Abstract
We have established that relatively simple calculations of the Coulomb interaction in the lattice of doped lithium niobate (LN, LiNbO3) can confirm the physical properties of real crystals. We have developed a method for the double adjustment of real XRD data for calculations of Coulomb interaction in a LN cluster. The study considers two crystals doped with boron (LN:B); LN:B(1) has been grown from a charge with 0.02 mol% B2O3, boron has been introduced by homogeneous doping, LN:B(2) has been grown from a charge with 0.547 mol% B2O3, and boron has been introduced by direct solid-state doping. XRD and Rietveld method data have been obtained for these crystals. The obtained data have been used to build a model of the LN cluster; the cluster in the calculations consists of six oxygen octahedra of the LN structure. The cluster configuration has been chosen in such a way that the structure contains two tetrahedral voids. We have studied 10 variants of filling a cluster with intrinsic cations (Li, Nb), defects, and vacancies. There are 10 of them because, in addition to the basic cations in their positions, defects are present in the structure. In terms of the defects used (NbLi, NbV), we have used only those that Rietveld found for these exact LN:B crystals, and the vacancy in the niobium octahedron (VNb) compensates for these defects, according to the models known for LN. The energy of the Coulomb interaction between the cluster structure of a real crystal and the boron cation localized in it in different positions has been calculated for each of the configurations. Calculations have demonstrated that B is more likely to be embedded near a defect than in a regular structure. This means that boron positively influences the local substructure of doped LN crystals, not only structures the melt during crystal growth. Calculations have shown that the type and location of structural defects affect the position of boron in the structure of a LN crystal. Calculations have also shown that LN:B(1) has a more stable structure, including optical damage resistance. The photoinduced light scattering (PILS) patterns and conoscopic patterns confirm this conclusion for the studied LN:B crystals. The information obtained in this study may be useful for interpreting the defective structure of LN crystals co-doped with boron and metals (Mg, Zn, etc.). This will supplement the knowledge available in the literature regarding models that describe the structure of complexly doped LN crystals.
1. Introduction
The requirements for the characteristics of functional materials of electron technics, based on a nonlinear optical single crystal of lithium niobate (LN, LiNbO3), are increasing all the time. This increase requires the modification of existing and the development of new technologies for obtaining nominally pure large crystals of good optical quality, high compositional uniformity, and with a low photorefractive effect. Stoichiometric (SLN, R = [Li]/[Nb] = 1) and near-stoichiometric (NSLN, R ≈ 1) LN crystals are promising, since they have a lower value of the coercive field (≈2.3 kV/mm) compared to congruent crystals (≈23 kV/mm, CLN, R = 0.946). Thus, SLN and NSLN crystals are suitable as frequency converters of laser radiation on periodically polarized domain structures.
Several technological approaches are currently applied to obtain SLN crystals. SLN crystals can be grown using the Czochralski method from a melt with a significant excess of lithium (58.6 mol% Li2O) [1]. The main disadvantage of the method is the low growth rate. It also requires suppressing concentration supercooling. The crystals grown using this technology are small, have a high photorefractive effect, and have a large inhomogeneity of the refractive index along the growth axis. The strong difference between the composition of the growing crystal and the melt determines the nonuniformity of the refractive index. Such crystals are rarely suitable for applications. SLN crystals can also be grown using the Czochralski method using a double crucible method. An LN charge enriched with lithium (~58–60 mol% Li2O) is melted in an external crucible, then the melt is transferred to the reactor with a movable platinum partition; the crystal is grown in the volume of the reactor. The added melt, or sometimes charge, should have a strictly stoichiometric composition. The composition of the melt should not change during the growth of an SLN crystal. The disadvantages of this method are the technical complexity of replenishing the charge in the crucible and the automation of its supply [2,3]. NSLN crystals can also be obtained using the Vapor Transport Equilibration (VTE) method. The method is based on the diffusion of gaseous lithium into the structure of the grown crystal. This increases the stoichiometry of the sample [4]. However, this method does not uniformly increase the stoichiometry throughout the crystal (especially large crystals), therefore, the method is only suitable for thin wafers.
