
Synthesis, Crystal Structure, and Computational Investigations of 2-(2-(4-Fluorophenyl)-2-oxoethyl)-6-methyl-5-(4-methylbenzyl)pyridazin-3(2H)-one as Antiviral Agent

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Instruments
2.2. Synthesis Procedure for the Preparation of FOMMP
2.3. X-ray Analysis
2.4. Computational Details
3. Results
3.1. Description of Crystal Structure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC Deposition Number | CCDC 1960994 |
|---|---|
| Chemical formula | C21H19FN2O2 |
| Mr | 350.38 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 296 |
| a, b, c (Å) | 13.7166 (15), 5.0232 (4), 27.016 (3) |
| β (°) | 103.413 (8) |
| V (Å3) | 1810.6 (3) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.09 |
| Crystal size (mm) | 0.78 × 0.34 × 0.05 |
| Data collection | |
| Diffractometer | STOE IPDS 2 |
| Absorption correction | Intergration (X-RED32 [39]) |
| Tmin, Tmax | 0.980, 0.993 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 10,673, 3174, 1320 |
| Rint | 0.084 |
| (sin θ/λ)max (Å−1) | 0.596 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.091, 0.334, 1.02 |
| No. of reflections | 3174 |
| No. of parameters | 236 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.21, −0.29 |
3.2. Optimized Molecular Geometry
3.3. FT-IR Spectra Analysis
3.4. NMR Spectra Analysis
3.5. ESI-MS Study
3.6. Hirshfeld Surface Analysis
3.7. Frontier Molecular Orbitals (FMOs) Analysis
3.7.1. Charge Transfer and Excitation Analysis
3.7.2. Natural Bond Orbital Analysis
- π (C9–C10) transition to π* (C11–C12) and π* (C13–C14) has stabilization energies of 18.36 and 19.08 kJ/mol, respectively;
- π (C11–C12) transition to π* (C9–C10) and π* (C13–C14) has stabilization energies of 20.39 and 18.68 kJ/mol, respectively;
- π (C13–C14) transition to π* (C9–C10) and π* (C13–C14) has stabilization energies of 20.41 and 21.05 kJ/mol, respectively;
- π (C20–C21) transition to π* (C22–C23) and π* (C24–C25) has stabilization energies of 18.35 and 20.27 kJ/mol, respectively;
- π (C22–C23) transition to π* (C20–C21) and π* (C24–C25) has stabilization energies of 20.48 and 16.09 kJ/mol, respectively;
- π (C24–C25) transition to π* (C20–C21) and π* (C22–C23) has stabilization energies of 17.83 and 23.12 kJ/mol, respectively.
- LP(1) N5 transition to σ* (C3–C4) with stabilization energy of 10.64 kJ/mol;
- LP(1) N6 transition to π* (C1–O7) and π* (C4–N5)with stabilization energy of 29.65 and 22.18 kJ/mol, respectively;
- LP(2) O7 transition to σ* (C1–C2) and σ* (C1–N6)with stabilization energy of 20.05 and 27.22 kJ/mol, respectively;
- LP(2) O19 transition to σ* (C17–C18) and σ* (C18–C20) with stabilization energy of 23 and 20.82 kJ/mol, respectively;
- LP(2) F26 transition to σ* (C22–C23) and σ* (C23–C24) with stabilization energy of 7.22 and 7.15 kJ/mol, respectively;
- LP(3) F26 transition to π* (C22–C23)with stabilization energy of 21.7 kJ/mol.
