
Effects of Tetrafluorocyclohexa-1,3-Diene Ring Position on Photoluminescence and Liquid-Crystalline Properties of Tricyclic π-Conjugated Molecules



Abstract
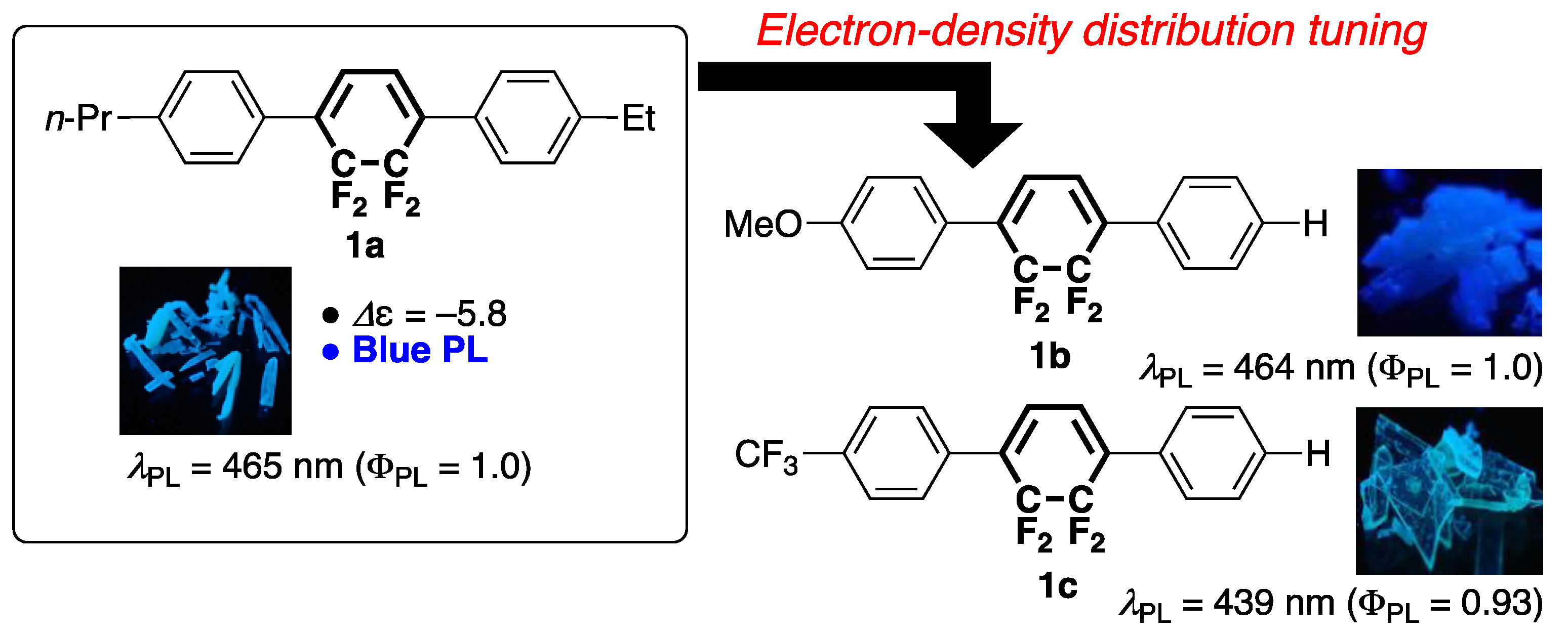
:1. Introduction
2. Materials and Methods
2.1. General Characterization
2.2. Materials
2.2.1. 4-Ethyl-5,5,6,6-tetrafluoro-1-[4-(4-n-propylphenyl)phenyl]cyclohexa-1,3-diene (2a)
2.2.2. 5,5,6,6-Tetrafluoro-1-(4-methoxyphenyl)phenylcyclohexa-1,3-diene (2b)
2.2.3. 5,5,6,6-Tetrafluoro-1-{4-(trifluoromethyl)phenyl}phenylcyclohexa-1,3-diene (2c)
2.2.4. 4-Ethyl-5,5,6,6-tetrafluoro-1-[4-{4-(n-octyloxy)phenyl}phenyl]cyclohexa-1,3-diene (2d)
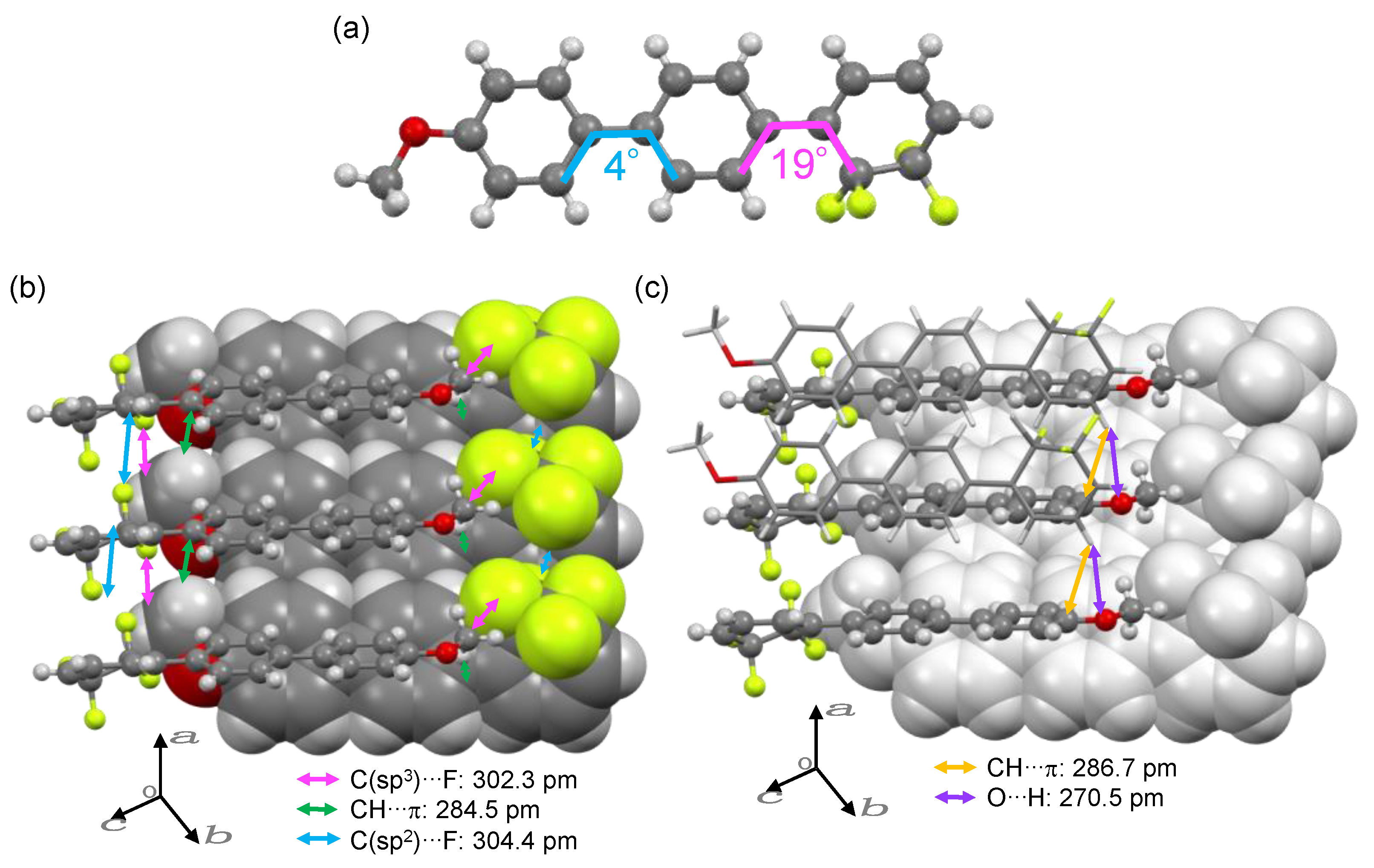
2.3. Single-Crystal X-ray Diffraction (XRD)
2.4. Photophysical Properties
2.5. LC Properties
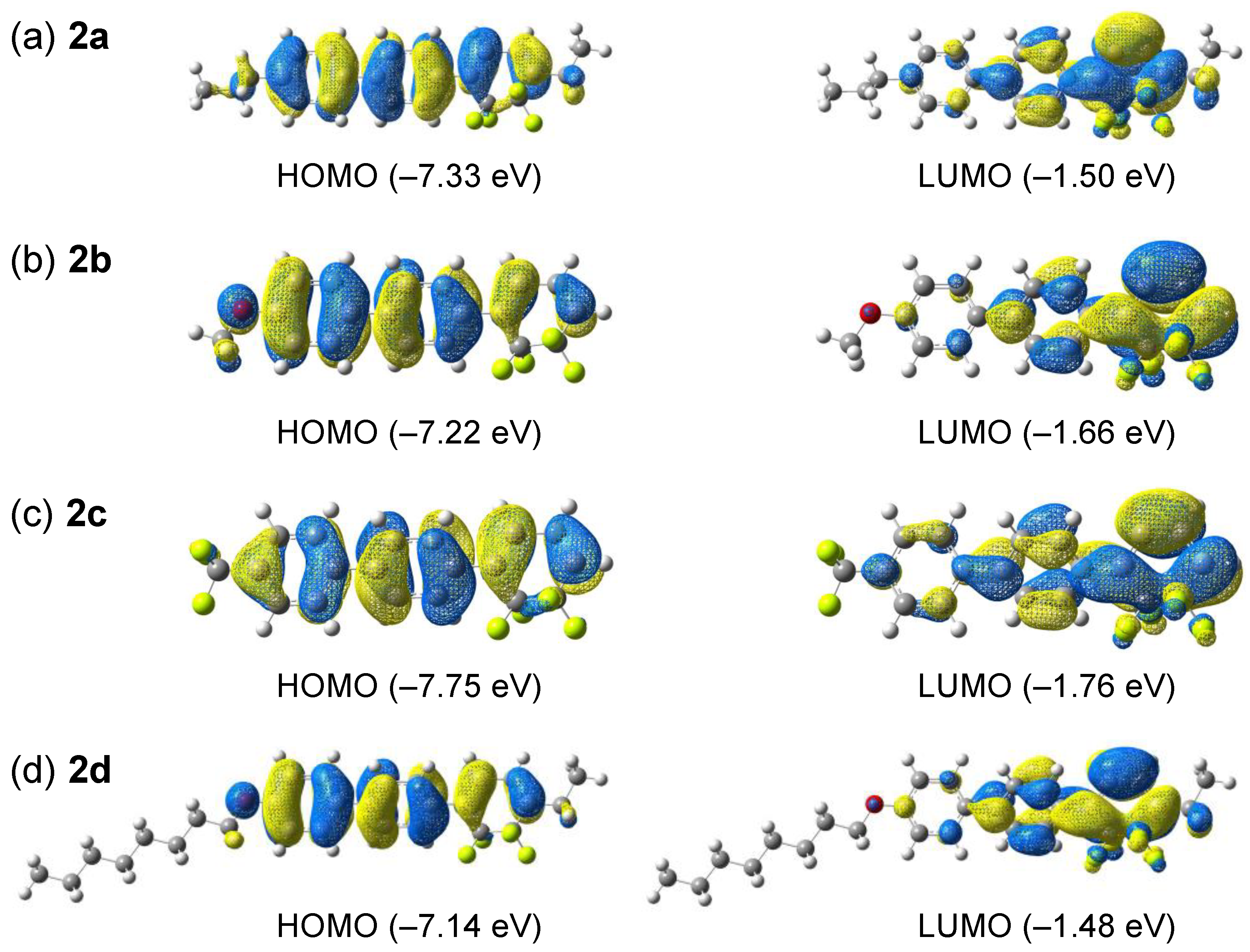
2.6. Theoretical Calculations
3. Results and Discussion
3.1. Synthesis
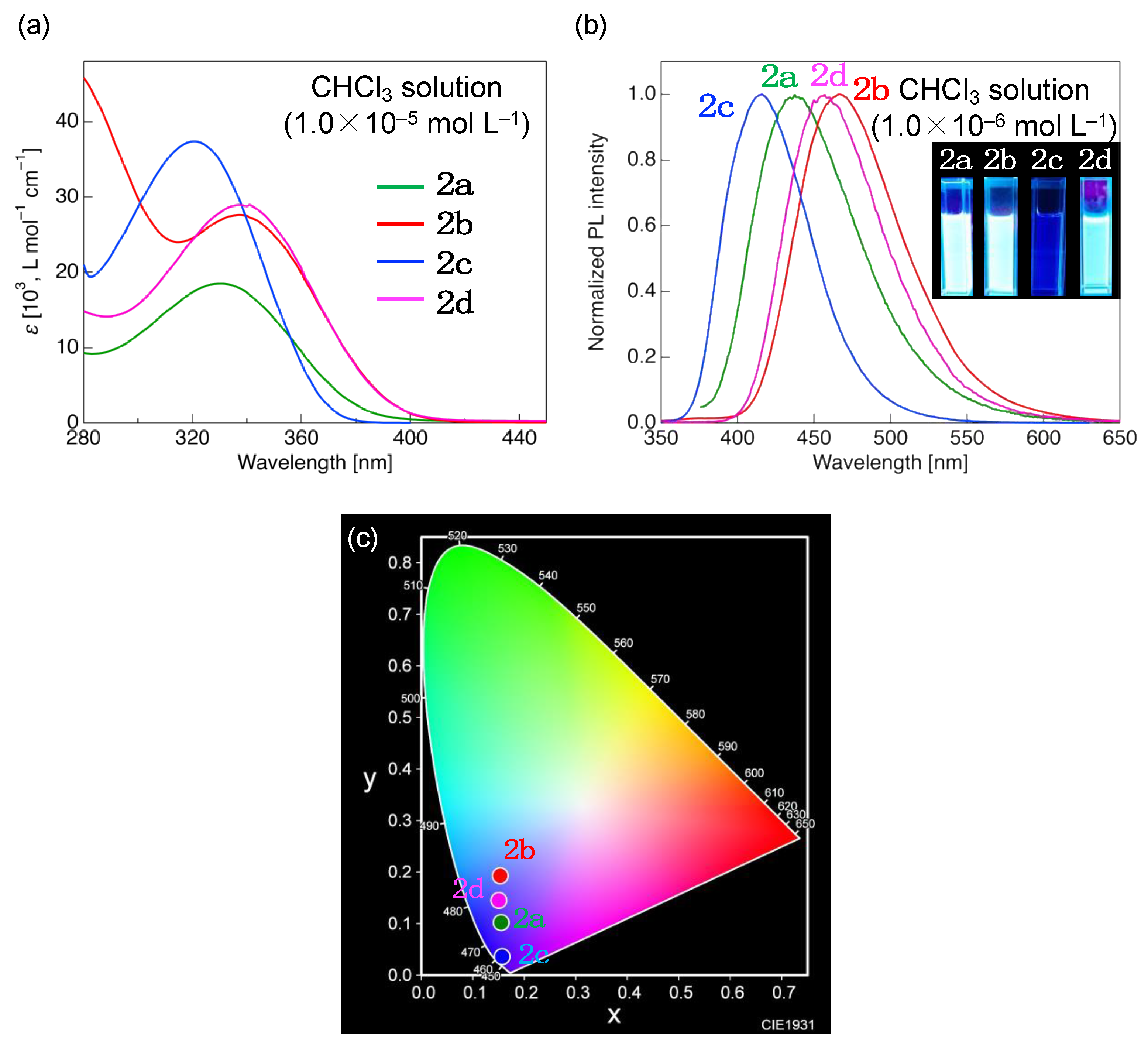
3.2. Photophysical Properties
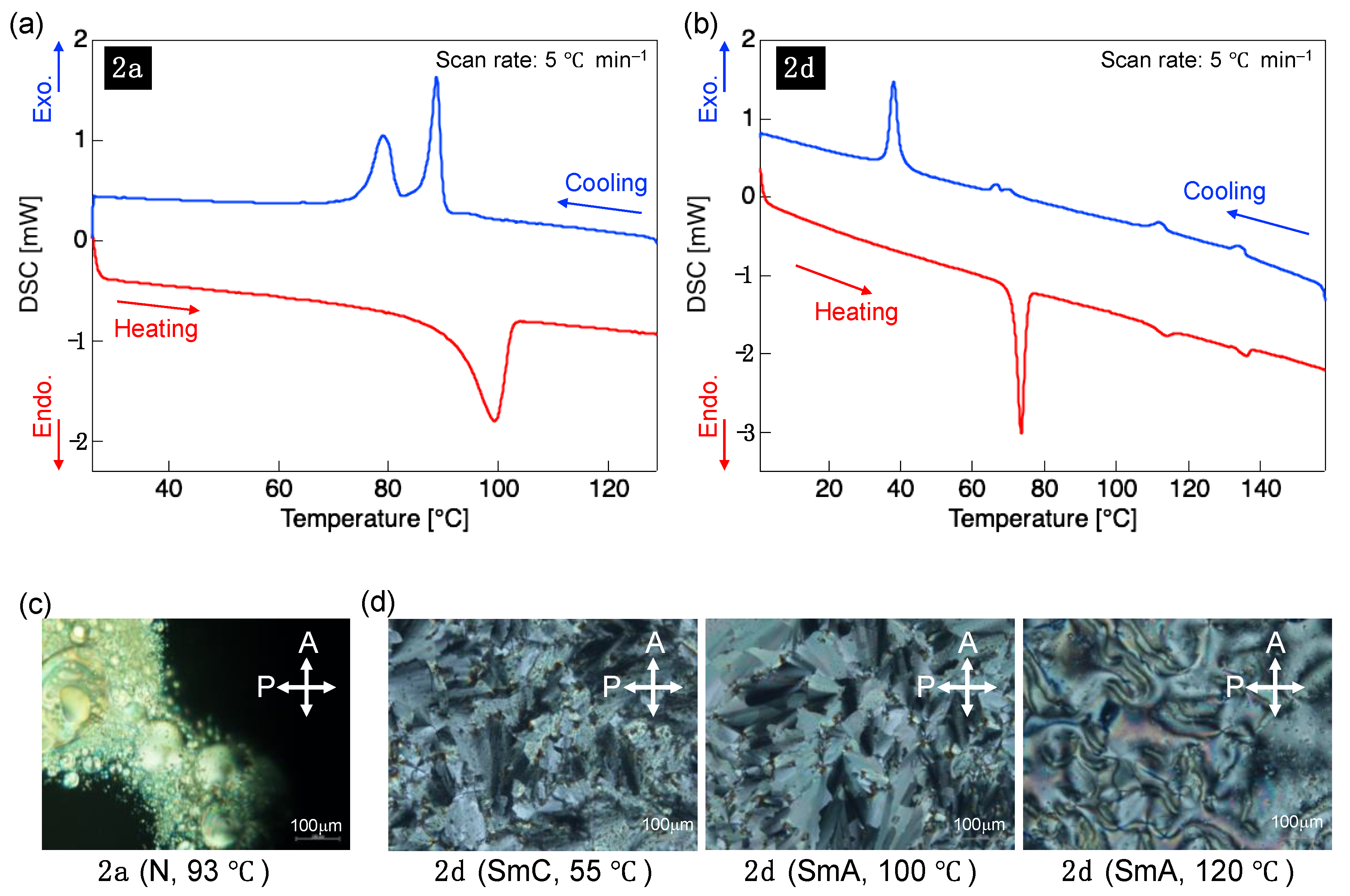
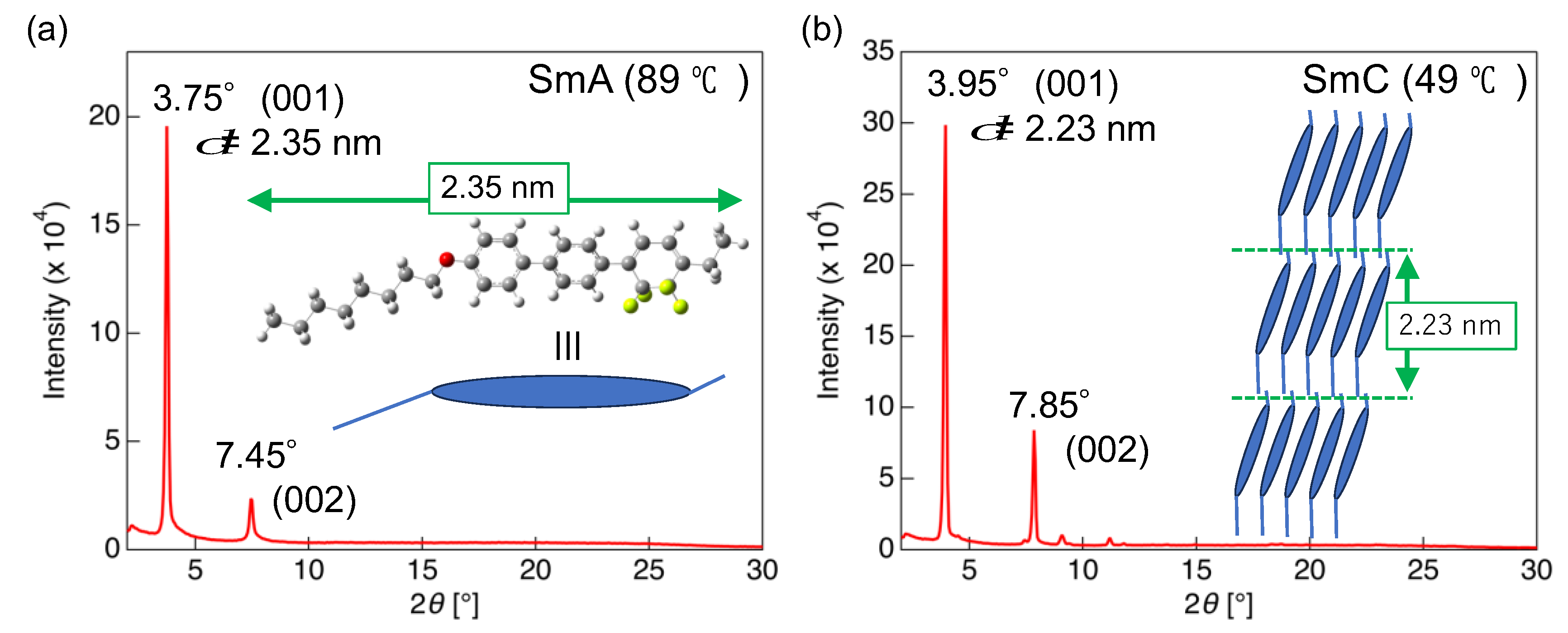
3.3. LC Properties
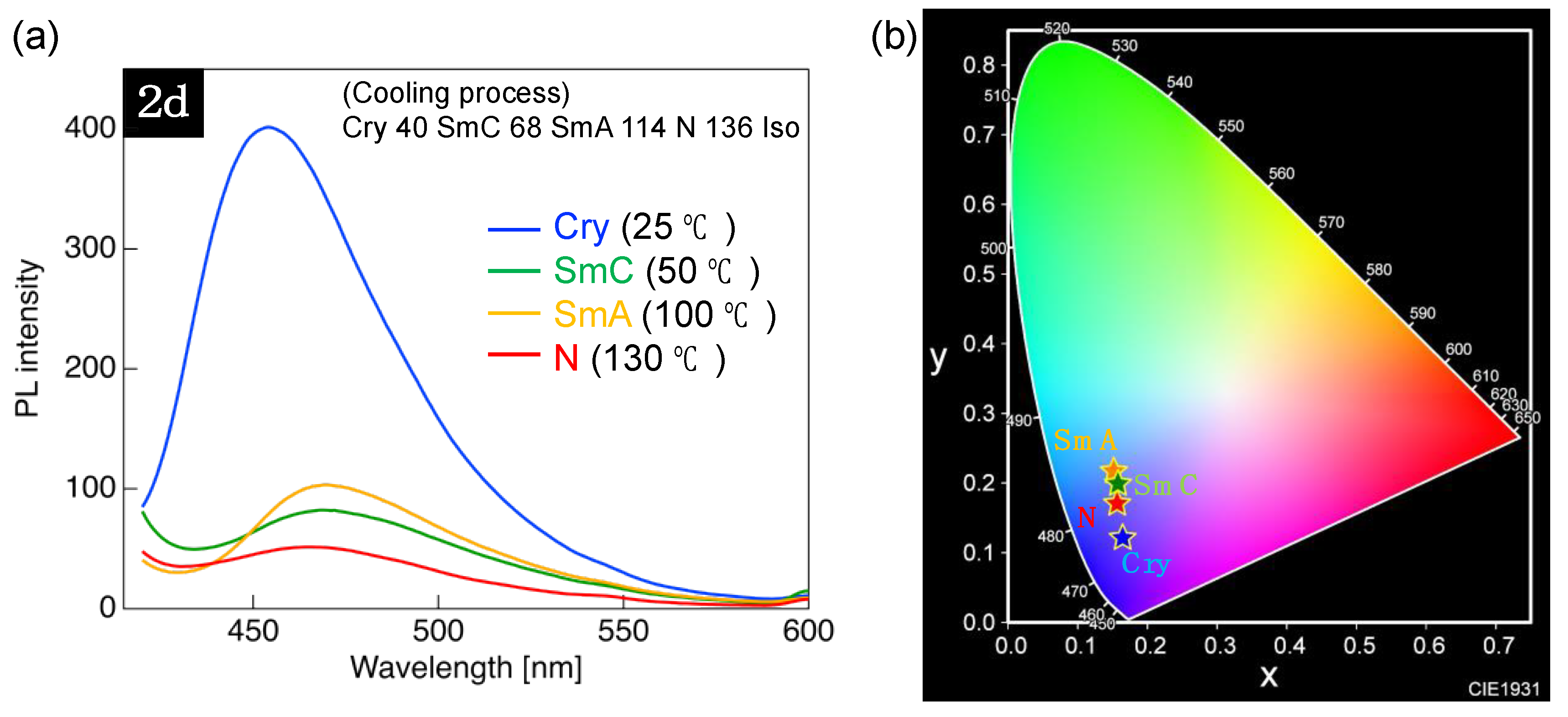
3.4. PL Properties of 2d in Various Molecular Aggregation States
