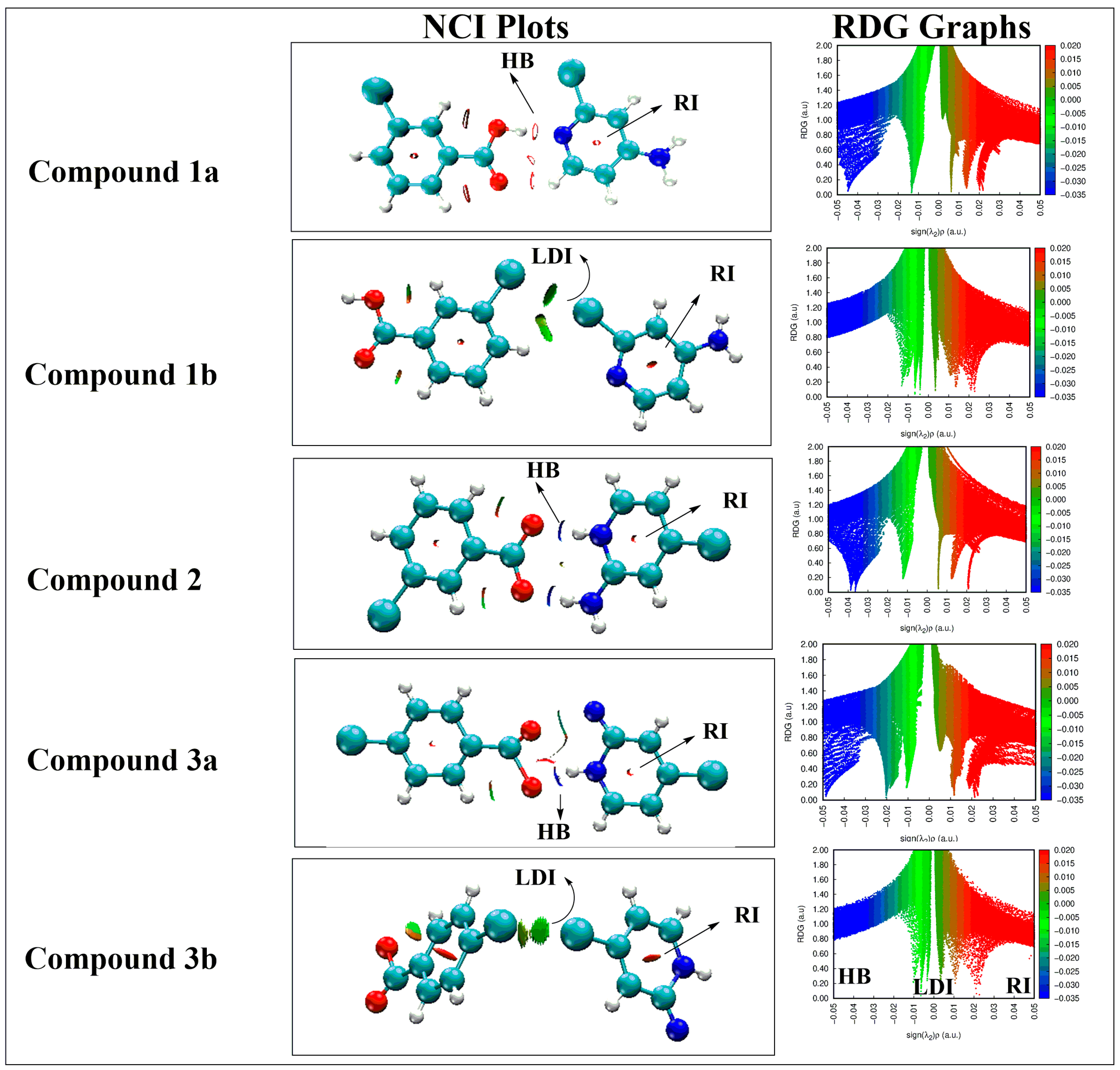
3.3. Noncovalent Interaction (NCI) Analysis
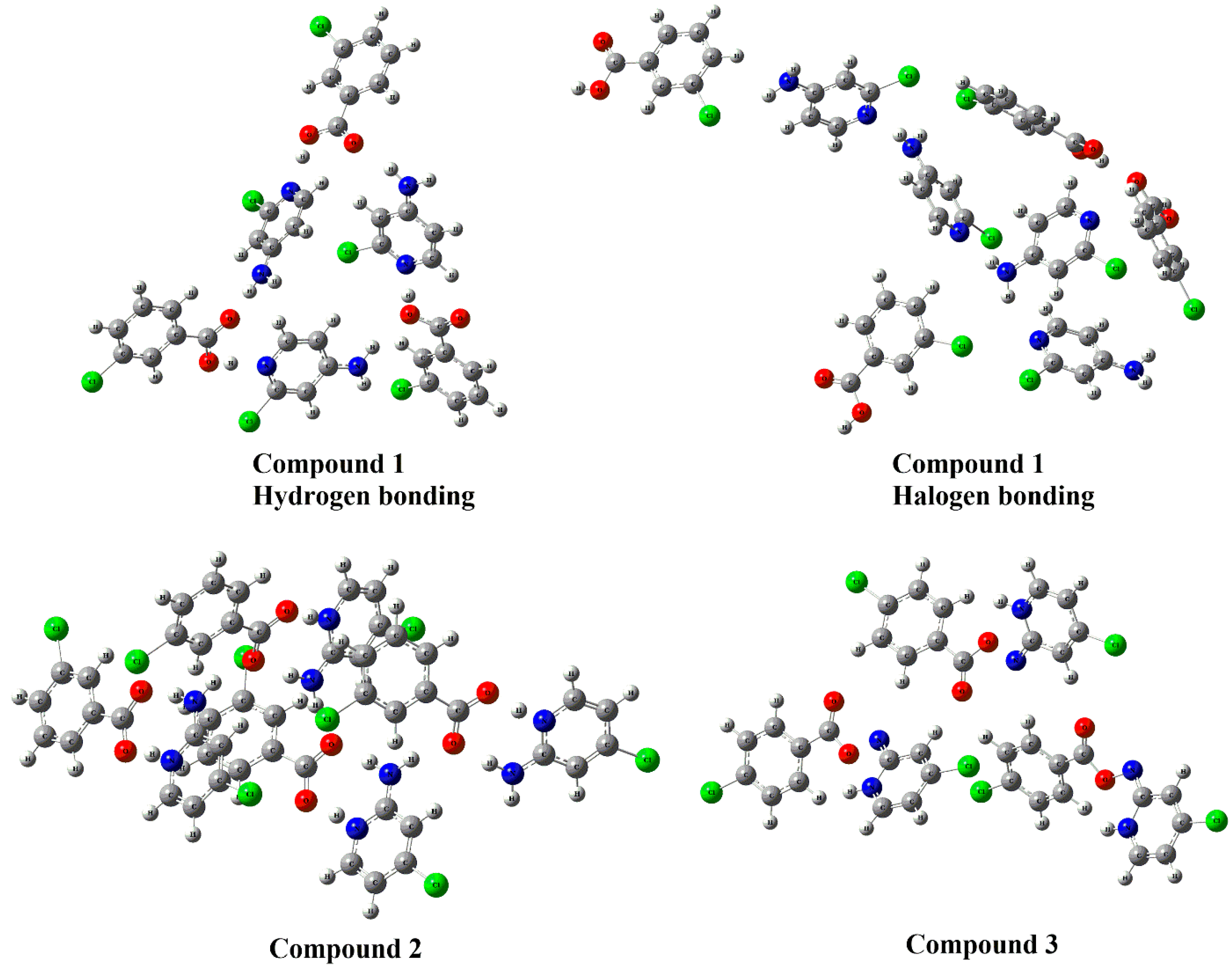
3.4. DFT Studies for Clusters of Compounds 1–3
3.5. UV Absorbance Spectra
4. Biological Applications of Compounds
4.1. Antioxidant Essay (DPPH)
4.2. Anti-Bacterial Study of Compounds 1, 2 and 3 and Their Comparison with Coformers
5. Materials and Methods
5.1. General Considerations and Instrumentations
5.2. Synthesis of Compound 1, (Cocrystal of 3-Chlorobenzoic Acid:4-Amino-2-Chloropyridine)
5.3. Synthesis of 2-Amino-4-Chloropyridinium 3-Chlorobenzoate (2) and 2-Amino-4-Chloropyridinium 4-Chlorobenzoate (3)
5.4. Computational Methodology
5.5. In Vitro Antibacterial Study
5.6. Determination of Antioxidant Potential
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Childs, S.L.; Stahly, G.P.; Park, A. The salt−cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in single-and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef]
- Stahl, P.H.; Wermuth, C.G. Handbook of pharmaceutical salts: Properties, selection and use. Chem. Int. 2002, 24, 21. [Google Scholar]
- Brittain, H.G. Strategy for the Prediction and Selection of Drug Substance Salt Forms. Pharm. Technol. 2007, 31, 78–84. [Google Scholar]
- Lemmerer, A.; Esterhuysen, C.; Bernstein, J. Synthesis characterization molecular modeling of a pharmaceutical co-crystal:(2-chloro-4-nitrobenzoic acid):(nicotinamide). J. Pharm. Sci. 2010, 99, 4054–4071. [Google Scholar] [CrossRef] [PubMed]
- Santra, R.; Ghosh, N.; Biradha, K. Crystal engineering with acid and pyridine heteromeric synthon: Neutral and ionic co-crystals. New J. Chem. 2008, 32, 1673–1676. [Google Scholar] [CrossRef]
- Bosch, E. Chain-link hydrogen-bonded capsules. CrystEngComm 2007, 9, 191–198. [Google Scholar] [CrossRef]
- Childs, S.L.; Hardcastle, K.I. Cocrystals of piroxicam with carboxylic acids. Cryst. Growth Des. 2007, 7, 1291–1304. [Google Scholar] [CrossRef]
- Wenger, M.; Bernstein, J. Designing a Cocrystal of γ-Amino Butyric Acid. Angew. Chem. Int. Ed. 2006, 45, 7966–7969. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. CrystEngComm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Proserpio, D.M.; Resnati, G. Highly Interpenetrated Supramolecular Networks Supported by N I Halogen Bonding. Chem. Eur. J. 2007, 13, 5765–5772. [Google Scholar] [CrossRef] [PubMed]
- Cincic, D.; Friscic, T.; Jones, W. A stepwise mechanism for the mechanochemical synthesis of halogen-bonded cocrystal architectures. J. Am. Chem. Soc. 2008, 130, 7524–7525. [Google Scholar] [CrossRef] [PubMed]