3.7.3. Electron Localization Function (ELF)
3.7.4. Molecular Electrostatic Potential Analysis
3.7.5. Molecular Docking and Drug-Likeness Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flefel, E.M.; Tantawy, W.A.; El-Sofany, W.I.; El-Shahat, M.; El-Sayed, A.A.; Abd-Elshafy, D.N. Synthesis of some new pyridazine derivatives for anti-HAV evaluation. Molecules 2017, 22, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhan, P.; Liu, H.; Pannecouque, C.; Balzarini, J.; De Clercq, E.; Liu, X. Synthesis and biological evaluation of pyridazine derivatives as novel HIV-1 NNRTIs. Bioorg. Med. Chem. 2013, 21, 2128–2134. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Bhatia, R.; Pani, B.; Gupta, D. Synthesis, crystal structures and biological evaluation of new pyridazine derivatives. J. Mol. Struct. 2020, 1200, 127084. [Google Scholar] [CrossRef]
- Daoui, S.; Direkel, Ş.; Ibrahim, M.M.; Tüzün, B.; Chelfi, T.; Al-Ghorbani, M.; Karrouchi, K. Synthesis, Spectroscopic Characterization, Antibacterial Activity, and Computational Studies of Novel Pyridazinone Derivatives. Molecules 2023, 28, 678. [Google Scholar] [CrossRef]
- Mantu, D.; Luca, M.C.; Moldoveanu, C.; Zbancioc, G.; Mangalagiu, I.I. Synthesis and antituberculosis activity of some new pyridazine derivatives. Part II. Eur. J. Med. Chem. 2010, 45, 5164. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Kassab, A.E.; El-Malah, A.A.; Hassan, M.S. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2019, 171, 25–37. [Google Scholar] [CrossRef]
- Barberot, C.; Moniot, A.; Allart-Simon, I.; Malleret, L.; Yegorova, T.; Laronze-Cochard, M.; Gérard, S. Synthesis and biological evaluation of pyridazinone derivatives as potential anti-inflammatory agents. Eur. J. Med. Chem. 2018, 146, 139–146. [Google Scholar] [CrossRef]
- Rafi, U.M.; Mahendiran, D.; Devi, V.G.; Doble, M.; Rahiman, A.K. Pyridazine-based heteroleptic copper (II) complexes as potent anticancer drugs by inducing apoptosis and S-phase arrest in breast cancer cell. Inorg. Chim. Acta 2018, 482, 160–169. [Google Scholar] [CrossRef]
- Livermore, D.G.; Bethell, R.C.; Cammack, N.; Hancock, A.P.; Hann, M.M.; Green, D.V.; Orr, D.C. Synthesis and anti-HIV-1 activity of a series of imidazo [1, 5-b] pyridazines. J. Med. Chem. 1993, 36, 3784–3794. [Google Scholar] [CrossRef]
- Zerroug, A.; Belaidi, S.; BenBrahim, I.; Sinha, L.; Chtita, S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. Sci. 2018, 31, 595–601. [Google Scholar] [CrossRef]
- Demirayak, S.; Karaburun, A.C.; Beis, R. Some pyrrole substituted aryl pyridazinone and phthalazinone derivatives and their antihypertensive activities. Eur. J. Med. Chem. 2004, 39, 1089–1095. [Google Scholar] [CrossRef]
- Partap, S.; Akhtar, M.J.; Yar, M.S.; Hassan, M.Z.; Siddiqui, A.A. Pyridazinone hybrids: Design, synthesis and evaluation as potential anticonvulsant agents. Bioorg. Chem. 2018, 77, 74–83. [Google Scholar] [CrossRef]
- Jacomini, A.P.; da Silva, M.J.; Poletto, J.; Ribeiro, G.M.; Yokoyama, J.T.; Bidóia, D.L.; Rosa, F.A. Potential antileishmanial activity of 4-N-Acylhydrazone Pyrazolo [3, 4-d] pyridazin-7-ones: Synthesis, in vitro biological evaluations and computational studies. J. Braz. Chem. Soc. 2018, 29, 2657–2668. [Google Scholar] [CrossRef]
- Lümmen, P. Complex I inhibitors as insecticides and acaricides. Biochim. Biophys. Acta Bioenerg. 1998, 1364, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Dekeyser, M.A. Acaricide mode of action. Pest. Manag. Sci. 2005, 61, 103–110. [Google Scholar] [CrossRef]
- Weissmuller, J.; Tietjen, K.G.; Stendel, W.; Wachendorff-Neumann, U. U.S. Patent No. 5,004,744, CI. 514-247.000; U.S. Patent and Trademark Office: Washington, DC, USA, 1991.