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of organofluorine compounds to pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Zhou, X.; Kang, S.W.; Kumar, S.; Li, Q. Self-assembly of discotic liquid crystal porphyrin into more controllable ordered nanostructure mediated by fluorophobic effect. Liq. Cryst. 2009, 36, 269–274. [Google Scholar] [CrossRef]
- Wang, H.; Bisoyi, H.K.; Urbas, A.M.; Bunning, T.J.; Li, Q. Reversible circularly polarized reflection in a self-organized helical superstructure enabled by a visible-light-driven axially chiral molecular switch. J. Am. Chem. Soc. 2019, 141, 8078–8082. [Google Scholar] [CrossRef]
- Wang, H.; Bisoyi, H.K.; Li, B.X.; McConney, M.E.; Bunning, T.J.; Li, Q. Visible-light-driven halogen bond donor based molecular switches: From reversible unwinding to handedness inversion in self-organized soft helical superstructures. Angew. Chem. Int. Ed. 2020, 59, 2684–2687. [Google Scholar] [CrossRef]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Kirsh, P., Ed.; WILEY-VCH: Weinheim, Germany, 2013; pp. 247–298. [Google Scholar]
- Hird, M. Fluorinated liquid crystals–properties and applications. Chem. Soc. Rev. 2007, 36, 2070–2095. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Konno, T. Trifluoromethylated internal alkynes: Versatile building blocks for the preparation of various fluorine-containing molecules. Synlett 2014, 25, 1350–1370, and references cited therein. [Google Scholar] [CrossRef]
- Ohsato, H.; Kawauchi, K.; Yamada, S.; Konno, T. Diverse synthetic transformations using 4-bromo-3,3,4,4-tetrafluorobut-1-ene and its applications in the preparation of CF2CF2-containing sugars, liquid crystals, and light-emitting materials. Chem. Rec. 2023, 23, e202300080, and references cited therein. [Google Scholar] [CrossRef]
- Yamada, S.; Konno, T. Development of donor-π-acceptor-type fluorinated tolanes as compact condensed phase luminophores and applications in photoluminescent liquid-crystalline molecules. Chem. Rec. 2023, 23, e202300094, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Design, synthesis and evaluation of new fluorinated liquid crystals bearing a CF2CF2 fragment with negative dielectric anisotropy. Org. Biomol. Chem. 2017, 15, 1495–1509. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. New entry for fluorinated carbocycles: Unprecedented 3,6-disubstituted 1,1,2,2-tetrafluorocyclohexane derivatives. J. Fluorine Chem. 2017, 200, 47–58. [Google Scholar] [CrossRef]
