
Two-Dimensional VSi2X2N2 (X = P, As, Sb, Bi) Janus Monolayers: Spin-Polarized Electronic Structure and Perpendicular Magnetic Anisotropy


Abstract
:1. Introduction
2. Computational Details
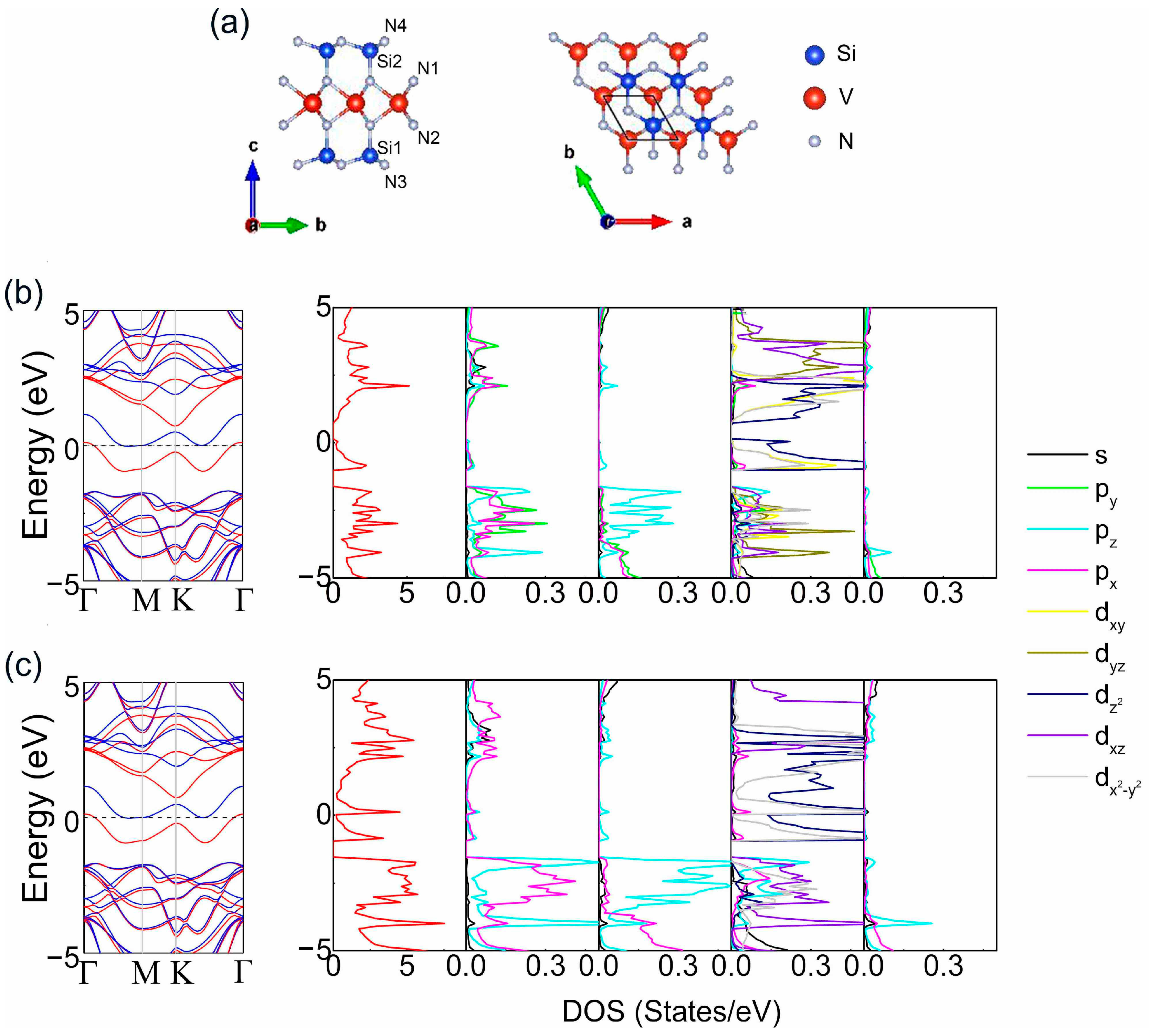
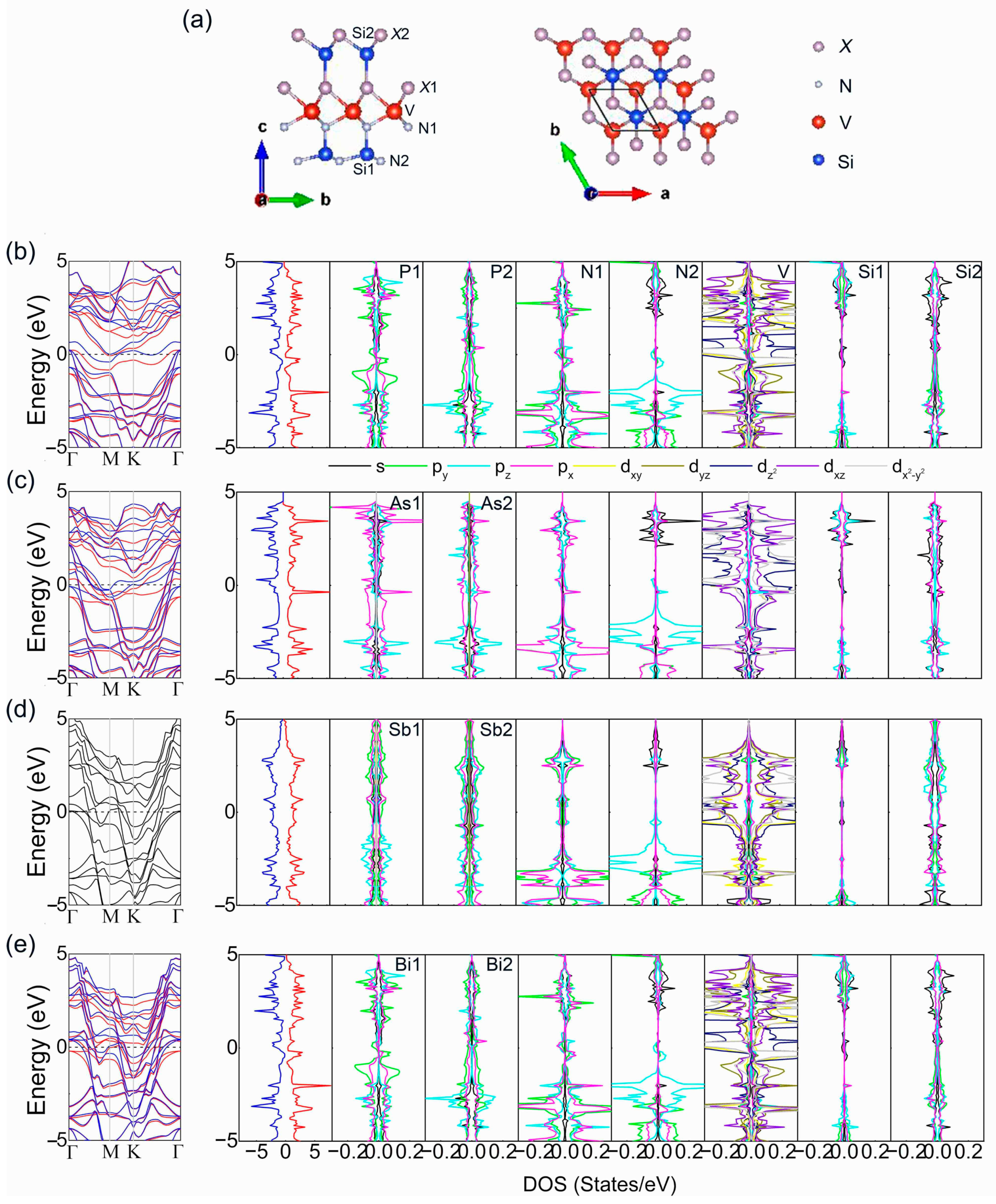
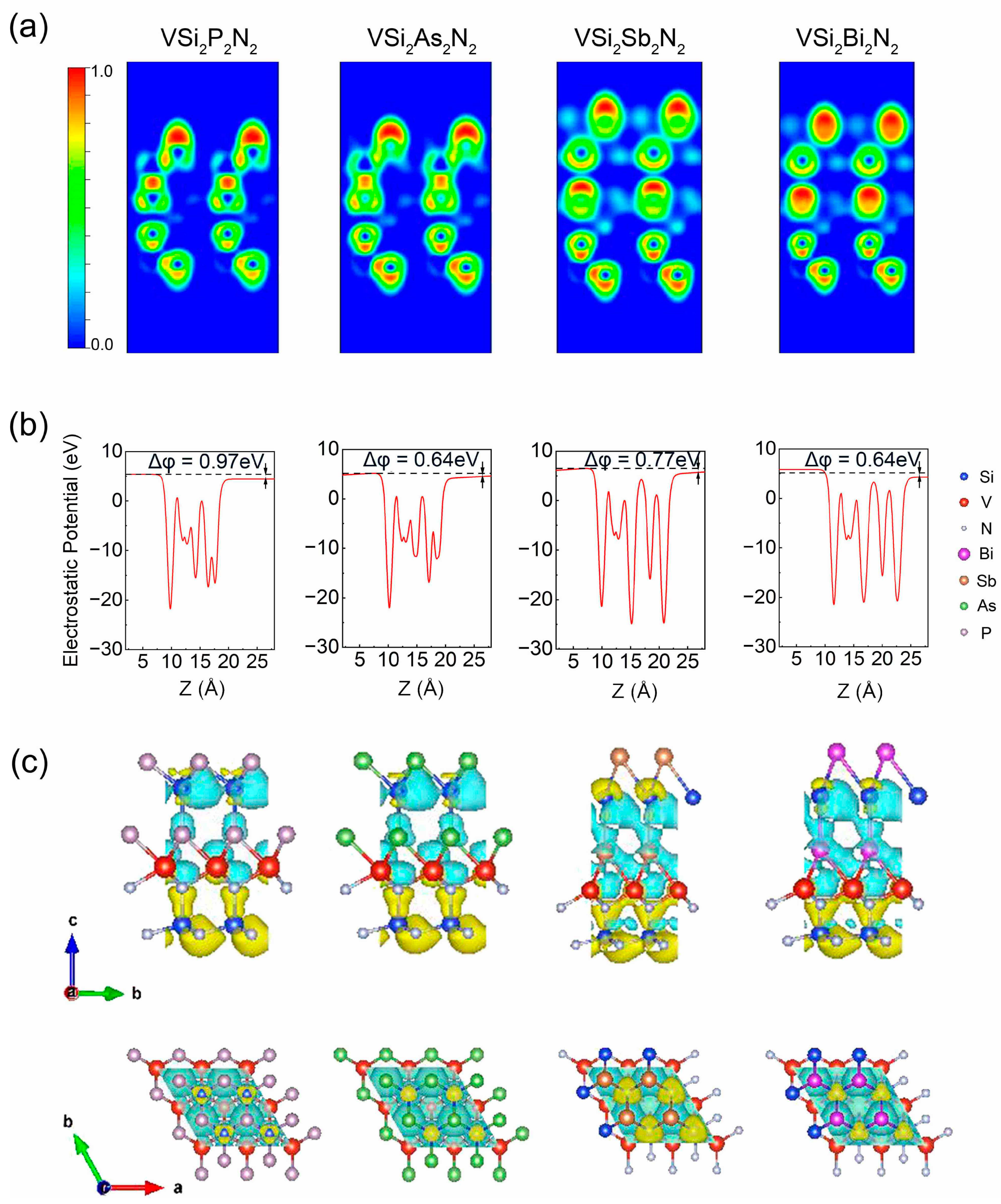
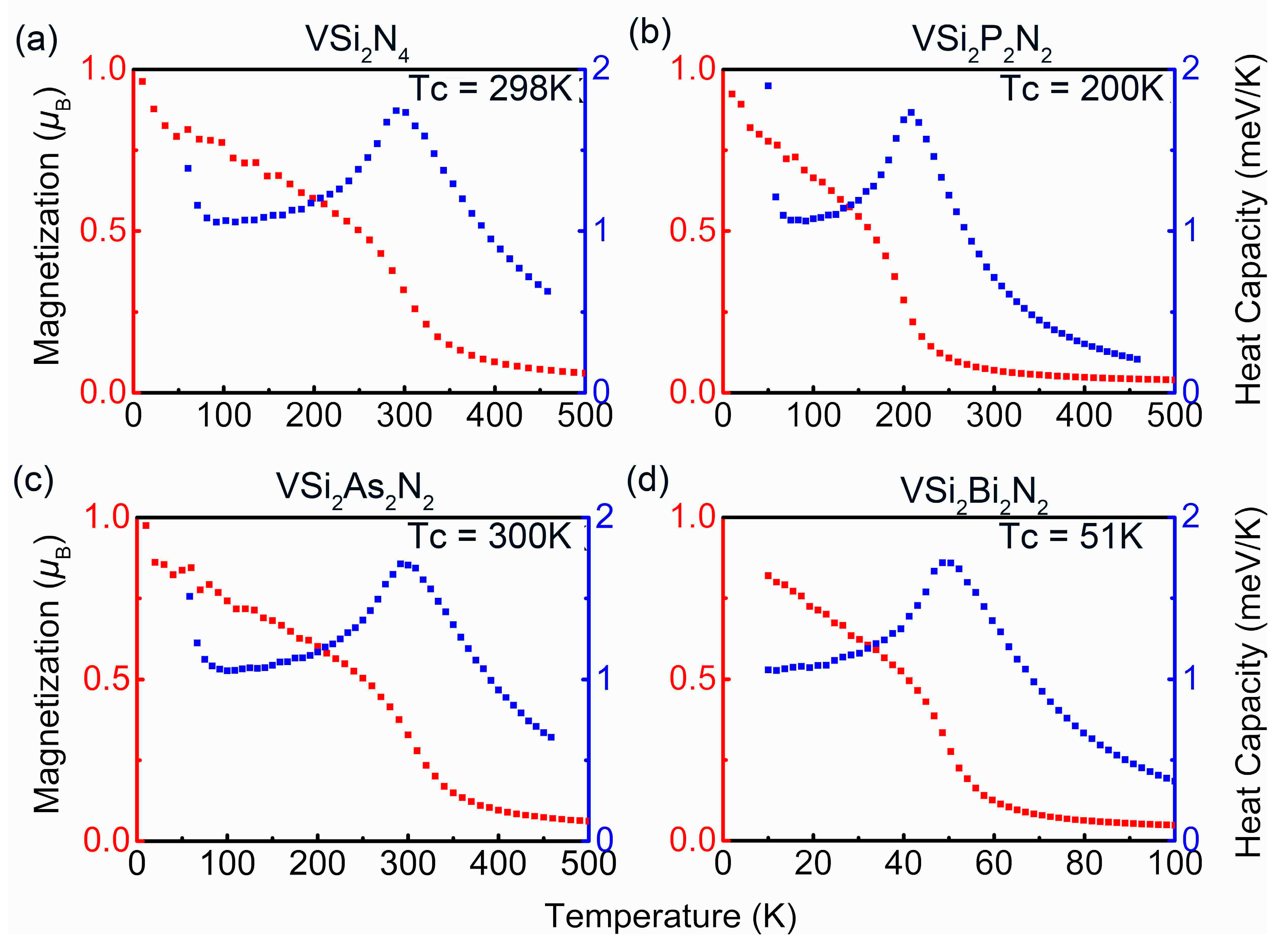
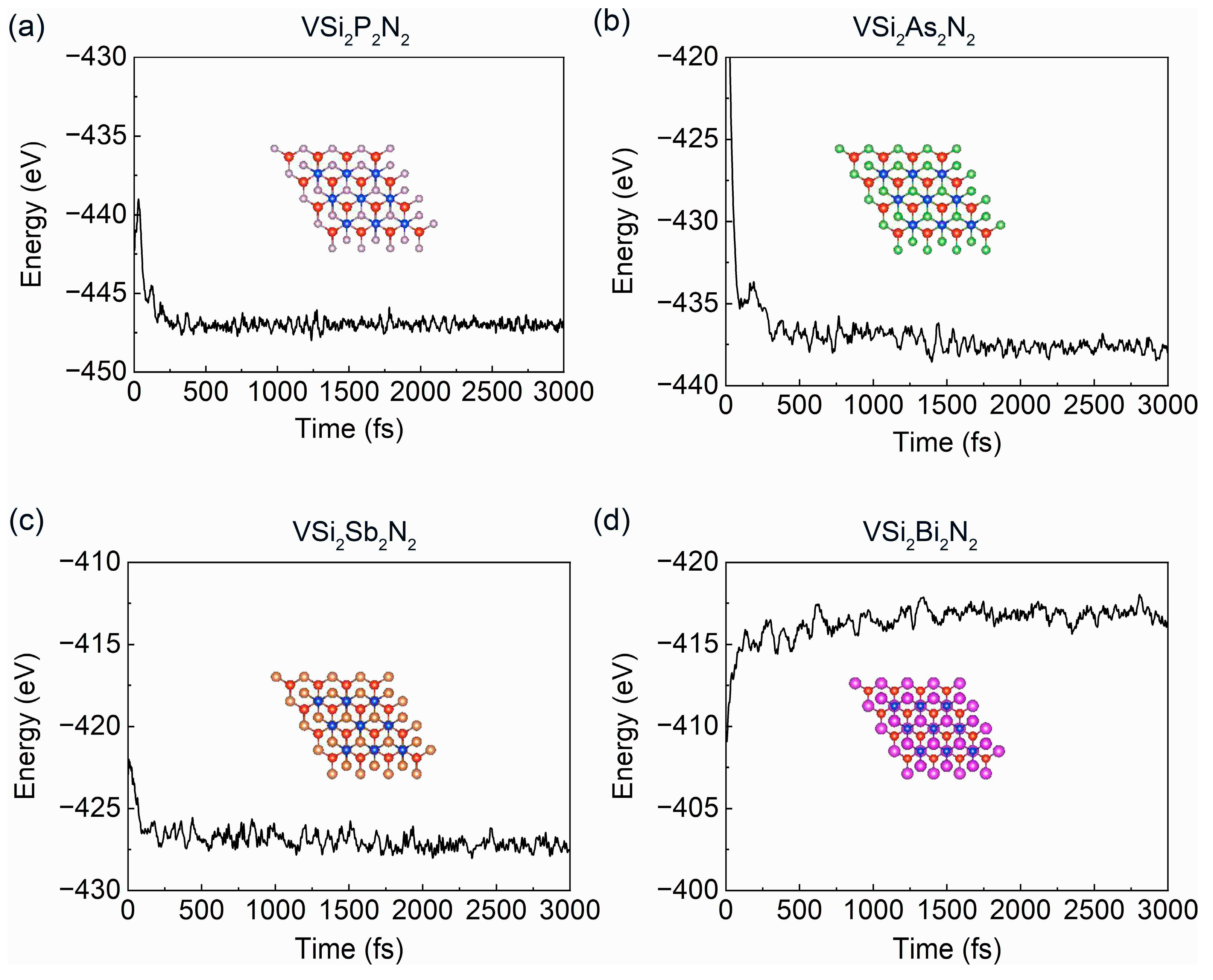
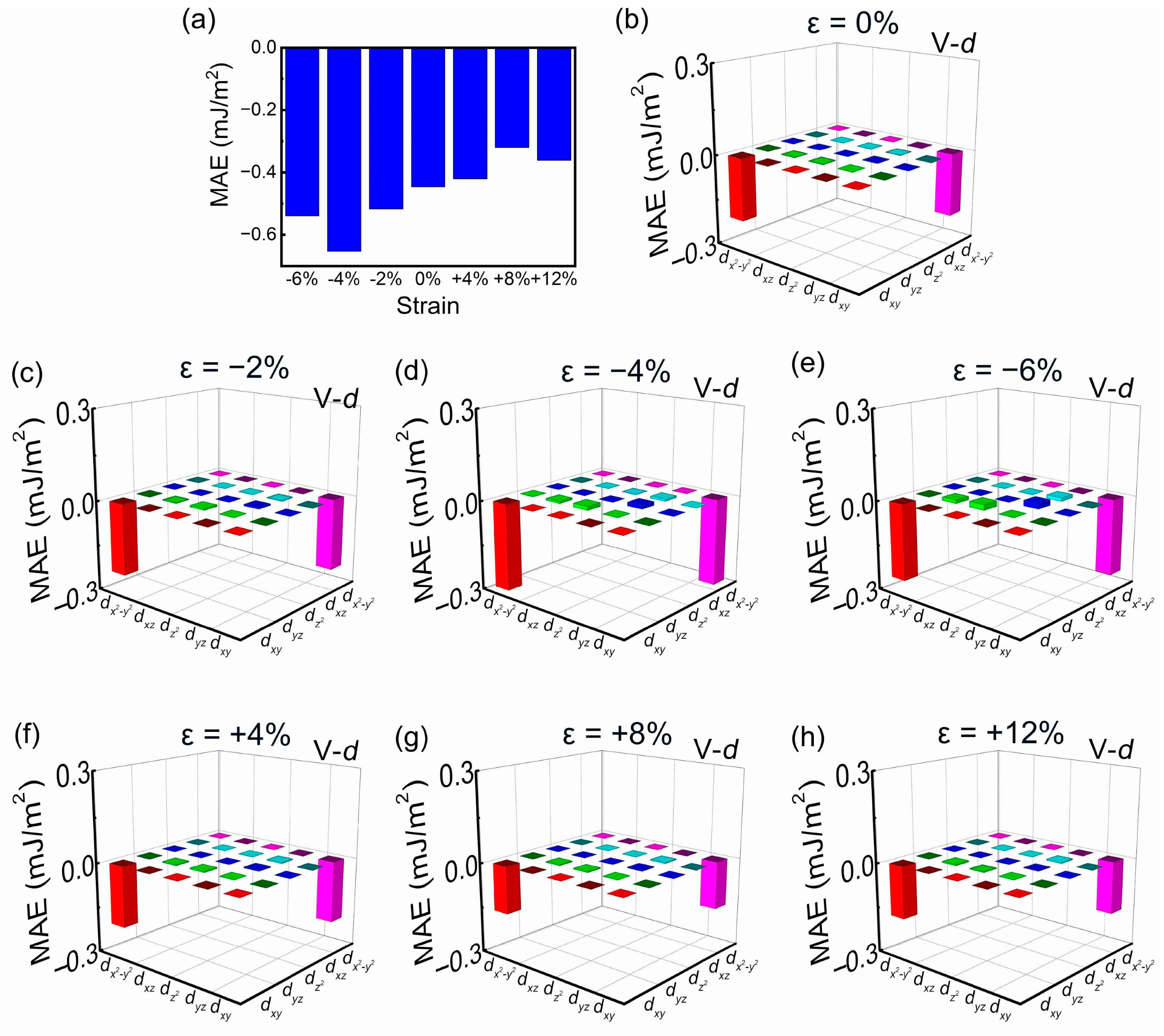
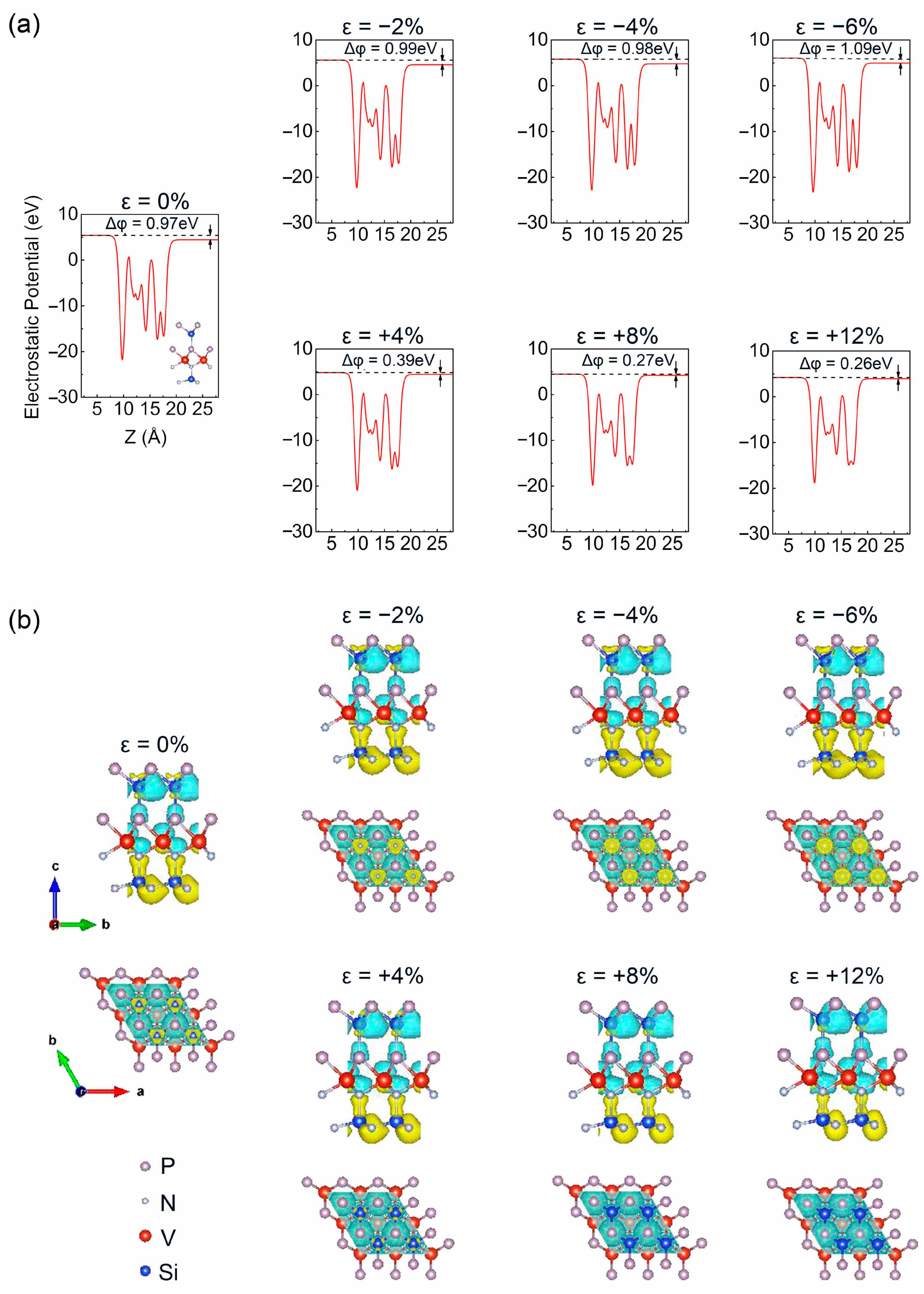
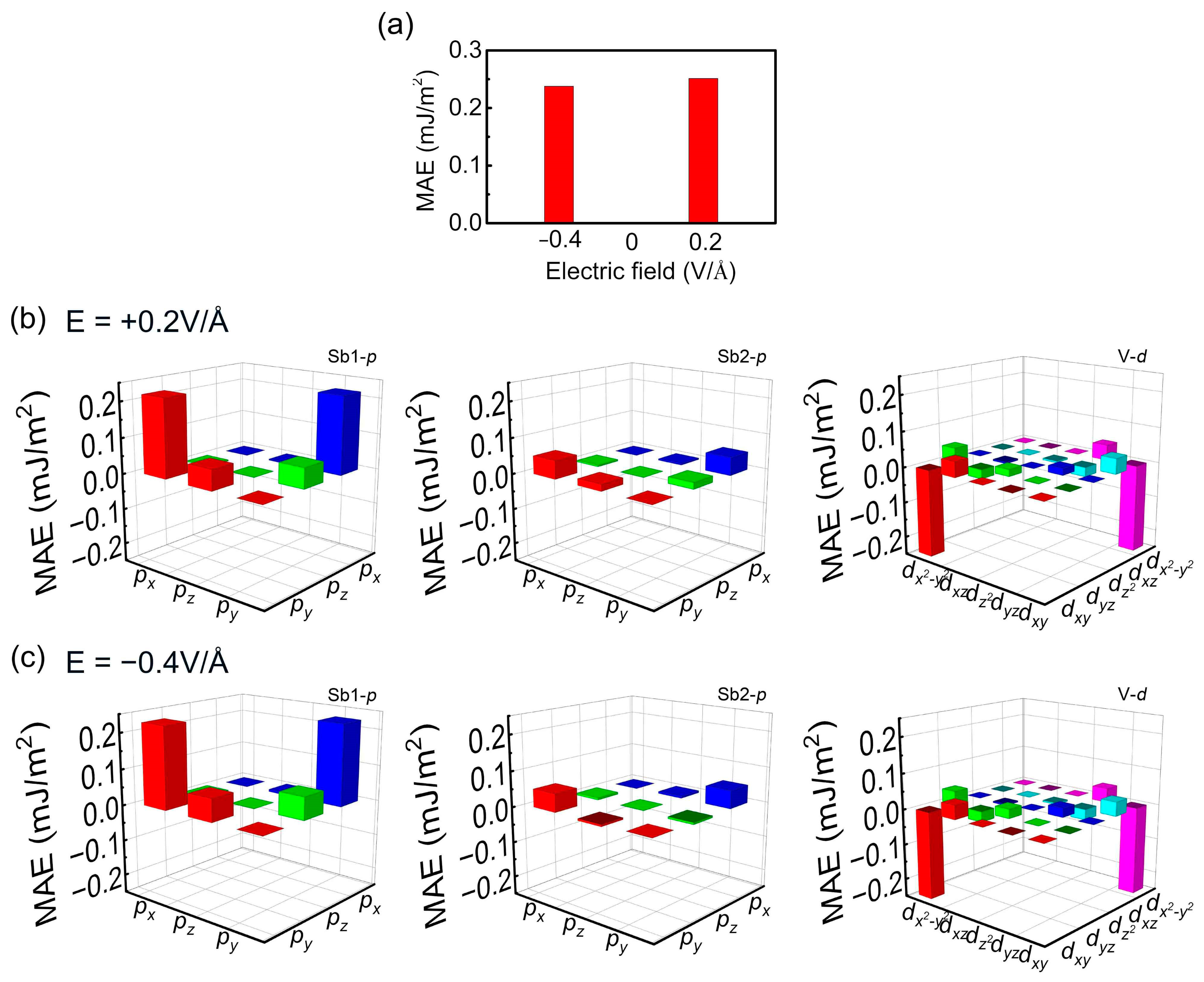
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Luo, M.; Li, Y.D.; Wang, K.J.; Shen, Y.H. Adsorption induced magnetic anisotropy in the two-dimensional magnet CrCl3. Solid State Commun. 2020, 321, 114048. [Google Scholar] [CrossRef]
- Zhang, W.; Hao, G.; Zhang, R.; Xu, J.; Ye, X.; Li, H. Effects of vertical strain and electrical field on electronic properties and Schottky contact of graphene/MoSe2 heterojunction. J. Phys. Chem. Solids 2021, 157, 110189. [Google Scholar] [CrossRef]
- Marjaoui, A.; Tamerd, M.A.; El Kasmi, A.; Diani, M.; Zanouni, M. First-principles calculations to investigate structural, electronic and optical properties of Janus AsMC3 (M: Sb, Bi) monolayers for optoelectronic applications. Solid State Commun. 2022, 343, 114667. [Google Scholar] [CrossRef]