- Cinčić, D.; Friščić, T.; Jones, W. Isostructural Materials Achieved by Using Structurally Equivalent Donors and Acceptors in Halogen-Bonded Cocrystals. Chem. A Eur. J. 2008, 14, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Shirman, T.; Freeman, D.; Posner, Y.D.; Feldman, I.; Facchetti, A.; van der Boom, M.E. Assembly of crystalline halogen-bonded materials by physical vapor deposition. J. Am. Chem. Soc. 2008, 130, 8162–8163. [Google Scholar] [CrossRef] [PubMed]
- Barooah, N.; Sarma, R.J.; Baruah, J.B. Solid-state hydrogen bonded assembly of N, N′-bis (glycinyl)-pyromellitic diimide with aromatic guests. CrystEngComm 2006, 8, 608–615. [Google Scholar] [CrossRef]
- Nishio, M.; Hirota, M.; Umezawa, Y. The CH/π Interaction: Evidence, Nature, and Consequences; John Wiley & Sons: Hoboken, NJ, USA, 1998; Volume 21. [Google Scholar]
- Fedorov, A.Y.; Rychkov, D. Comparison of different computational approaches for unveiling the high-pressure behavior of organic crystals at a molecular level. Case study of tolazamide polymorphs. J. Struct. Chem. 2020, 61, 1356–1366. [Google Scholar] [CrossRef]
- Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478. [Google Scholar]
- Aakeroy, C.B.; Chopade, P.D.; Desper, J. Establishing a hierarchy of halogen bonding by engineering crystals without disorder. Cryst. Growth Des. 2013, 13, 4145–4150. [Google Scholar] [CrossRef]
- Karanam, M.; Choudhury, A.R. Study of halogen-mediated weak interactions in a series of halogen-substituted azobenzenes. Cryst. Growth Des. 2013, 13, 4803–4814. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen versus hydrogen. Science 2008, 321, 918–919. [Google Scholar] [CrossRef]
- Cardillo, P.; Corradi, E.; Lunghi, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G. The N···I intermolecular interaction as a general protocol for the formation of perfluorocarbon–hydrocarbon supramolecular architectures. Tetrahedron 2000, 56, 5535–5550. [Google Scholar] [CrossRef]
- Jennifer, S.J.; Muthiah, P.T. Design of co-crystals/salts of some Nitrogenous bases and some derivatives of thiophene carboxylic acids through a combination of hydrogen and halogen bonds. Chem. Cent. J. 2014, 8, 1–22. [Google Scholar] [CrossRef]
- Prri, A.; Cabriella, G.; Metrabgolo, P.; Resbati, G. The halogen bond in the design of functional supramolecular materials: Recent advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar]
