
A Probe to Surface Reactivity, Crystal Structure, and DFT Investigations for Newly Synthesized 4,5-bis(4-Nitrophenyl)-8a-phenyl-decahydro-[1,3]diazino[4,5-d]pyrimidine-2,7-dione: A Combined Theoretical and Experimental Study

 ,
, 
Abstract
:1. Introduction
2. Materials and Method
2.1. Chemicals
2.2. X-ray Analysis
2.3. Hirshfeld Surface Analysis
2.4. DFT Study
3. Results and Discussion
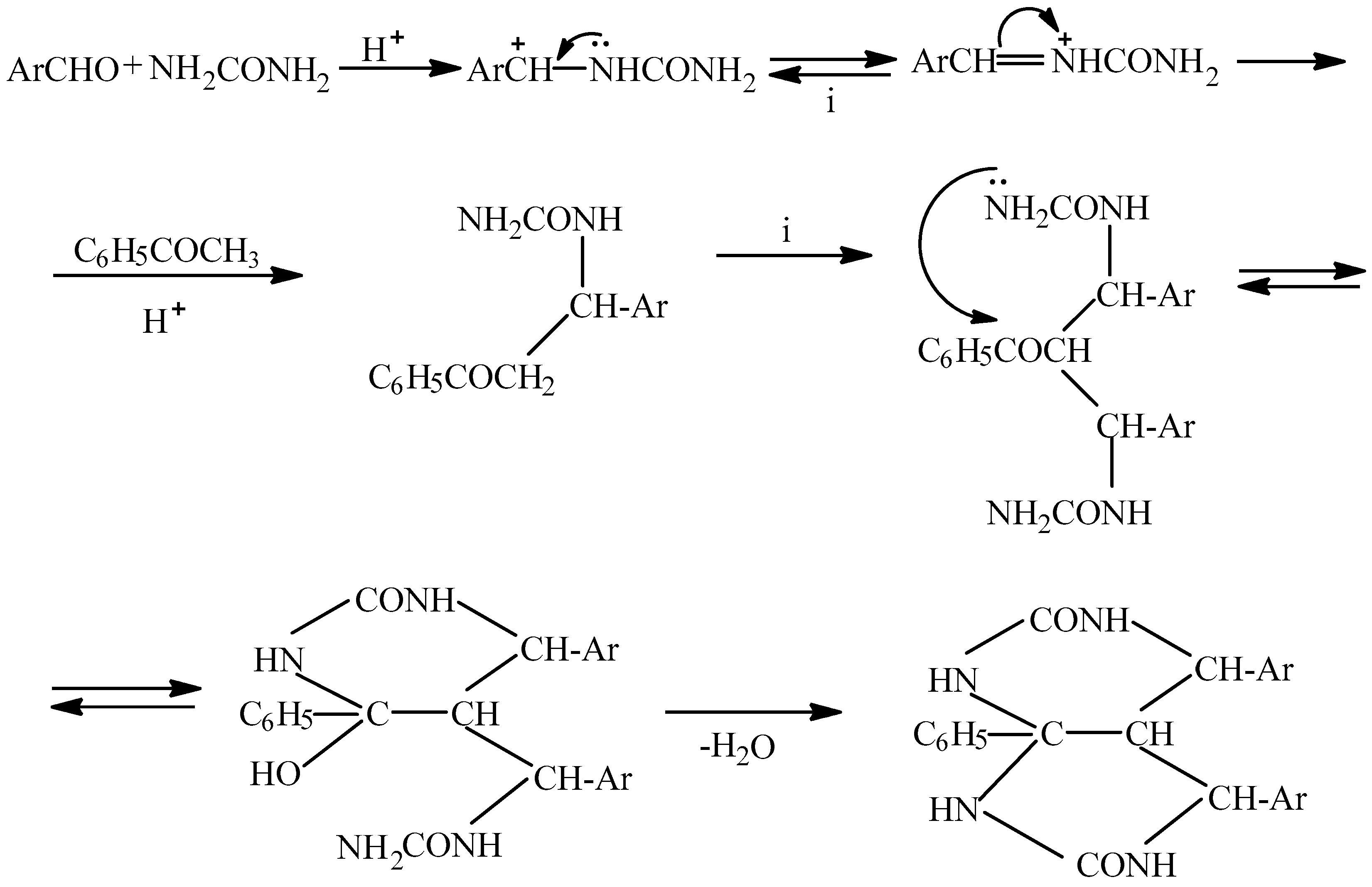
3.1. Chemical Synthesis
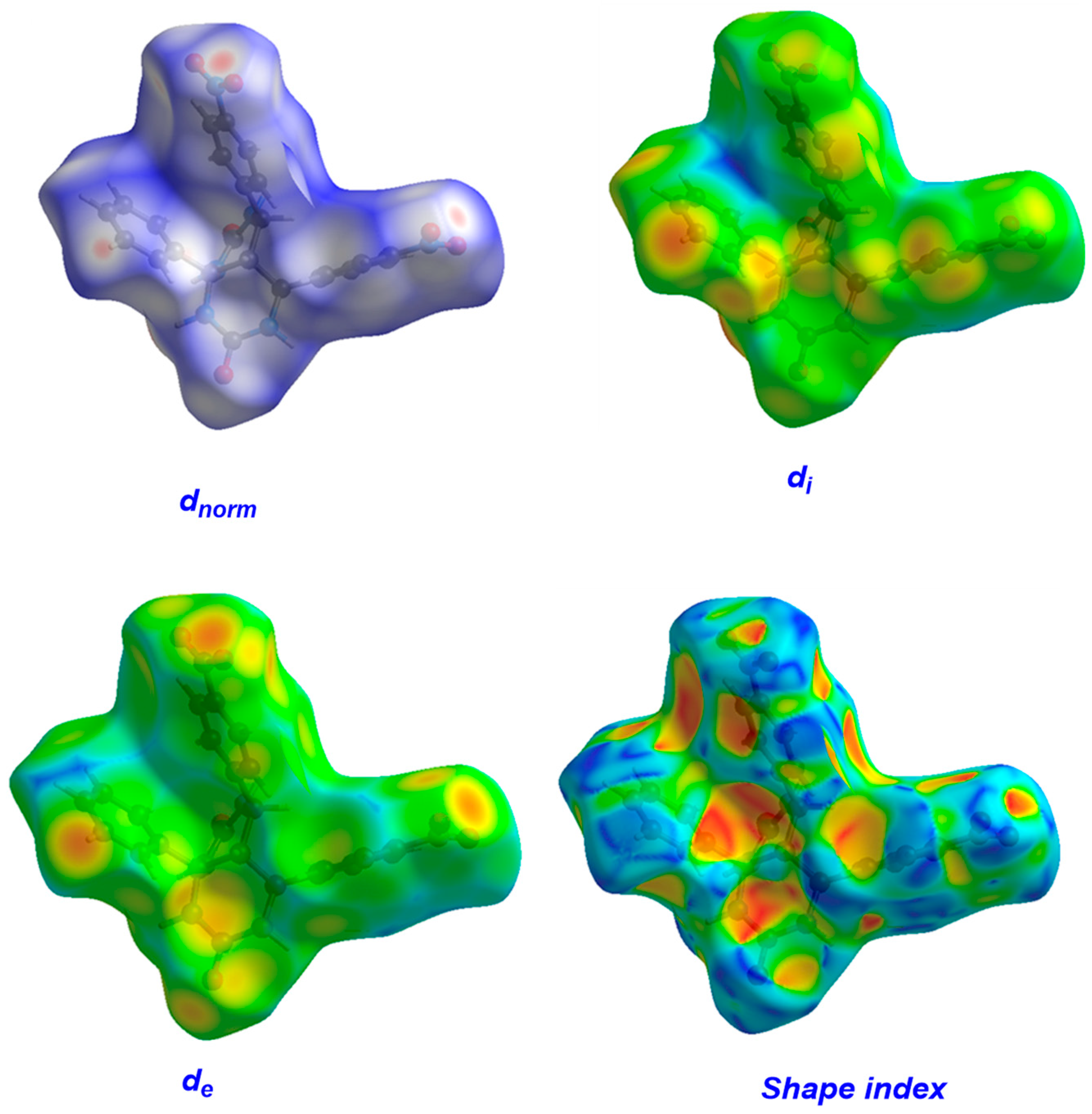
3.2. Hirshfeld Surfaces (HS) Analysis
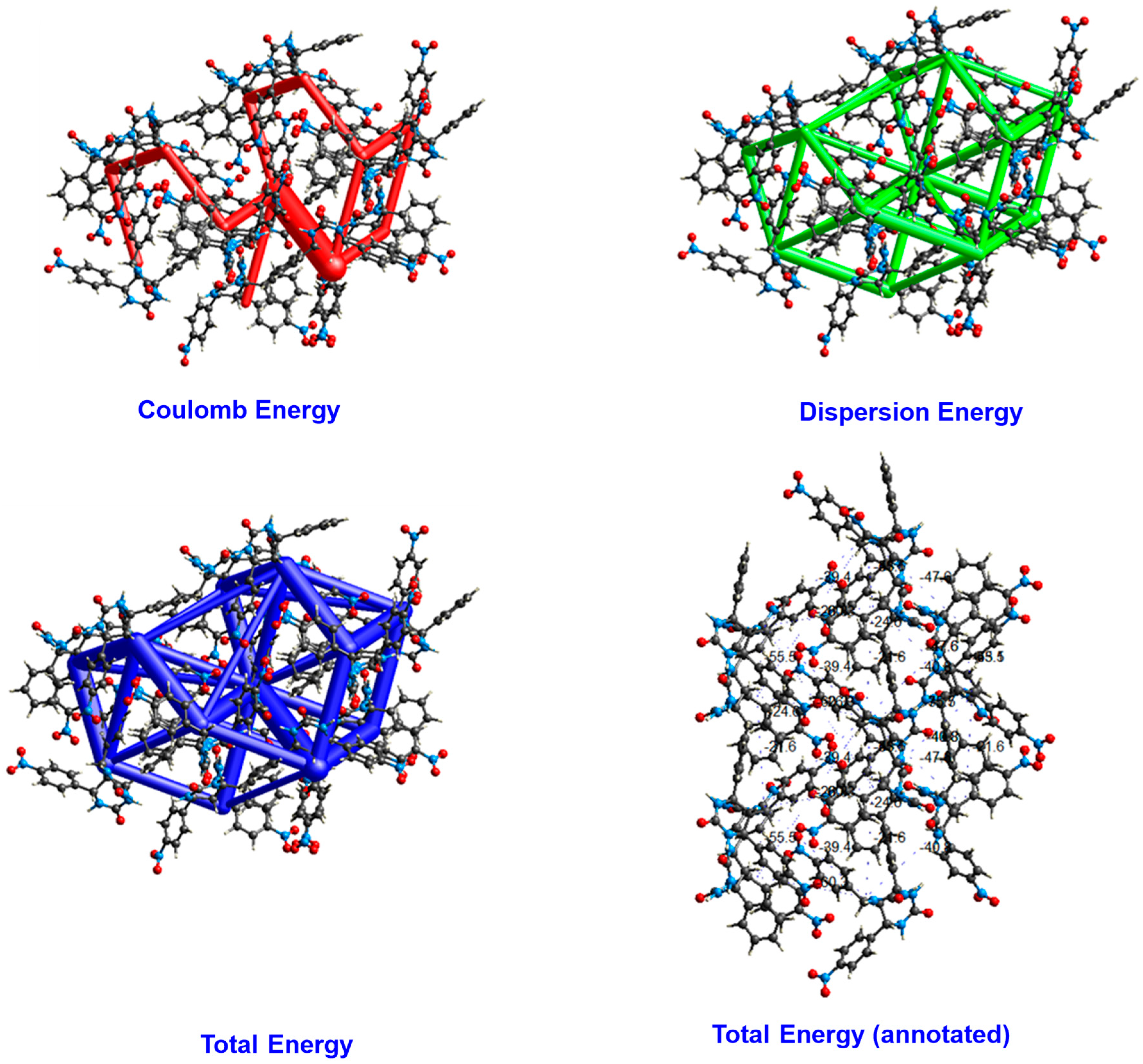
3.3. Energy Framework Analysis
3.4. DFT Analysis
3.4.1. Optimized Geometry of Studied Compound
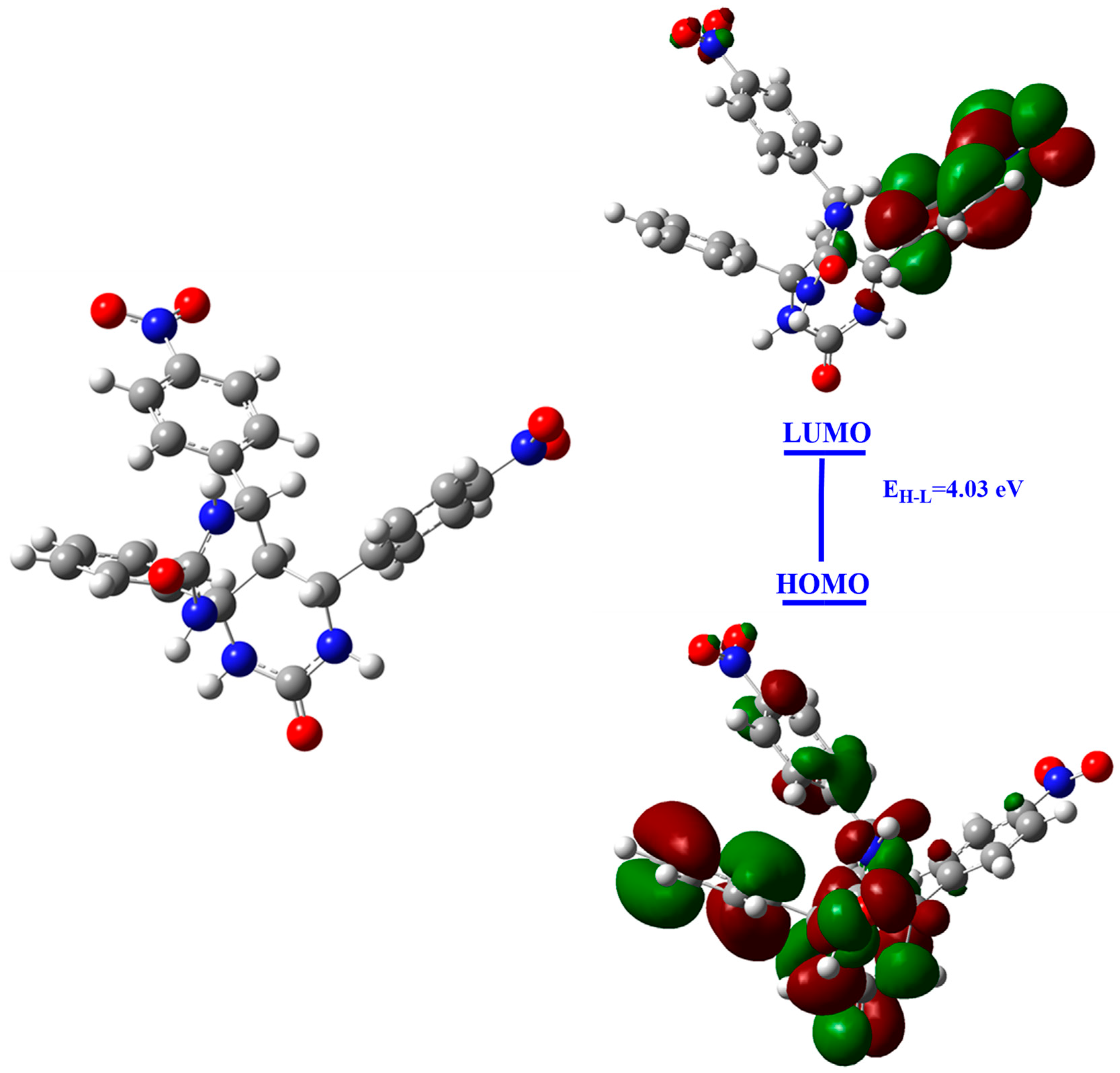
3.4.2. Mulliken’s Atomic Charges and FMO Analysis
3.4.3. MESP and DOS Analysis
3.4.4. TD-DFT Study and Dipole Moment
3.4.5. Nonlinear Optical Properties of the Compound
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef] [PubMed]
- El Bakri, Y.; Kurbanova, M.M.; Siddique, S.A.; Ahmad, S.; Goumri-Said, S. One-pot synthesis, X-ray crystal structure, and identification of potential molecules against COVID-19 main protease through structure-guided modeling and simulation approach. Arab. J. Chem. 2022, 15, 104230. [Google Scholar] [CrossRef] [PubMed]