Crystals of high compositional uniformity with different ratios of Li/Nb (including SLN crystals) can be obtained with High Temperature Top Seeded Solution Growth (HTTSSG) technology. NSLN crystals grow from a congruent charge (R = 0.946) with the addition of alkali metals fluxes; flux limits the evaporation of lithium from the melt surface, and this ensures the growth of Me2O (Me–Na, K, Rb, Cs) using HTTSSG [5,6]. K2O flux is the most commonly used flux. The advantages of HTTSSG are a decrease in the crystallization temperature and the presence of an alkaline flux. The large-sized NSLN crystals have a high compositional uniformity. The disadvantage of the HTTSSG is the high concentration of potassium in the grown crystals (1 × 10−2 – 2 × 10−2 wt%). It can be compared with the concentration of traces of numerous impurities in a crystal. Potassium ions have a large radius (1.38 Å) and are unable to enter the O6 oxygen octahedra of the LN crystal structure. Potassium becomes a mechanically trapped impurity localized on crystal defects.
Existing technologies for growing LN single crystals have disadvantages. This is why, today, a lot of studies have been dedicated to the search for new technologies for growing and doping LN crystals. All of these works are aimed at a better understanding of the connection between the growth technology and crystal properties. Such studies help to create LN-based materials with the given properties in a fairly wide range. The relationship between the melt and solid crystal was considered in reference [7], in MgO-doped LN prepared using the micro-pulling-down method. The authors of reference [8] prepared pressed pellets of LN:Pr, LN:Mg,Pr, obtained from nanopowders; the powders were prepared by high-energy ball milling with subsequent annealing. An interface electric field was applied during the growth of CLN and LN:Mg crystals using the micro-pulling-down technique in [9]. Fano-resonant Si nanoparticles were introduced into a co-doped LN:Mg:Er single crystal using ion implantation and subsequent thermal annealing in reference [10]. A series of MgO-doped LN crystals were simultaneously grown in one furnace using the modified vertical Bridgman method in reference [11]. Mg-doped NSLN crystals were prepared by Li-rich growth and VTE in reference [12]. The authors of reference [13] showed the growth of LN:Pr:Mg co-doped crystal using the Bridgman method and studied the optical properties of the crystals. These are only a few examples of the most recent studies. Thus, the search for new LN growth methods is still highly relevant.
A new technological approach to obtaining NSLN crystals was recently developed in the ICT KSC RAS. The method essence is doping the LN charge with boron-containing compounds [14,15]. The concentration of boron cations in LN crystals grown from such a charge is at a trace level (~4 × 10−4 mol%). The concentration is comparable with that of numerous uncontrollable impurities of other elements [14]. The nonmetallic element B has a small ionic radius (0.15 Å for B(III), 0.25 Å for B(IV), [16]); it is not a mechanically trapped impurity (like K, for example). Boron occupies the faces of the tetrahedral O4 voids of the LN structure as a part of [BO3]3− groups [14,17]. Boron can also incorporate into the oxygen plane of the tetrahedral voids. The O4 voids are much smaller than the O6 ones. The main (Li+ and Nb5+) impurities and dopant metallic elements localize in the O6 voids of the LN crystal structure. Metal cations with relatively large ionic radii are unable to localize in the O4. Thus, only small-radius cations can localize in tetrahedral voids.
The ICT KSC RAS has developed the technology for growing boron-containing NSLN crystals. The technology includes three different ways of introducing the B3+ dopant. The first method is a direct solid-phase doping of a congruent charge with boron oxide (B2O3) [18]. The second method is a direct solid-phase doping with boric acid (H3BO3) [19]. The third method is a homogeneous doping of Nb2O5 with boric acid resulting in the doped Nb2O5:B precursor [20]. LN:B crystals obtained using these technologies have a high compositional and structural uniformity, and a low photorefractive effect [14,15,17]. Their composition and the coercive field magnitude are close to those of a SLN crystal. This is important to create functional nonlinear optical materials for converting laser radiation on periodically polarized micron and submicron domains [21]. Such materials are currently fabricated with heavily magnesium-doped (≈5.0 mol% MgO) LN crystals [22,23]. However, usually, crystals doped with magnesium near the main concentration threshold (≈5.5 mol% MgO [24]) have a low compositional uniformity [25] and thus a lot of structural defects [26].