- De Santis, A.; Forni, A.; Liantonio, R.; Metrangolo, P.; Pilati, T.; Resnati, G. N···Br Halogen Bonding: One-Dimensional Infinite Chains through the Self-Assembly of Dibromotetrafluorobenzenes with Dipyridyl Derivatives. Chem.—A Eur. J. 2003, 9, 3974–3983. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bilewicz, E.; Rybarczyk-Pirek, A.J.; Dubis, A.T.; Grabowski, S.J. Halogen bonding in crystal structure of 1-methylpyrrol-2-yl trichloromethyl ketone. J. Mol. Struct. 2007, 829, 208–211. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole bond vs π-hole bond: A comparison based on halogen bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. σ-Hole bonding: A physical interpretation. In Halogen Bonding I; Springer International Publishing A.G. Switzerland: Cham, Switzerland, 2014; pp. 19–42. [Google Scholar]
- Bui, T.T.T.; Dahaoui, S.; Lecomte, C.; Desiraju, G.R.; Espinosa, E. The Nature of Halogen···Halogen Interactions: A Model Derived from Experimental Charge-Density Analysis. Angew. Chem. 2009, 121, 3896–3899. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Moussa, N.A.M. Unconventional Type III Halogen···Halogen Interactions: A Quantum Mechanical Elucidation of σ-Hole···σ-Hole and Di-σ-Hole Interactions. ACS Omega 2020, 5, 21824–21835. [Google Scholar] [CrossRef] [PubMed]
- Baldrighi, M.; Cavallo, G.; Chierotti, M.R.; Gobetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding and pharmaceutical cocrystals: The case of a widely used preservative. Mol. Pharm. 2013, 10, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- La Motta, C.; Sartini, S.; Mugnaini, L.; Salerno, S.; Simorini, F.; Taliani, S.; Marini, A.M.; Da Settimo, F.; Lavecchia, A.; Novellino, E. Exploiting the pyrazolo [3, 4-d] pyrimidin-4-one ring system as a useful template to obtain potent adenosine deaminase inhibitors. J. Med. Chem. 2009, 52, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Khan, E. Pyridine Derivatives as Biologically Active Precursors; Organics and Selected Coordination Complexes. ChemistrySelect 2021, 6, 3041–3064. [Google Scholar] [CrossRef]
- Ullah, I.; Khan, E.; Zhang, Z.; Xiang, S.; Chen, C.; Li, L. Synthesis, crystal growth and supramolecular chemistry of 4-dimethylaminopyridinium salts of benzoates and a phenolate ion. Z. Phys. Chem. 2023, 237, 1381–1408. [Google Scholar] [CrossRef]
- Khan, E.; Khan, A.; Gul, Z.; Ullah, F.; Tahir, M.N.; Khalid, M.; Asif, H.M.; Asim, S.; Braga, A.A.C. Molecular salts of terephthalic acids with 2-aminopyridine and 2-aminothiazole derivatives as potential antioxidant agents; Base-Acid-Base type architectures. J. Mol. Struct. 2020, 1200, 127126. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Smirnov, S.N.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. NMR studies of solid state—Solvent and H/D isotope effects on hydrogen bond geometries of 1: 1 complexes of collidine with carboxylic acids. J. Mol. Struct. 2004, 700, 19–27. [Google Scholar] [CrossRef]