- Zabihollahi, R.; Fassihi, A.; Aghasadeghi, M.R.; Memarian, H.R.; Soleimani, M.; Majidzadeh-A, K. Inhibitory effect and structure–activity relationship of some Biginelli-type pyrimidines against HSV-1. Med. Chem. Res. 2013, 22, 1270–1276. [Google Scholar] [CrossRef]
- Kappe, C.O. Biologically active dihydropyrimidones of the Biginelli-type—A literature survey. Eur. J. Med. Chem. 2000, 35, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- El Bakri, Y.; Kurbanova, M.; Siddique, S.A.; Karthikeyan, S.; Maharramov, A.; Ramazanzade, N. Synthesis, virtual screening, and computational approach of 6-(4-metoxyphenyl)-4-phenyl-1, 2-dihydropyrimidin-2-one as a potential target for thioredoxin glutathione reductase (TGR). J. Mol. Struct. 2023, 1286, 135623. [Google Scholar] [CrossRef]
- Kurbanova, M.; Ashfaq, M.; Tahir, M.N.; Maharramov, A.; Dege, N.; Ramazanzade, N.; Cinar, E.B. Synthesis, Crystal Structure, Supramolecular Assembly Inspection by Hirshfeld Surface Analysis and Computational Exploration of 4-Phenyl-6-(p-Tolyl) Pyrimidin-2 (1H)-One (PPTP). J. Struct. Chem. 2023, 64, 437–449. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Zhou, Y.; Zhang, Q.; Han, T.; Tang, C.; Fan, W. Pyrazolo [1, 5-a] pyrimidine based Trk inhibitors: Design, synthesis, biological activity evaluation. Bioorg. Med. Chem. Lett. 2021, 31, 127712. [Google Scholar] [CrossRef] [PubMed]