Direct experimental methods that determine the localization of trace amounts of light non-metallic elements in the crystals structure are absent. For example, the influence of boron on the LN crystals’ structure was studied by XRD, but not directly. The study has compared the structural details of two crystals: NSLN grown by HTTSSG with 5.5 wt% K2O (NSLN(HTTSSG 5.5 wt% K2O)) and NSLN doped with boron (LN:B) [15]. The structural particularities studied in reference [15] are as follows: the distortion of the anionic sublattice, and the type, number, and location of point defects. During the Rietveld adjustment, we have used vacancy split models, a review of which is given in reference [27]. Rietveld refinement determines the distribution of basic and doping metal cations, and point defects in the form of irregularly located cations. In addition, Raman spectroscopy revealed the distortion of the anionic lattice of LN:B crystals, the distortion was absent from nominally pure SLN and CLN crystals [14,17].
Model calculations provide important information about the fine features of the structure, including the structure of lightly doped crystals. Reference [28] is dedicated to theoretical studies of an anti-site defect cluster NbLi-4VLi of pure and Mg2+-, Sc3+-, and Zr4+-doped LN. The authors of reference [29] calculated the Bi site and position in octahedrons; the dependence of optical properties and electronic structures on a dopant concentration in LN:Bi was considered. The authors of reference [30] calculated the dependence of cations sites and unit cell parameters in differently doped LN crystals. In reference [31], the authors used ab initio calculations to describe free and defect-bound polarons and bipolarons in LN. They used the calculations to explain the different spectroscopic properties of the crystals under study. Reference [32] considered polarons at a level of separate defect using calculations. The results were confirmed by the optical properties of real crystals.
Coulomb energy is an easy but effective way to reveal details of the LN structure that are impossible to detect with other methods. A computer simulation of the influence of the doping Mg2+ cations’ concentration on the formation of intrinsic defects in LN crystals, such as NbLi, VLi, and VNb, is presented in reference [33]. The Coulomb interaction in model calculations is an integral part of modern works devoted to the study of the fine features of the defect structure of a LN crystal [34,35]. The mutual Coulomb attraction of small-radius polarons (Nb4+ and O−–VLi) was considered in reference [34] using the example of a LN:Mg(6.5 mol% MgO) crystal. The authors of reference [35] determined that a multiply charged dopant (Fe2+/3+) localized in the LN structure in a lithium octahedron is capable of capturing an electron with a certain force; the force is a mixture of the Coulomb attraction and deformation of the polaron lattice.
Thus, it is possible to qualitatively evaluate the localization of trace amounts of B3+ in O4 tetrahedra using model calculations of the electrostatic interaction energy of point charges in a rather small cluster built from several oxygen octahedra (LiO6, NbO6, and VO6). The boron cation can localize in different faces of O4 tetrahedra. Model calculations of the features of localization of point defects (NbLi, NbV, VNb) in oxygen–octahedral clusters are carried out in this study using the previously obtained structural data. Calculations are made according to the method proposed in reference [17].
In our work, we subject the B3+ cations’ positions to the same model calculations in LN:B crystals obtained via different doping methods. These results are compared with the results for a nominally pure NSLN(HTTSSG 5.5 wt% K2O, [Li2O] ≈ 49.7 mol% [36]) crystal. It should be noted that the features of MeO6 oxygen–octahedral clusters and O4 tetrahedral voids significantly affect the compositional uniformity, nonlinear optical, and ferroelectric characteristics of the LiNbO3 crystal.
2. Materials and Methods
LN:B(1) was grown from a charge with 0.02 mol% B2O3 and LN:B(2) was grown from a charge with 0.547 mol% B2O3. Both single crystals were grown in the (001) direction using the Czochralski method on a growth setup Crystall-2 (Voroshilovgradsky zavod electronnogo mashinostroeniya, Voroshilovgrad, USSR). The setup was equipped with inductive heating and a system that is able to control a crystal diameter automatically. Crystals were grown in air from platinum crucibles. Initial components for both syntheses were Li2CO3, Nb2O5, and H3BO3 (99.9, Solikamsk Magnesium Plant, Solikamsk, Russia). Homogeneous doping required HF (99.99, Vekton Ltd., Saint Petersburg, Russia) and NH4OH (25% solution, Komponent-reaktiv Ltd., Moscow, Russia).