- Yamada, S.; Tamamoto, K.; Kida, T.; Asai, T.; Ishihara, T.; Konno, T. Rational design and synthesis of a novel laterally-tetrafluorinated tricyclic mesogen with large negative dielectric anisotropy. Org. Biomol. Chem. 2017, 15, 9442–9454. [Google Scholar] [CrossRef] [PubMed]
- Kumon, T.; Hashishita, S.; Kida, T.; Yamada, S.; Ishihara, T.; Konno, T. Gram-scale preparation of negative-type liquid crystals with a CF2CF2-carbocycle unit via an improved short-step synthetic protocol. Beilstein J. Org. Chem. 2018, 14, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Ohsato, H.; Morita, M.; Yamada, S.; Agou, T.; Fukumoto, H.; Konno, T. Aggregation-induced enhanced fluorescence by hydrogen bonding in π-conjugated tricarbocycles with a CF2CF2-containing cyclohexa-1,3-diene skeleton. Mol. Syst. Des. Eng. 2022, 7, 1129–1137. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement, and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the performance of the M05-2X and M06-2X exchange-correlation functionals for noncovalent interactions in biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jensen, J.H. Improving the efficiency and convergence of geometry optimization with the polarizable continuum model: New energy gradients and molecular surface tesselation. J. Comput. Chem. 2004, 25, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Rybalova, T.V.; Bagryanskaya, I.Y. C–F···π, F···H, and F···F intermolecular interactions and F-aggregations: Role in crystal engineering of fluoroorganic compounds. J. Struct. Chem. 2009, 50, 741–753. [Google Scholar] [CrossRef]
- Kawahara, S.; Tsuzuki, S.; Uchimaru, T. Theoretical study of the C–F/π interaction: Attractive interaction between fluorinated alkane and an electron-deficient π-system. J. Phys. Chem. A 2004, 108, 6744–6749. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Fujii, A. Nature and physical origin of CH/π interaction: Significant difference from conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2008, 10, 2584–2594. [Google Scholar] [CrossRef] [PubMed]