- Aydin, B.O.; Anil, D.; Demir, Y. Synthesis of N-alkylated pyrazolo [3, 4-d] pyrimidine analogs and evaluation of acetylcholinesterase and carbonic anhydrase inhibition properties. Arch. Pharm. 2021, 354, 2000330. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.H.S.; Masson, F.T.; Simeoni, L.A.; Homem-de-Mello, M. Biological activity of dihydropyrimidinone (DHPM) derivatives. A Syst. Rev. 2018, 143, 1779. [Google Scholar]
- Khasimbi, S.; Ali, F.; Manda, K.; Sharma, A.; Chauhan, G.; Wakode, S. Dihydropyrimidinones scaffold as a promising nucleus for synthetic profile and various therapeutic targets: A Review. Curr. Org. Synth. 2021, 18, 270–293. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Madhankumar, S.; Muthuraja, P.; Dhandapani, M. Structural characterization, quantum chemical calculations and Hirshfeld surface analysis of a new third order harmonic organic crystal: 2-Amino-4-methylpyridinium benzilate. J. Mol. Struct. 2020, 1201, 127151. [Google Scholar] [CrossRef]
- Anju, L.S.; Aruldhas, D.; Joe, I.H.; Balachandran, S. Density functional theory, spectroscopic and hydrogen bonding analysis of fenoxycarb–water complexes. J. Mol. Struct. 2020, 1201, 127201. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, UK, 2010. [Google Scholar]