- Ishida, H.; Fukunaga, T. 3-Cyanopyridine–2-chloro-4-nitrobenzoic acid (1/1). Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, o1664–o1665. [Google Scholar] [CrossRef]
- Oruganti, M.; Nechipadappu, S.K.; Khade, P.A.; Trivedi, D.R. Solid-state versatility of the molecular salts/cocrystals of 2-chloro-4-nitrobenzoic acid: A case study on halogen bonds. ACS Omega 2017, 2, 7146–7162. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the p K a rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Delori, A.; Galek, P.T.; Pidcock, E.; Patni, M.; Jones, W. Knowledge-based hydrogen bond prediction and the synthesis of salts and cocrystals of the anti-malarial drug pyrimethamine with various drug and GRAS molecules. CrystEngComm 2013, 15, 2916–2928. [Google Scholar] [CrossRef]
- Morissette, S.L.; Almarsson, Ö.; Peterson, M.L.; Remenar, J.F.; Read, M.J.; Lemmo, A.V.; Ellis, S.; Cima, M.J.; Gardner, C.R. High-throughput crystallization: Polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Deliv. Rev. 2004, 56, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Patra, R.; Titi, H.M.; Goldberg, I. Crystal engineering of molecular networks: Tailoring hydrogen-bonding self-assembly of tin-tetrapyridylporphyrins with multidentate carboxylic acids as axial ligands. Cryst. Growth Des. 2013, 13, 1342–1349. [Google Scholar] [CrossRef]
- Hanif, M.; Khan, E.; Khalid, M.; Tahir, M.N.; de Alcântara Morais, S.F.; Braga, A.A.C. 2-Amino-3-methylpyridinium, 2-amino-4-methylbenzothiazolium and 2-amino-5-chloropyridinium salts. Experimental and theoretical findings. J. Mol. Struct. 2020, 1222, 128914. [Google Scholar] [CrossRef]
- Khan, E.; Khalid, M.; Gul, Z.; Shahzad, A.; Tahir, M.N.; Asif, H.M.; Asim, S.; Braga, A.A.C. Molecular structure of 1, 4-bis (substituted-carbonyl) benzene: A combined experimental and theoretical approach. J. Mol. Struct. 2020, 1205, 127633. [Google Scholar] [CrossRef]
- Khan, S.; Zahoor, M.; Rahman, M.U.; Gul, Z. Cocrystals; basic concepts, properties and formation strategies. Z. Für Phys. Chem. 2023, 237, 273–332. [Google Scholar] [CrossRef]
- Yamashita, H.; Hirakura, M.Y.; Yuda, T.; Teramura, K. Terada, Detection of cocrystal formation based on binary phase diagrams using thermal analysis. Pharm. Res. 2013, 30, 70–80. [Google Scholar] [CrossRef]
- Islam, N.U.; Umar, M.N.; Khan, E.; Al-Joufi, F.A.; Abed, S.N.; Said, M.; Ullah, H.; Iftikhar, M.; Zahoor, M.; Khan, F.A. Levofloxacin Cocrystal/Salt with Phthalimide and Caffeic Acid as Promising Solid-State Approach to Improve Antimicrobial Efficiency. Antibiotics 2022, 11, 797. [Google Scholar] [CrossRef]