- Asif, M. Antifeedant, herbicidal and molluscicidal activities of pyridazinone compounds. Mini-Rev. Org. Chem. 2013, 10, 113–122. [Google Scholar] [CrossRef]
- Abraham, C.S.; Prasana, J.C.; Muthu, S. Quantum mechanical, spectroscopic and docking studies of 2-amino-3-bromo-5-nitropyridine by density functional method. Spectrochim. Acta A 2017, 181, 153–163. [Google Scholar] [CrossRef]
- Pillai, R.R.; Karrouchi, K.; Fettach, S.; Armaković, S.; Armaković, S.J.; Brik, Y.; Faouzi, M.E.A. Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamics simulations and biological evaluation of Schiff bases tethered 1, 2, 4-triazole and pyrazole rings. J. Mol. Struct. 2019, 1177, 47–54. [Google Scholar] [CrossRef]
- Tighadouini, S.; Radi, S.; Abrigach, F.; Benabbes, R.; Eddike, D.; Tillard, M. Novel β-keto–enol pyrazolic compounds as potent antifungal agents. Design, synthesis, crystal structure, DFT, homology modeling, and docking studies. J. Chem. Inf. Model. 2019, 59, 1398–1409. [Google Scholar] [CrossRef]
- Karrouchi, K.; Yousfi, E.B.; Sebbar, N.K.; Ramli, Y.; Taoufik, J.; Ouzidan, Y.; Radi, S. New pyrazole-hydrazone derivatives: X-ray analysis, molecular structure investigation via density functional theory (DFT) and their high in-situ catecholase activity. Int. J. Mol. Sci. 2017, 18, 2215. [Google Scholar] [CrossRef] [Green Version]
- Abraham, C.S.; Muthu, S.; Prasana, J.C.; Rizwana, B.F.; Armaković, S.; Armaković, S.J. Vibrational and electronic absorption spectroscopic profiling, natural hybrid orbital, charge transfer, electron localization function and molecular docking analysis on 3-amino-3-(2-nitrophenyl) propionic acid. J. Mol. Struct. 2018, 1171, 733–746. [Google Scholar] [CrossRef]
- Abraham, C.S.; Muthu, S.; Prasana, J.C.; Armaković, S.J.; Armaković, S.; AS, B.G. Spectroscopic profiling (FT-IR, FT-Raman, NMR and UV-Vis), autoxidation mechanism (H-BDE) and molecular docking investigation of 3-(4-chlorophenyl)-N, N-dimethyl-3-pyridin-2-ylpropan-1-amine by DFT/TD-DFT and molecular dynamics: A potential SSRI drug. Comput. Biol. Chem. 2018, 77, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Bouzian, Y.; Karrouchi, K.; Sert, Y.; Lai, C.H.; Mahi, L.; Ahabchane, N.H.; Essassi, E.M. Synthesis, spectroscopic characterization, crystal structure, DFT, molecular docking and in vitro antibacterial potential of novel quinoline derivatives. J. Mol. Struct. 2020, 1209, 127940. [Google Scholar] [CrossRef]
- El Kalai, F.; Çınar, E.B.; Lai, C.H.; Daoui, S.; Chelfi, T.; Allali, M.; Benchat, N. Synthesis, spectroscopy, crystal structure, TGA/DTA study, DFT and molecular docking investigations of (E)-4-(4-methylbenzyl)-6-styrylpyridazin-3 (2H)-one. J. Mol. Struct. 2021, 1228, 12943. [Google Scholar] [CrossRef]
- Daoui, S.; Faizi, M.S.H.; Kalai, F.E.; Saddik, R.; Dege, N.; Karrouchi, K.; Benchat, N. Crystal structure and the DFT and MEP study of 4-benzyl-2-[2-(4-fluorophenyl)-2-oxoethyl]-6-phenylpyridazin-3 (2H)-one. Acta Cryst. 2019, E75, 1030. [Google Scholar] [CrossRef] [Green Version]
- El Kalai, F.; Baydere, C.; Daoui, S.; Saddik, R.; Dege, N.; Karrouchi, K.; Benchat, N. Crystal structure and Hirshfeld surface analysis of ethyl 2-[5-(3-chlorobenzyl)-6-oxo-3-phenyl-1, 6-dihydropyridazin-1-yl] acetate. Acta Cryst. 2019, E75, 892–895. [Google Scholar] [CrossRef]
- Dadou, S.; Kansiz, S.; Daoui, S.; El Kalai, F.; Baydere, C.; Saddik, R.; Benchat, N. Crystal structures and Hirshfeld surface analyses of 4-benzyl-6-phenyl-4, 5-dihydropyridazin-3 (2H)-one and methyl 2-[5-(2, 6-dichlorobenzyl)-6-oxo-3-phenyl-1, 4, 5, 6-tetrahydropyridazin-1-yl] acetate. Acta Cryst. 2019, E75, 1679–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kalai, F.; Chelfi, T.; Benchat, N.; Bouklah, M.; Daoui, S.; Karrouchi, K.; Ben Hadda, T. New heterocyclic compounds based on pyridazinones scaffold as efficient inhibitor of corrosion of mild steel in acidic solution 1 M HCl. J. Bio-Tribo-Corros. 2020, 6, 89. [Google Scholar] [CrossRef]
- Bruker. APEX3, SAINT, SADABS & SHELXTL; Bruker AXS, Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Brandenburg, K.; Berndt, M. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2012. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09, Revision E.01; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5656. [Google Scholar] [CrossRef] [Green Version]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2018.