- Arivazhagan, M.; Kumar, J.S. Molecular structure, vibrational spectral assignments, HOMO–LUMO, MESP, Mulliken analysis and thermodynamic properties of 2,6-xylenol and 2,5-dimethyl cyclohexanol based on DFT calculation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 137, 490–502. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Value |
|---|---|
| I | |
| Empirical formula | C24H20N6O6 |
| M | 488.46 |
| Temperature, K | 293 |
| Crystal size, mm3 | 0.19 × 0.18 × 0.16 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a, Å | 12.6302 (10) |
| b, Å | 12.5115 (13) |
| c, Å | 29.265 (2) |
| β, deg | 100.174 (6) |
| V, Å3 | 4551.8 (7) |
| Z | 8 |
| ρcalcd, g/cm3 | 1.426 |
| µ, mm–1 | 0.11 |
| F (000) | 2032 |
| Nref | 7076 |
| h, k, lmax | 14, 14, 33 |
| S | 0.818 |
| Npar | 681 |
| Data completeness | 0.993 |
| R (reflections) | 0.0465 (2455) |
| wR2 (reflections) | 0.0924 (7076) |
| Theta (max) | 23.968 |
| Δρmax, Δρmin (e Å−3) | 0.16, −0.16 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| C33—H33···O5 | 0.98 | 2.51 | 3.423 (5) | 156 |
| C2—H2···O10 i | 0.93 | 2.56 | 3.480 (6) | 173 |
| C31—H31···O7 ii | 0.98 | 2.38 | 3.121 (6) | 132 |
| C23—H23···O1 iii | 0.93 | 2.63 | 3.401 (6) | 140 |
| N6—H6···O6 iv | 0.78 (3) | 2.11 (3) | 2.893 (5) | 178 (3) |
| N3—H3···O11 | 0.79 (3) | 2.17 (3) | 2.940 (5) | 162 (3) |
| N5—H5···O5 v | 0.77 (3) | 2.30 (3) | 2.998 (5) | 151 (3) |
| N12—H12A···O6 v | 0.83 (3) | 2.15 (3) | 2.961 (5) | 169 (3) |
| N10—H10···O11 vi | 0.84 (4) | 2.03 (4) | 2.870 (5) | 178 (4) |