- Chiarella, R.A.; Davey, R.J.; Peterson, M.L. Making co-crystals the utility of ternary phase diagrams. Cryst. Growth Des. 2007, 7, 1223–1226. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Stare, J.; Boldyreva, E.V. Pressure-driven phase transition mechanisms revealed by quantum chemistry: L-serine polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 6671–6676. [Google Scholar] [CrossRef] [PubMed]
- Rychkov, D.; Arkhipov, S.; Boldyreva, E. Structure-forming units of amino acid maleates. Case study of l-valinium hydrogen maleate. Acta Crystallogr. Sect. B 2016, 72, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Kosar, N.; Gul, S.; Ayub, K.; Bahader, A.; Gilani, M.A.; Arshad, M.; Mahmood, T. Significant nonlinear optical response of alkaline earth metals doped beryllium and magnesium oxide nanocages. Mater. Chem. Phys. 2020, 242, 122507. [Google Scholar] [CrossRef]
- Hanif, M.; Kosar, N.; Mahmood, T.; Muhammad, M.; Ullah, F.; Tahir, M.N.; Ribeiro, A.I.; Khan, E. Schiff Bases Derived from 2-Amino-6-methylbenzothiazole, 2-Amino-5-chloropyridine and 4-Chlorobenzaldehyde: Structure, Computational Studies and Evaluation of Biological Activity. Chem. Sel. 2022, 7, e202203386. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An Overview of Strengths and Directionalities of Noncovalent Interactions: σ-Holes and π-Holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. Sect. E 2019, 75, 308–318. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin. J. Sci. Technol. 2004, 26, 211–219. [Google Scholar]
- Liu, L.; Zou, D.; Zhang, Y.; Zhang, Q.; Feng, Y.; Guo, Y.; Liu, Y.; Zhang, X.; Cheng, G.; Wang, C. Pharmaceutical salts/cocrystals of enoxacin with dicarboxylic acids: Enhancing in vitro antibacterial activity of enoxacin by improving the solubility and permeability. Eur. J. Pharm. Biopharm. 2020, 154, 62–73. [Google Scholar] [CrossRef]
- Bruker. APEX3, SAINT and SADABS; Crystallography Software Suite, Bruker AXS, Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Fox Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- John, D. Roy, Keith, Todd, Millam, GaussView 5.0; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Tretyakova, I.S.; Rychkov, D.A.; Lomovskiy, I.O. Computational study of chemical phenol glycosylation mechanism in the gas phase for modeling direct glycoconjugate formation in raw plant material. Comput. Theor. Chem. 2023, 1225, 114182. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- Asghar, A.; Tan, Y.C.; Zahoor, M.; Zainal Abidin, S.A.; Yow, Y.Y.; Khan, E.; Lahiri, C. A scaffolded approach to unearth potential antibacterial components from epicarp of Malaysian Nephelium lappaceum L. Sci. Rep. 2021, 11, 13859. [Google Scholar] [CrossRef]