- Mataga, N.; Kaifu, Y.; Koizumi, M. The solvent effect on the fluorescence spectrum changes of solute-solvent interactions during the lifetime of the excited solute molecule. Bull. Chem. Soc. Jpn. 1955, 28, 690–691. [Google Scholar] [CrossRef] [Green Version]
- Mataga, N.; Kaifu, Y.; Koizumi, M. Solvent effects on fluorescence spectra and dipole moments of excited molecules. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | λabs [nm] 1 (ε [103, L·mol−1·cm−1]) | λPL [nm] 2 (ΦPL) 3 | τ [ns] | kr [108, s−1] 4 | knr [108, s−1] 5 | CIE (x, y) |
|---|---|---|---|---|---|---|
| 2a | 330 (18.5) | 437 (0.94) | 2.08 | 4.52 | 0.28 | (0.155, 0.102) |
| 2b | 337 (27.6) | 463 (0.60) | 1.84 | 3.29 | 2.15 | (0.153, 0.193) |
| 2c | 320 (37.4) | 416 (0.24) | 0.73 | 3.30 | 10.44 | (0.157, 0.036) |
| 2d | 337 (28.9) | 463 (0.89) | 2.11 | 4.21 | 0.53 | (0.150, 0.145) |
| Molecule | HOMO Energy [eV] | LUMO Energy [eV] | Theoretical λcalcd [nm] | Oscillator Strength (f) | Theoretical Transition (Probability) |
|---|---|---|---|---|---|
| 2a | −7.33 | −1.50 | 331 | 1.00 | HOMO→LUMO (85%) HOMO–1→LUMO (12%) |
| 2b | −7.22 | −1.66 | 338 | 0.90 | HOMO→LUMO (77%) HOMO–1→LUMO (20%) |
| 2c | −7.75 | −1.76 | 325 | 0.87 | HOMO→LUMO (91%) HOMO–2→LUMO (6%) |