- Li, F.; Yang, D.; Qiao, L.; Eglitis, R.I.; Jia, R.; Yi, Z.; Zhang, H. Novel 2D boron nitride with optimal direct band gap: A theoretical prediction. Appl. Surf. Sci. 2022, 578, 151929. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Bafekry, A.; Faraji, M.; Fadlallah, M.M.; Bagheri Khatibani, A.; Abdolahzadeh Ziabari, A.; Ghergherehchi, M.; Nedaei, S.; Shayesteh, S.F.; Gogova, D. Tunable electronic and magnetic properties of MoSi2N4 monolayer via vacancy defects, atomic adsorption and atomic doping. Appl. Surf. Sci. 2021, 559, 149862. [Google Scholar] [CrossRef]
- Zhang, C.; Ji, W.; Li, S.; Li, P.; Zhang, C.; Wang, P. 2D ternary nitrides XNY (X =Ti, Zr, Hf; Y = F, Cl, Br) with applications as photoelectric and photocatalytic materials featuring mechanical and optical anisotropy: A DFT study. Solid State Chem. 2021, 303, 122517. [Google Scholar] [CrossRef]
- Srivastava, M.; Srivastava, A.; Pandey, S. Suitability of graphene monolayer as sensor for carcinogenic heavy metals in water: A DFT investigation. Appl. Surf. Sci. 2020, 517, 146021. [Google Scholar] [CrossRef]
- Sun, Q.; Li, J.; Yang, Z.; Wu, R. Cr2NX2 MXene (X = O, F, OH): A 2D ferromagnetic half-metal. Appl. Phys. Lett. 2021, 119, 062404. [Google Scholar] [CrossRef]
- Kanahashi, K.; Pu, J.; Takenobu, T. 2D Materials for large-area flexible thermoelectric devices. Adv. Energy Mater. 2020, 10, 1902842. [Google Scholar] [CrossRef]