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Mod. 1999, 17, 57. [Google Scholar]
- Stoe, C. X-AREA (Version 1.18) and X-RED (Version 1.04); Stoe & Cie: Darmstadt, Germany, 2002. [Google Scholar]
- Bak, B.; Christensen, D.; Dixon, W.B.; Hansen-Nygaard, L.; Rastrup-Andersen, J. Benzene ring distortion by one substituent. microwave determination of the complete structure of benzonitrile. J. Chem. Phys. 1962, 37, 2027. [Google Scholar] [CrossRef]
- Bailey, A.S.; Prout, C.K. 899. Molecular complexes. Part III. The crystal and molecular structure of the 1:2 molecular compound of bis-8-hydroxyquinolinatocopper (II) and picryl azide. J. Chem. Soc 1965, 4867–4881. [Google Scholar] [CrossRef]
- Schomaker, V.T.; Pauling, L. The electron diffraction investigation of the structure of benzene, pyridine, pyrazine, butadiene-1, 3, cyclopentadiene, furan, pyrrole, and thiophene. J. Am. Chem. Soc. 1939, 61, 1769. [Google Scholar] [CrossRef]
- James, M.N.G.; Williams, G.J.B. Structural studies of histamine H1 effector molecules: The crystal structure of the antihistamine drug (+)-chlorpheniramine maleate;[(+)-S-1-(p-chlorophenyl)-1-(2-pyridyl)-3-N, N-dimethylpropylamine maleate]. J. Chem. 1974, 52, 1872–1879. [Google Scholar] [CrossRef]
- Srinivasan, R. On the method of least squares as applied to the refinement of crystal structures. Acta Cryst. 1961, 14, 1163–1167. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandán, S.A.; Sert, Y.; El-Marzouqi, H.; Radi, S.; Ferbinteanu, M.; Garcia, Y. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N′-(4-(dimethylamino) benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2020, 1219, 128541. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandán, S.A.; Sert, Y.; El Karbane, M.; Radi, S.; Ferbinteanu, M.; Garcia, Y. Synthesis, structural, molecular docking and spectroscopic studies of (E)-N′-(4-methoxybenzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2021, 1225, 129072. [Google Scholar] [CrossRef]
- Karrouchi, K.; Brandán, S.A.; Hassan, M.; Bougrin, K.; Radi, S.; Ferbinteanu, M.; Garcia, Y. Synthesis, X-ray, spectroscopy, molecular docking and DFT calculations of (E)-N′-(2, 4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2021, 1228, 129714. [Google Scholar] [CrossRef]