| 2d | −7.14 | −1.48 | 335 | 1.04 | HOMO→LUMO (78%) HOMO–1→LUMO (19%) |
| Molecule | λPL [nm] (ΦPL) 1 | τave [ns] | τ1 [ns] | τ2 [ns] | kr [108, s−1] 2 | knr [108, s−1] 3 | CIE (x, y) |
|---|---|---|---|---|---|---|---|
| 2a | 509 (0.99) | 1.84 | – | – | 5.38 | 0.054 | (0.265, 0.570) |
| 2b | 463 (0.63) | 2.43 | 2.06 | 4.47 | 2.56 | 1.55 | (0.162, 0.200) |
| 2c | 413 (0.31) | 3.04 | – | – | 1.03 | 2.26 | (0.155, 0.175) |
| 2d | 458 (0.93) | 3.19 | – | – | 2.92 | 0.21 | (0.182, 0.423) |
| Molecule | Process | Phase Transition Temperatures [°C] and Enthalpies [kJ·mol−1] 1 |
|---|---|---|
| 2a | Heating | Cry 92 (14.0) Iso |
| Cooling | Cry 81 (−4.9) N 90 (−6.2) Iso | |
| 2b | Heating | Cry 130 (16.4) Iso |
| Cooling | Cry 93 (−12.1) Iso | |
| 2c | Heating | Cry 138 (11.8) Iso |
| Cooling | Cry 98 (−8.2) Iso | |
| 2d | Heating | Cry 71 (12.6) SmA 110 (0.88) N 133 (0.72) Iso |
| Cooling | Cry 40 (−8.1) SmC 68 (−1.1) SmA 114 (−1.0) N 136 (−0.85) Iso |
| Temp. [°C]/Phase | λPL [nm] | I/IN 1 | CIE (x, y) |
|---|---|---|---|
| 130/N | 466 | 7.8 | (0.156, 0.171) |
| 100/SmA | 470 | 1.6 | (0.152, 0.217) |
| 50/SmC | 468 | 2.0 | (0.158, 0.199) |