- Wolf, S.A.; Awschalom, D.D.; Buhrman, R.A.; Daughton, J.M.; von Molnar, S.; Roukes, M.L.; Chtchelkanova, A.Y.; Treger, D.M. Spintronics: A spin-based electronics vision for the future. Science 2001, 294, 1488–1495. [Google Scholar] [CrossRef] [Green Version]
- Meng, R.S.; Pereira, L.D.C.; Locquet, J.P.; Afananev, V.; Pourtois, G.; Houssa, M. Hole-doping induced ferromagnetism in 2D materials. NPJ Comput. Mater. 2022, 8, 230. [Google Scholar] [CrossRef]
- Molle, A.; Goldberger, J.; Houssa, M.; Xu, Y.; Zhang, S.C.; Akinwande, D. Buckled two-dimensional Xene sheets. Nat. Mater. 2017, 16, 163–169. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Zhang, S.C. Two-dimensional time-reversal-invariant topological superconductivity in a doped quantum spin-Hall insulator. Phys. Rev. B 2014, 90, 054503. [Google Scholar] [CrossRef] [Green Version]
- Qi, S.; Jiang, J.; Wang, X.; Mi, W. Valley polarization, magnetic anisotropy and Dzyaloshinskii-Moriya interaction of two-dimensional graphene/Janus 2H-VSeX (X = S, Te) heterostructures. Carbon 2021, 174, 540–555. [Google Scholar] [CrossRef]
- Chauhan, P.; Singh, J.; Kumar, A. Mechanical, optical and thermoelectric properties of Janus BiTeCl monolayer. J. Phys. Chem. Solids 2022, 167, 110758. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, M.; Lin, H.; Hou, T.; Wang, L.; Li, Y.; Lee, S.T. Janus structures of transition metal dichalcogenides as the heterojunction photocatalysts for water splitting. J. Phys. Chem. C 2018, 122, 3123–3129. [Google Scholar] [CrossRef]