- Baliyan, S.; Mukherjee, R.; Priyadarshini, A.; Vibhuti, A.; Gupta, A.; Pandey, R.P.; Chang, C.M. Determination of Antioxidants by DPPH Radical Scavenging Activity and Quantitative Phytochemical Analysis of Ficus religiosa. Molecules 2022, 27, 1326. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Compound 2 | Compound 3 | |
|---|---|---|---|
| Chemical formula | C7H5ClO2·C5H5ClN2 | C7H4ClO2·C5H6ClN2 | 0.5(C7H4ClO2)·0.5(C5H4ClN2) |
| Mr | 285.13 | 285.13 | 141.56 |
| Crystal system, space group | Monoclinic, P21 | Monoclinic, P21 | Monoclinic, Pc |
| Temperature (K) | 150 | 150 | 150 |
| a, b, c (Å) | 3.7979 (4), 11.4738 (11), 14.0722 (15) | 8.0024 (16), 6.8306 (12), 11.1631 (18) | 10.0269 (10), 5.1405 (6), 12.1494 (15) |
| β (°) | 92.088 (4) | 93.656 (5) | 90.678 (4) |
| V (Å3) | 612.81 (11) | 608.95 (19) | 626.18 (12) |
| Z | 2 | 2 | 4 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα |
| µ (mm−1) | 0.52 | 0.53 | 0.51 |
| Crystal size (mm) | 0.17 × 0.16 × 0.11 | 0.24 × 0.16 × 0.12 | 0.27 × 0.33 × 0.37 |
| Data collection | |||
| Diffractometer | Bruker D8 VENTURE PHOTON II with Oxford COBRA Cryosystem | ||
| Absorption correction | Multi-scan | Multi-scan | Multi-scan |
| No. of measured, independent, and observed [I ≥ 2u(I)] reflections | 2460, 2460, 2048 | 12465, 2473, 1653 | 9501, 2433, 1516 |
| Rint | 0.022 | 0.126 | 0.059 |
| (sin θ/λ)max (Å−1) | 0.624 | 0.624 | 0.626 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.046, 0.110, 1.08 | 0.051, 0.112, 1.08 | 0.125, 0.445, 1.71 |
| No. of reflections | 2460 | 2473 | 2433 |
| No. of parameters | 167 | 173 | 163 |
| No. of restraints | 1 | 1 | 2 |
| H-atom treatment | H-atom parameters constrained | ||
| Δρmax, Δρmin (e Å−3) | 0.58, −0.33 | 0.50, −0.56 | 0.85, −0.91 |
| Absolute structure parameter | −0.04 (10) | 0.02 (12) | 0.56 (6) |
| Compounds | Eint | Ecp | ELUMO | EHOMO | EL-H |
|---|---|---|---|---|---|
| 1a | −17.81 | −12.91 | −1.38 | −6.76 | 5.38 |
| 1b | −8.20 | −2.10 | −1.72 | −6.72 | 5.00 |
| 2 | −101.32 | −96.45 | −3.01 | −6.10 | 3.09 |
| 3a | 29.99 | 36.37 | −0.47 | −5.93 | 5.46 |
| 3b | 46.12 | 52.63 | −0.65 | −5.82 | 5.17 |
| Compound | λmax | Molar Absorptivity Coefficient (ε) |
|---|---|---|
| 1 | 345 ± 1 | 782 |
| 2 | 243 ± 1.5 | 604 |
| 3 | 344 ± 1 | 632 |
| Compounds | Conc. µg/mL | Sample | Control | %RSA | IC50 |
|---|---|---|---|---|---|
| 3-Chlorobenzoic acid | 50 | 0.355 | 0.52 | 31.73 | 4.85 |
| 100 | 0.316 | 39.23 | |||
| 150 | 0.289 | 44.42 | |||
| 200 | 0.269 | 48.27 | |||