| Compound (C24H20N6O6) | |||
|---|---|---|---|
| Minimum | Mean | Maximum | |
| di (Å) | 0.7564 | 1.7575 | 2.9874 |
| de (Å) | 0.7593 | 1.7764 | 2.8371 |
| dnorm | −0.5783 | 0.5396 | 1.4363 |
| Shape index | −0.9957 | 0.2180 | 0.9990 |
| curvedness | −4.2126 | −1.0243 | 0.2362 |
| N | Symop | R | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|---|---|
| 1 | - | 7.55 | 0.5 | −12.0 | −57.6 | 19.9 | −43.1 |
| 0 | −x, −y, −z | 10.49 | −21.7 | −7.3 | −12.9 | 3.7 | −35.5 |
| 1 | - | 9.59 | −8.6 | −8.0 | −47.5 | 19.7 | −40.8 |
| 1 | - | 10.08 | 6.9 | −2.7 | −16.5 | 7.7 | −3.3 |
| 0 | - | 8.09 | −33.0 | −9.5 | −35.5 | 14.1 | −60.3 |
| 0 | - | 9.57 | −29.4 | −13.7 | −30.3 | 22.9 | −47.6 |
| 0 | - | 13.32 | −18.6 | −2.5 | −6.4 | 0.0 | −26.3 |
| 0 | x, y, z | 12.51 | −9.1 | −2.5 | −15.1 | 0.0 | −24.6 |
| 0 | - | 7.98 | −37.2 | −21.0 | −37.6 | 37.0 | −55.5 |
| 0 | −x, −y, −z | 9.97 | −84.8 | −26.1 | −24.9 | 61.9 | −75.7 |
| 0 | −x, y + 1/2, −z + 1/2 | 9.65 | −19.4 | −5.0 | −27.5 | 10.4 | −39.4 |
| 0 | - | 10.40 | −1.9 | −3.6 | −26.5 | 8.1 | −21.6 |
| Total energy | −387.8 |
| Atoms | Charge | Atoms | Charge | Atoms | Charge |
|---|---|---|---|---|---|
| 1O | −0.429510 | 19C | 0.126098 | 36C | 0.164288 |
| 2O | −0.439976 | 20H | 0.141425 | 37O | −0.024438 |
| 3N | −0.372600 | 21C | 0.589265 | 38O | 0.000959 |
| 4N | −0.575703 | 22H | 0.139331 | 39C | −0.033118 |