- Bao, J.; Qiu, J.; Liu, X. Large in-plane piezoelectricity of Janus Bi2X2Y (X = S, Se, Te; Y = S, Se, Te; X≠Y) monolayers with polyatomic thickness. Mater. Lett. 2021, 296, 129878. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, S.; Kholmanov, I.; Dong, L.; Er, D.; Chen, W.; Guo, H.; Jin, Z.; Shenoy, V.B.; Shi, L.; et al. Janus monolayer transition-metal dichalcogenides. ACS Nano 2017, 11, 8192–8198. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lin, Z.; Du, Y.; Qiu, J.; Chen, X.; Yu, J. The piezoelectricity of 2D Janus ZnBrI: Multiscale prediction. Chem. Phys. Lett. 2022, 794, 139506. [Google Scholar] [CrossRef]
- Ma, X.; Tian, Y.; Zhao, P.; Wu, X.; Jing, T.; Zhang, J. Janus MoCrSSe monolayer: A strong two-dimensional polar antiferromagnet. Appl. Surf. Sci. 2022, 581, 152420. [Google Scholar] [CrossRef]
- Manchon, A.; Koo, H.C.; Nitta, J.; Frolov, S.M.; Duine, R.A. New perspectives for Rashba spin-orbit coupling. Nat. Mater. 2015, 14, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Chen, J.; Zhang, B.; Duan, H.; Ouyang, F. Manipulation of the Rashba spin-orbit coupling of a distorted 1T-Phase Janus WSSe monolayer: Dominant role of charge transfer and orbital components. Phys. Rev. B 2021, 103, 195114. [Google Scholar] [CrossRef]
- Sengupta, A. First principles design of 2 dimensional Nickel dichalcogenide Janus materials NiXY (X,Y = S, Se, Te). Comp. Mater. Sci. 2022, 206, 111278. [Google Scholar] [CrossRef]