| 250 | 0.21 | 59.62 | |||
| 4-Amino-2-chloropyridine | 50 | 0.455 | 0.52 | 12.5 | 9.53 |
| 100 | 0.436 | 16.15 | |||
| 150 | 0.399 | 23.27 | |||
| 200 | 0.389 | 25.19 | |||
| 250 | 0.28 | 46.15 | |||
| 2-Amino-4-chloropyridine | 50 | 0.45 | 0.52 | 13.46 | 8.27 |
| 100 | 0.416 | 20 | |||
| 150 | 0.369 | 29.04 | |||
| 200 | 0.315 | 39.42 | |||
| 250 | 0.26 | 50 | |||
| 1 | 50 | 0.35 | 0.52 | 32.69 | 4.92 |
| 100 | 0.32 | 38.46 | |||
| 150 | 0.29 | 44.23 | |||
| 200 | 0.26 | 50 | |||
| 250 | 0.21 | 59.62 | |||
| 2 | 50 | 0.31 | 0.52 | 40.38 | 4.05 |
| 100 | 0.28 | 46.15 | |||
| 150 | 0.24 | 53.85 | |||
| 200 | 0.21 | 59.62 | |||
| 250 | 0.18 | 65.38 | |||
| 3 | 50 | 0.31 | 0.52 | 40.38 | 3.55 |
| 100 | 0.29 | 44.23 | |||
| 150 | 0.26 | 50 | |||
| 200 | 0.22 | 57.69 | |||
| 250 | 0.21 | 59.62 |
| Compound No. | Average Zone of Inhibition (mm) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gram Positive | Gram Negative | |||||||||||
| S. Aureus | S. Epidermis | K. Pneumonia | P. Mirabilis | |||||||||
| Acid | Base | Product | Acid | Base | Product | Acid | Base | Product | Acid | Base | Product | |
| 1 | 12 | 10 | 14 | 11 | 18 | 13 | 11 | 06 | 16 | 11 | 14 | 13 |
| 2 | 12 | 09 | 16 | 11 | 08 | 14 | 11 | 05 | 17 | 11 | 07 | 14 |
| 3 | 11 | 09 | 15 | 10 | 08 | 14 | 15 | 05 | 16 | 13 | 07 | 15 |
| Standard Azithromycin | 28 | 27 | 30 | 27 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, T.; Kosar, N.; Said, M.; Ahmed, M.; Mahmood, T.; Khan, E. Supramolecular Assemblies of 3/4-Chlorobenzoic Acid and Amino-Chloropyridine Derivatives: Synthesis, X-ray Diffraction, DFT Calculations, and Biological Screening. Crystals 2023, 13, 1663. https://doi.org/10.3390/cryst13121663
Ahmad T, Kosar N, Said M, Ahmed M, Mahmood T, Khan E. Supramolecular Assemblies of 3/4-Chlorobenzoic Acid and Amino-Chloropyridine Derivatives: Synthesis, X-ray Diffraction, DFT Calculations, and Biological Screening. Crystals. 2023; 13(12):1663. https://doi.org/10.3390/cryst13121663
Chicago/Turabian StyleAhmad, Tufail, Naveen Kosar, Muhammad Said, Maqsood Ahmed, Tariq Mahmood, and Ezzat Khan. 2023. "Supramolecular Assemblies of 3/4-Chlorobenzoic Acid and Amino-Chloropyridine Derivatives: Synthesis, X-ray Diffraction, DFT Calculations, and Biological Screening" Crystals 13, no. 12: 1663. https://doi.org/10.3390/cryst13121663
APA StyleAhmad, T., Kosar, N., Said, M., Ahmed, M., Mahmood, T., & Khan, E. (2023). Supramolecular Assemblies of 3/4-Chlorobenzoic Acid and Amino-Chloropyridine Derivatives: Synthesis, X-ray Diffraction, DFT Calculations, and Biological Screening. Crystals, 13(12), 1663. https://doi.org/10.3390/cryst13121663