| 5N | −0.608828 | 23C | 0.305970 | 40H | 0.103148 |
| 6N | −0.009338 | 24C | −0.545560 | 41C | −0.363958 |
| 7O | 0.116045 | 25H | 0.091652 | 42H | 0.101357 |
| 8C | 0.138310 | 26C | 0.140567 | 43C | −0.237432 |
| 9H | 0.005341 | 27H | 0.115648 | 44H | 0.088397 |
| 10 O | 0.267182 | 28C | −0.172610 | 45C | −0.412813 |
| 11C | 0.356575 | 29H | 0.114955 | 46H | 0.066000 |
| 12C | −0.378345 | 30C | −0.145976 | 47C | 0.111632 |
| 13N | 0.963146 | 31H | 0.066267 | 48H | 0.079510 |
| 14C | 0.129359 | 32C | 0.092904 | 49C | −0.045811 |
| 16C | 0.397683 | 33C | −0.367934 | 50H | 0.085517 |
| 17C | −0.215797 | 34H | 0.088038 | 51C | 0.047120 |
| 18H | 0.080191 | 35N | −0.302980 | 52H | 0.075031 |
| 53H | 0.250129 | ||||
| 54H | 0.220666 | ||||
| 55H | 0.219381 | ||||
| 56H | 0.067541 |
| Point Group Symmetry | C1 |
|---|---|
| Total electronic energy | −1706.413579 |
| Imaginary frequency | 0 |
| Q (O/N/) | −0.65/−0.71 |
| EH | −7.415 |
| EL | −3.384 |
| EH-L | 4.03 |
| Ionization energy (IE) | 7.45 |
| Electron affinity (EA) | 3.38 |
| Chemical hardness (Ƞ) | 2.03 |
| Chemical potential (µ) | −5.41 |
| Electronegativity (χ) | 5.41 |
| Excitation Energy (ΔE) | 4.2141 |
|---|---|
| Absorbance wavelength (λ) | 294.21 nm |
| Oscillator strength (fo) | 0.0466 |
| Orbital contribution | HOMO → LUMO + 4 (23%) HOMO → LUMO + 3 (6%) HOMO → LUMO + 5 (9%) HOMO → LUMO + 2 (43%) |