- Guo, S.; Guo, X.; Han, R.; Deng, Y. Predicted Janus SnSSe monolayer: A comprehensive first-principles study. Phys. Chem. Chem. Phys. 2019, 21, 24620–24628. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Wang, Z. Mechanical and electronic properties of Janus monolayer transition metal dichalcogenides. J. Phys. Condens. Matter 2018, 30, 215301. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhu, Z.; Tahir, M.; Schwingenschlögl, U. Spin-orbit–induced spin splittings in polar transition metal dichalcogenide monolayers. Europhys. Lett. 2013, 102, 57001. [Google Scholar] [CrossRef]
- Novoselov, K.S. Discovery of 2D Van Der Waals layered MoSi2N4 family. Natl. Sci. Rev. 2020, 7, 1842–1844. [Google Scholar] [CrossRef]
- Cui, Z.; Luo, Y.; Yu, J.; Xu, Y. Tuning the electronic properties of MoSi2N4 by molecular doping: A first principles investigation. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 134, 114873. [Google Scholar] [CrossRef]
- Hong, Y.; Liu, Z.; Wang, L.; Zhou, T.; MA, W.; Xu, C.; Feng, S.; Chen, L.; Chen, M.; Sun, D.; et al. Chemical vapor deposition of layered two-dimensional MoSi2N4 materials. Science 2020, 369, 670–674. [Google Scholar] [CrossRef]
- Yang, J.S.; Zhao, L.; Li, S.Q.; Liu, H.; Wang, L.; Chen, M.; Gao, J.; Zhao, J. Accurate electronic properties and non-linear optical response of two-dimensional MA2Z4. Nanoscale 2021, 13, 5479–5488. [Google Scholar] [CrossRef]