- Karrouchi, K.; Fettach, S.; Jotani, M.M.; Sagaama, A.; Radi, S.; Ghabbour, H.A.; Issaoui, N. Synthesis, crystal structure, hirshfeld surface analysis, DFT calculations, anti-diabetic activity and molecular docking studies of (E)-N′-(5-bromo-2-hydroxybenzylidene) isonicotinohydrazide. J. Mol. Struct. 2020, 1221, 128800. [Google Scholar] [CrossRef]
- Daoui, S.; Baydere, C.; Akman, F.; El Kalai, F.; Mahi, L.; Dege, N.; Benchat, N. Synthesis, X-ray crystallography, vibrational spectroscopy, thermal and DFT studies of (E)-6-(4-methylstyryl)-4, 5-dihydropyridazin-3 (2H)-one. J. Mol. Struct. 2021, 1225, 129180. [Google Scholar] [CrossRef]
- El Kalai, F.; Karrouchi, K.; Baydere, C.; Daoui, S.; Allali, M.; Dege, N.; Brandan, S.A. Synthesis, crystal structure, spectroscopic studies, NBO, AIM and SQMFF calculations of new pyridazinone derivative. J. Mol. Struct. 2021, 1223, 129213. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–2020. [Google Scholar] [CrossRef]
- Demircioğlu, Z.; Kaştaş, G.; Kaştaş, Ç.A.; Frank, R. Spectroscopic, XRD, Hirshfeld surface and DFT approach (chemical activity, ECT, NBO, FFA, NLO, MEP, NPA& MPA) of (E)-4-bromo-2-[(4-bromophenylimino) methyl]-6-ethoxyphenol. J. Mol. Struct. 2019, 1191, 129–137. [Google Scholar]
- Uzun, S.; Demircioğlu, Z.; Taşdoğan, M.; Ağar, E. Quantum chemical and X-ray diffraction studies of (E)-3-(((3, 4-dimethoxybenzyl) imino) methyl) benzene-1, 2-diol. J. Mol. Struct. 2020, 1206, 127749. [Google Scholar] [CrossRef]
- Padmaja, L.; Ravikumar, C.; Sajan, D.; Joe, I.H.; Jayakumar, V.S.; Pettit, G.R.; Neilsen, F.O. Density functional study on the structural conformations and intramolecular charge transfer from the vibrational spectra of the anticancer drug combretastatin-A2. J. Raman. Spectrosc. 2020, 40, 419–428. [Google Scholar] [CrossRef]
- Ravikumar, C.; Joe, I.H.; Jayakumar, V.S. Charge transfer interactions and nonlinear optical properties of push–pull chromophore benzaldehyde phenylhydrazone: A vibrational approach. Chem. Phys. Lett. 2008, 460, 552–558. [Google Scholar] [CrossRef]
- Zhou, Z.; Parr, R.G. Activation hardness: New index for describing the orientation of electrophilic aromatic substitution. J. Am. Chem. Soc. 1990, 112, 5720–5724. [Google Scholar] [CrossRef]