The charge for growing LN:B(1) crystal was obtained via homogeneous doping. The Nb2O5:B precursor was obtained via introducing a calculated amount of boric acid into a niobium pentoxide fluoride solution [20,35]. The solution was prepared by dissolving the Nb2O5 in HF. Niobium hydroxide was then precipitated by adding NH4OH. The precipitate was then mixed with the H3BO3. Details are given in reference [20].
The ratios of the main components must correspond to the congruent composition. The concentration of boron was not taken into account due to its low value (≈10−4 wt%) while calculating the amount of Li2CO3 required for congruent composition with Nb2O5:B precursor. A detailed technological scheme of the homogeneous doping is described in reference [20]. A boron-containing mixture was synthesized using Nb2O5:B precursor; LN:B(1) was grown from the congruent charge, synthesized using the precursor.
The charge for growing a LN:B(2) crystal was doped by a direct solid-phase method. A mixture of Li2CO3:Nb2O5:H3BO3 was used in this case [19]. The amount of Li2CO3 and Nb2O5 required for the synthesis of the LN:B charge was taken in the ratio corresponding to the congruent composition, and the amount of H3BO3 was calculated for the nominally pure Nb2O5.
The amount of impurities in charges and samples LN:B(1) and LN:B(2), the exact details of crystal growth can be found in [15]. Micro- and macro-defects images, and linear dimensions characteristic of boron-doped LN crystals can also be found in references [14,17].
XRD patterns of powder crystal samples were recorded on a diffractometer DRON-6 (NPP Burevestnik, Sankt-Petersburg, Russia). A pyrolytic graphite monochromator was located in the primary beams (CuKα radiation, voltage 45 kV, current 25 mA). The XRD patterns were taken in more detail with a step of 0.02° in the reflection areas, and with a step of 0.2° in the background areas. The stability of the registration scheme was controlled when an XRD pattern was obtained. The accuracy of determining the intensity at each point of the diffraction line was no less than 3%. The XRD data of a control NSLN(HTTSSG 5.5 wt% K2O) crystal are given in reference [15]. The calculation of the profile characteristics of the XRD patterns was performed by the Pauli method: the XRD patterns were decomposed into the sum of the integral intensities. The structural characteristics—atomic coordinates, thermal motion parameters, site population factors—were refined using the Rietveld method (i.e., the full profile analysis). The following programs were used: MRIA and FULL PROF. XRD patterns of powder samples and structural characteristics of the studied LN:B(1) and LN:B(2) crystals were first given in reference [15].
The calculation of the total energy of the Coulomb interaction of point charges (U, eV) of a LN oxygen–octahedral structure fragment, with the B3+ cation considered in the sp2-hybrid state, was performed according to the Coulomb potential [17]:
where z1 and z2 are the values of charge of interacting particles; r12 is the distance between centers of interacting charges [Å]; and k is a constant. It is expressed by the formula (eV⋅Å) [17]:
where e is electron charge and ε0 is vacuum permittivity.
U = (k × z1 × z2)/r12
k = e2/(4 × π × ε0 × 10−10) = 14.41971
The considered system (cluster) consists of six oxygen octahedra in two layers (I and II): LiIO6, NbIO6, VIO6, LiIIO6, NbIIO6, and VIIO6. Lithium, niobium, and vacant octahedra lay in the first (I) and second (II) layers of the cluster, respectively. These oxygen octahedra form two tetrahedral voids in this configuration. The system is not electrically neutral. Model calculations consider a cluster “pulled out” from a large electrically neutral system. This was made to study the changes in the energy of the B3+ cation interaction with the closest surrounding fragment of the LiNbO3 crystal structure (including NbLi, NbV, and VNb defects in the crystal structure). We moved the position of B3+ in the faces of vacant tetrahedral voids and calculated the corresponding interaction energy. The position of the boron cation is considered to be equidistant from each of the vertices of the occupied face of the vacant tetrahedral void in the calculations. The framework of the cluster is formed by 20 oxygen anions O2−. The number and localization of the main metal cations in the cluster—lithium (Li+) and niobium (Nb5+)—varies and depends on the type and number of defects introduced into the cluster. The structural defects NbLi and NbV were detected in real LN:B(1) and LN:B(2) crystals by XRD analysis. These defects determined the choice of models for describing the localization of intrinsic point defects in the studied crystals: the model of niobium vacancies (M2 [27,37]) and the empty octahedra filling model (M3 [1,27]). We considered the formation of several types of defects in this work: NbLi4+ (niobium antisite, a niobium cation localized in a lithium octahedron), NbV5+ (a niobium cation localized in a vacant octahedron), and VNb5− (a vacancy in a niobium octahedron formed as a part of the charge compensation for defects NbLi4+ and NbV5+).