- Yang, C.; Song, Z.; Sun, X.; Lu, J. Valley pseudospin in monolayer MoSi2N4 and MoSi2As4. Phys. Rev. B 2021, 103, 035308. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, G.; Wang, Y.; Huang, C.; Liu, Y.; Ouyang, C.; Hu, J. Heavy 2D VSi2N4: High capacity and full battery open-circuit voltage as Li/Na-ion batteries anode. Appl. Surf. Sci. 2022, 593, 153354. [Google Scholar] [CrossRef]
- Mortazavi, B.; Javvaji, B.; Shojaei, F.; Rabczuk, T.; Shapeev, A.V.; Zhuang, X. Exceptional piezoelectricity, high thermal conductivity and stiffness and promising photocatalysis in two-dimensional MoSi2N4 family confirmed by first-principles. Nano Energy 2021, 82, 105716. [Google Scholar] [CrossRef]
- Akanda, M.R.K.; Lake, R.K. Magnetic properties of NbSi2N4, VSi2N4, and VSi2P4 monolayers. Appl. Phys. Lett. 2021, 119, 052402. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal- amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Semiempirical, J. GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Blöchl, P. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.Q.; Wang, X.C.; Mi, W.B. Spin-dependent electronic structure and magnetic properties of 2D Janus Mn2CFCl/CuBiP2Se6 van der Waals multiferroic heterostructures. Adv. Theor. Simul. 2021, 4, 2100302. [Google Scholar] [CrossRef]