- Pearson, R.G. Recent advances in the concept of hard and soft acids and bases. J. Chem. Ed. 1987, 64, 561. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Kovačević, N.; Kokalj, A. Analysis of molecular electronic structure of imidazole-and benzimidazole-based inhibitors: A simple recipe for qualitative estimation of chemical hardness. Corros. Sci. 2011, 53, 909–921. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Subramanian, V.; Roy, D.R.; Chattaraj, P.K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004, 12, 5533. [Google Scholar] [CrossRef]
- Guido, C.A.; Cortona, P.; Mennucci, B.; Adamo, C. On the metric of charge transfer molecular excitations: A simple chemical descriptor. J. Chem. Theory Comput. 2013, 9, 3118–3126. [Google Scholar] [CrossRef]
- Mandal, S.; Nandi, S.; Anoop, A.; Chattaraj, P.K. Viability of aromatic all-pnictogen anions. Phys. Chem. Chem. Phys. 2016, 18, 11738–11745. [Google Scholar] [CrossRef]
- Williams, D.E. Comprehensive Chemical Kinetics. Rev. Comp. Chem. 1991, 2, 226. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 4–17. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.; Powell, A.J.; Douangamath, A.; Protein Data Bank. PanDDA Analysis Group Deposition—Crystal Structure of COVID-19 Main Protease in Complex with Z219104216; Protein Data Bank Japan: Suita, Japan, 2020. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]

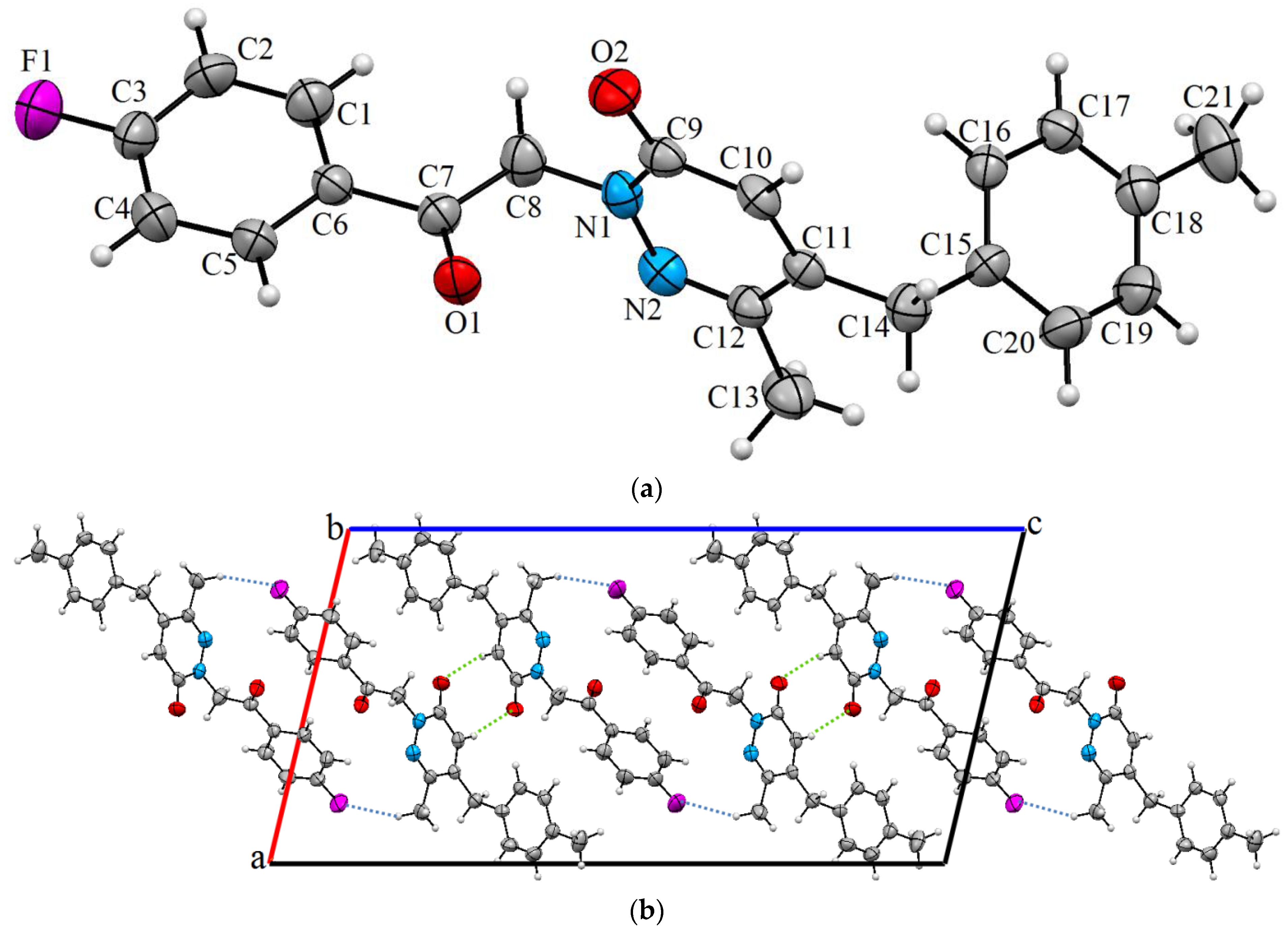











| D–H···A | D–H | H···A | D···A |
|---|---|---|---|
| C10–H10···O2 i | 0.93 | 2.59 | 3.496(8) |
| C8–H8A···O1 ii | 0.97 | 2.62 | 3.543(9) |
| C13–H13B···F1 iii | 0.96 | 2.46 | 3.357(6) |
| Molecular Properties | EHOMO | ELOMO | EHOMO−1 | ELUMO+1 | EHUMO−2 | ELUMO+2 |
|---|---|---|---|---|---|---|
| Energy (eV) | −6.19 | −1.863 | 6.460 | 1.777 | 6.701 | 1.163 |
| Energy Gap (eV) | 4.327 | 4.683 | 5.538 | |||
| Ionization Potential (I) | 6.190 | 6.460 | 6.701 | |||
| Electron Affinity (A) | 1.863 | 1.777 | 1.162 | |||
| Global Hardness (η) | 2.164 | 2.341 | 2.770 | |||
| Electro negativity (χ) | 4.027 | 4.118 | 3.932 | |||
| Global Softness (σ) | 0.231 | 0.214 | 0.181 | |||
| Chemical Potential (µ) | −4.027 | −4.118 | −3.932 | |||
| Global Electrophilicity (ω) | 3.747 | 3.621 | 2.792 |
| Parameter | Excited States | ||
|---|---|---|---|
| S1 | S2 | S3 | |
| Overlap integral of electron–hole, S | 0.02 | 0.08 | 0.08 |