The steps of adjustment of real XRD data in the calculation are described in the Results and Discussion section.
3. Results and Discussion
The XRD patterns of LN:B(1) and LN:B(2) samples are similar and correspond to the XRD pattern of a nominally pure LN crystal, as can be seen in Figure 1. LN is characterized by the R3c symmetry space group (two formula units for a rhombohedral cell and six for a hexagonal one) [1,15,38,39,40]. The results of refinement of the unit cell periods of the studied LN:B(1) and LN:B(2) crystal samples, and a control NSLN(HTTSSG 5.5 wt% K2O), are given in Table 1 [15].
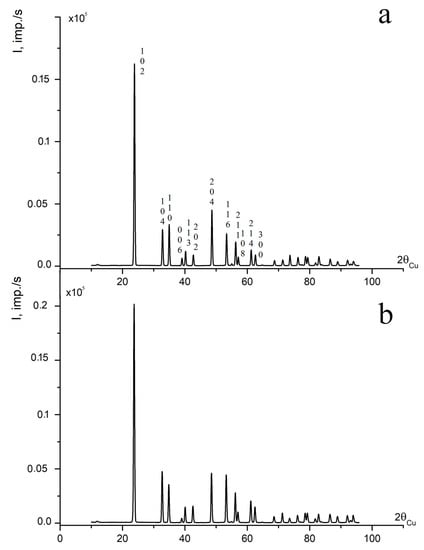
Figure 1.
XRD patterns of powdered samples of crystals (a) LN:B(2) and (b) LN:B(1) [15].

Table 1.
Unit cell parameters of NSLN(HTTSSG 5.5 wt% K2O), LN:B(1), and LN:B(2) crystals [15].
Table 1 shows that the values of the unit cell parameters of LN:B(1) and LN:B(2) crystals are close to each other and approach the values for the NSLN(HTTSSG 5.5 wt% K2O) crystal. This may indicate that the structure of LN:B(1) and LN:B(2) crystals is close to that of the LN crystal of near-stoichiometric composition.
The structural characteristics of LN:B crystals (atomic coordinates and site population factors G) characterize the distribution of cations over O6 oxygen octahedra. These characteristics are given in Table 2 [15].
Table 2.
Refined atom coordinates (x/a, y/b, z/c) and site population factors G of LN:B(1) and LN:B(2) crystals [15].
Table 2 shows that NbLi and NbV point structural defects are present in LN:B crystals regardless of the doping method. The site populations of these defects are close in LN:B crystals of different genesis: for the LN:B(1) G(NbLi) is 0.016 and G(NbV) is 0.009; for the LN:B(2) crystal G(NbLi) is 0.018 and G(NbV) is 0.01. The situation is quite different with the population of the lithium G(Li) and niobium G(Nb) sites of these crystals. G(Li) is almost identical for LN:B(1) and LN:B(2) crystals (0.98 and 0.99), but G(Nb) differs more significantly: 0.97 for a LN:B(1) crystal and 0.93 for the LN:B (2) crystal, as shown in Table 2. The difference can be explained by the high complexing ability of boron. Boron derivatives have a complex effect on the crystal-melt system. In particular, they bind impurity metal cations, form borates, and prevent their transition into a growing crystal. This has been confirmed by the calculation of the isobaric-isothermal potential for the formation of impurity metal borates in a boron-containing LN melt [15]. Boron derivatives also equalize the distribution coefficients of lithium and niobium when the crystal grows; they bind excess niobium cations in a boron-containing congruent melt [14]. The concentration of boron cations in the LN:B(2) melt was probably able to bind a larger amount of niobium cations than the concentration in the LN:B(1) melt.