- Hu, J.; Ouyang, C.; Yang, S.A.; Yang, H.Y. Germagraphene as a promising anode material for lithium-ion batteries predicted from first-principles calculations. Nanoscale Horiz. 2019, 4, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Chadi, D.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1977, 16, 1746–1747. [Google Scholar] [CrossRef]
- Daalderop, G.H.O.; Kelly, P.J.; Schuurmsans, M.F.H. First-principles calculation of the magnetocrystalline anisotropy energy of iron, cobalt, and nickel. Phys. Rev. B 1990, 41, 11919. [Google Scholar] [CrossRef]
- Gao, Y.Q.; Zhou, B.Z.; Wang, X.C. Biaxial strain, electric field and interlayer distance-tailored electronic structure and magnetic properties of two-dimensional g-C3N4/Li-adsorbed Cr2Ge2Te6 van der Waals heterostructures. Phys. Chem. Chem. Phys. 2021, 23, 6171–6181. [Google Scholar] [CrossRef]
- Koumpouras, K.; Larsson, J.A. Distinguishing between chemical bonding and physical binding using electron localization function (ELF). J. Phys. Condens. Matter 2020, 32, 315502. [Google Scholar] [CrossRef]
- Li, S.; Zhou, M.; Wang, X.; Zheng, F.; Shao, X.; Zhang, P. Two-dimensional uranium halide monolayers UX3 (X = Cl, Br) with high Curie temperatures. Phys. Lett. A 2021, 394, 127078. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a = b (Å) | M (μB) | MAE | |
|---|---|---|---|
| VSi2N4 | 2.87 | 0.91 | IMA |
| VSi2P2N2 | 3.09 | 0.68 | IMA |