| Dipole moment (µo) | 2.491573 |
| Polarizability (αo) | 342.53 |
| Polarizability volime | 50.75 |
| Hyperpolarizability (βo) | 2047.668 |
| (βvec) | 1931.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Bakri, Y.; Kurbanova, M.; Ahsin, A.; Ramazanzade, N.; Al-Salahi, R. A Probe to Surface Reactivity, Crystal Structure, and DFT Investigations for Newly Synthesized 4,5-bis(4-Nitrophenyl)-8a-phenyl-decahydro-[1,3]diazino[4,5-d]pyrimidine-2,7-dione: A Combined Theoretical and Experimental Study. Crystals 2023, 13, 942. https://doi.org/10.3390/cryst13060942
El Bakri Y, Kurbanova M, Ahsin A, Ramazanzade N, Al-Salahi R. A Probe to Surface Reactivity, Crystal Structure, and DFT Investigations for Newly Synthesized 4,5-bis(4-Nitrophenyl)-8a-phenyl-decahydro-[1,3]diazino[4,5-d]pyrimidine-2,7-dione: A Combined Theoretical and Experimental Study. Crystals. 2023; 13(6):942. https://doi.org/10.3390/cryst13060942
Chicago/Turabian StyleEl Bakri, Youness, Malahat Kurbanova, Atazaz Ahsin, Nacaf Ramazanzade, and Rashad Al-Salahi. 2023. "A Probe to Surface Reactivity, Crystal Structure, and DFT Investigations for Newly Synthesized 4,5-bis(4-Nitrophenyl)-8a-phenyl-decahydro-[1,3]diazino[4,5-d]pyrimidine-2,7-dione: A Combined Theoretical and Experimental Study" Crystals 13, no. 6: 942. https://doi.org/10.3390/cryst13060942