| 25/Cry | 454 | 1.0 | (0.164, 0.121) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohsato, H.; Yamada, S.; Yasui, M.; Konno, T. Effects of Tetrafluorocyclohexa-1,3-Diene Ring Position on Photoluminescence and Liquid-Crystalline Properties of Tricyclic π-Conjugated Molecules. Crystals 2023, 13, 1208. https://doi.org/10.3390/cryst13081208
Ohsato H, Yamada S, Yasui M, Konno T. Effects of Tetrafluorocyclohexa-1,3-Diene Ring Position on Photoluminescence and Liquid-Crystalline Properties of Tricyclic π-Conjugated Molecules. Crystals. 2023; 13(8):1208. https://doi.org/10.3390/cryst13081208
Chicago/Turabian StyleOhsato, Haruka, Shigeyuki Yamada, Motohiro Yasui, and Tsutomu Konno. 2023. "Effects of Tetrafluorocyclohexa-1,3-Diene Ring Position on Photoluminescence and Liquid-Crystalline Properties of Tricyclic π-Conjugated Molecules" Crystals 13, no. 8: 1208. https://doi.org/10.3390/cryst13081208
APA StyleOhsato, H., Yamada, S., Yasui, M., & Konno, T. (2023). Effects of Tetrafluorocyclohexa-1,3-Diene Ring Position on Photoluminescence and Liquid-Crystalline Properties of Tricyclic π-Conjugated Molecules. Crystals, 13(8), 1208. https://doi.org/10.3390/cryst13081208