| Charge transfer length, D (Å) | 11.09 | 6.00 | 8.16 |
| Excitation energy, ΔE (eV) | 3.45 | 3.72 | 3.93 |
| RMSD of electron | |||
| X | 1.4 | 2.613 | 2.20 |
| Y | 1.33 | 1.30 | 1.34 |
| Z | 0.86 | 1.02 | 0.91 |
| RMSD of hole | |||
| X | 2.52 | 2.03 | 3.08 |
| Y | 0.98 | 0.99 | 1.25 |
| Z | 0.78 | 0.83 | 1.00 |
| t index | |||
| X | 9.131 | 3.61 | 5.52 |
| Y | −1.09 | −0.65 | −1.22 |
| Z | −0.67 | −0.15 | −0.94 |
| Excited States | Band Gap (eV) | Wavelength (nm) | Energy (cm−1) | Osc. Strength |
|---|---|---|---|---|
| 1 | 3.452 | 359.445 | 27,820.67 | 0.029 |
| 2 | 3.719 | 333.629 | 29,973.38 | 0.021 |
| 3 | 3.937 | 315.142 | 31,731.68 | 0.010 |
| Descriptor | Values |
|---|---|
| Hydrogen bond donor (HBD) | 4 |
| Hydrogen bond acceptor (HBA) | 0 |
| AlogP | 4.01 |
| Polar surface area (PSA) [Å2] | 51.96 |
| Molar refractivity | 98.82 |
| Number of rotatable bonds | 5 |
| Ligand | Protein PDB ID | Binding Amino Acid Residues | Bond Distance (Å) | Binding Energy (kcal/mol) | Inhibition Constants (uM) |
|---|---|---|---|---|---|
| FOMMP | 5R82 | Arg217(A) | 2.99 | −4.90 | 257.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Kalai, F.; Abraham, C.S.; Kansiz, S.; Oulmidi, A.; Muthu, S.; Prasana, J.C.; Dege, N.; Abuelizz, H.A.; Al-Salahi, R.; Benchat, N.; et al. Synthesis, Crystal Structure, and Computational Investigations of 2-(2-(4-Fluorophenyl)-2-oxoethyl)-6-methyl-5-(4-methylbenzyl)pyridazin-3(2H)-one as Antiviral Agent. Crystals 2023, 13, 1098. https://doi.org/10.3390/cryst13071098
El Kalai F, Abraham CS, Kansiz S, Oulmidi A, Muthu S, Prasana JC, Dege N, Abuelizz HA, Al-Salahi R, Benchat N, et al. Synthesis, Crystal Structure, and Computational Investigations of 2-(2-(4-Fluorophenyl)-2-oxoethyl)-6-methyl-5-(4-methylbenzyl)pyridazin-3(2H)-one as Antiviral Agent. Crystals. 2023; 13(7):1098. https://doi.org/10.3390/cryst13071098
Chicago/Turabian StyleEl Kalai, Fouad, Christina Susan Abraham, Sevgi Kansiz, Afaf Oulmidi, Sambantham Muthu, Johanan Christian Prasana, Necmi Dege, Hatem A. Abuelizz, Rashad Al-Salahi, Noureddine Benchat, and et al. 2023. "Synthesis, Crystal Structure, and Computational Investigations of 2-(2-(4-Fluorophenyl)-2-oxoethyl)-6-methyl-5-(4-methylbenzyl)pyridazin-3(2H)-one as Antiviral Agent" Crystals 13, no. 7: 1098. https://doi.org/10.3390/cryst13071098