| VSi2As2N2 | 3.08 | 0.97 | IMA |
| VSi2Sb2N2 | 3.01 | / | / |
| VSi2Bi2N2 | 3.08 | 0.59 | PMA |
| ΔEFM (eV) | ΔENéel-AFM (eV) | ΔEstripy-AFM (eV) | ΔEzigzag-AFM (eV) | Magnetic Ground State | |
|---|---|---|---|---|---|
| VSi2P2N2 | 0 | 0.124 | 0.124 | 0.124 | FM |
| VSi2As2N2 | 0 | 0.191 | 0.191 | 0.191 | FM |
| VSi2Sb2N2 | / | / | / | / | / |
| VSi2Bi2N2 | 0 | 0.031 | 0.025 | 0.030 | FM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Wang, X.; Mi, W. Two-Dimensional VSi2X2N2 (X = P, As, Sb, Bi) Janus Monolayers: Spin-Polarized Electronic Structure and Perpendicular Magnetic Anisotropy. Crystals 2023, 13, 1007. https://doi.org/10.3390/cryst13071007
Zhao Z, Wang X, Mi W. Two-Dimensional VSi2X2N2 (X = P, As, Sb, Bi) Janus Monolayers: Spin-Polarized Electronic Structure and Perpendicular Magnetic Anisotropy. Crystals. 2023; 13(7):1007. https://doi.org/10.3390/cryst13071007
Chicago/Turabian StyleZhao, Zhenxian, Xiaocha Wang, and Wenbo Mi. 2023. "Two-Dimensional VSi2X2N2 (X = P, As, Sb, Bi) Janus Monolayers: Spin-Polarized Electronic Structure and Perpendicular Magnetic Anisotropy" Crystals 13, no. 7: 1007. https://doi.org/10.3390/cryst13071007
APA StyleZhao, Z., Wang, X., & Mi, W. (2023). Two-Dimensional VSi2X2N2 (X = P, As, Sb, Bi) Janus Monolayers: Spin-Polarized Electronic Structure and Perpendicular Magnetic Anisotropy. Crystals, 13(7), 1007. https://doi.org/10.3390/cryst13071007