The R values in the LN:B(1) and LN:B(2) crystals were calculated using the site population factors (G(Li), G(Nb), G(NbLi), G(NbV)); R = 0.985 and 1.033, respectively. Thus, doping of a congruent charge with boron increases the stoichiometry of the grown LN crystal and R approaches unity. Moreover, the population of regular lithium sites in the structure is higher than the population of niobium sites for both variants of doping of LN:B crystals, as shown in Table 2. However, according to the G(Li)/G(Nb) ratio, the LN:B(1) crystal is closer to the stoichiometric composition than the LN:B(2) crystal. Table 2 also shows that niobium cations incorporate into lithium and vacant octahedra and form NbLi and NbV structural defects. This distorts the ideal order of alternation of the main cations and vacancies along the polar axis and makes the cation sublattice less stoichiometric. This additionally increases the defectiveness of the cationic sublattice of LN:B crystals [14].
The interatomic distances of metal–oxygen (Me-O, Å) was calculated in oxygen–octahedral MeO6 clusters in NSLN(HTTSSG 5.5 wt% K2O) and in LN:B(1) and LN:B(2) crystals using refined atoms coordinates and unit cell periods, as shown in Table 3 [15].
Table 3.
Interatomic distances (Å) in NSLN(HTTSSG 5.5 wt% K2O), LN:B(1), and LN:B(2) crystals [15].
Table 3 shows that long and short Me-O distances in LiO6 and NbO6 octahedra of the LN:B(1) crystal correlate with the corresponding values in the NSLN(HTTSSG 5.5 wt% K2O) crystal. Me-O bond lengths change the most noticeably in the LN:B(2) crystal. This indicates a greater distortion of O6 oxygen octahedra in this crystal than in the other one, as shown in Table 3. The authors of reference [41] found that six lithium borates (LiB3O5, Li2B4O7, LiBO2, Li6B4O9, Li4B2O5, and Li3BO3), one niobium borate (Nb3BO9), and three phases of lithium niobate (LiNb3O8, LiNbO3, and Li3NbO4) can be formed in the ternary phase system Li2O-Nb2O5-B2O3. The homogeneous doping includes the step of introducing a boron-containing agent (H3BO3) directly into niobium pentoxide while Nb2O5 is separated from a fluoride niobium-containing solution. This ensures the formation of only one borate (Nb3BO9) during the preparation of the doped precursor. The B-O and Nb-O bonds in this borate will differ less in length, because the formation of seven different borates is possible during direct solid-phase doping [41]. Certain boron- and niobium-containing complexes form in the boron-containing melt during the solid-phase synthesis of the Li2CO3:Nb2O5:B mixture. The complexes are interconnected by covalent bonds. The lithium niobate melt consists of clusters (mostly polyhedra, octahedra, and tetrahedra [1]). They determine such structural characteristics of the grown crystal as the geometry and composition of MeO6 oxygen–octahedral clusters [14]. According to the experimental data (Table 3), the LiO6 and NbO6 polyhedra are distorted to the least extent in the LN:B(1) crystal in comparison to the LN:B(2) crystal. Thus, homogeneous doping is more advantageous than the solid-phase method. Homogeneous doping can provide more perfect LN crystals. Their composition is close to stoichiometric, and the photorefractive effect in them is low. Thus, samples 1 and 2, grown from a boron-doped melt, are expected to have optical homogeneity at least as good as CLN crystal and noticeably better than NSLN(HTTSSG 5.5 wt% K2O) crystal.
The laser conoscopy and photoinduced light scattering (PILS) patterns data [15] have confirmed the high optical quality of the LN:B(1) and LN:B(2) crystals; the photorefractive effect in the crystals is much lower than in NSLN(HTTSSG 5.5 wt% K2O). A detailed analysis of laser conoscopy and PILS patterns has revealed that the LN:B(1) is more optically homogeneous crystal than LN:B(2) [15].
Growing LiNbO3 from a charge containing boron equalizes the distribution coefficients of lithium and niobium in the single crystals. This increases their stoichiometry and reduces the concentration of traces of uncontrolled metal impurities (Al, Ca, Pb, etc.) in the bulk of the crystal since metal borates form in the melt with the impurities [15]. Moreover, boron cations incorporate into the faces of tetrahedral voids in the LN crystal structure, and reduce the concentration of NbLi point structural defects. These defects are the deepest electron traps in nominally pure LN crystals. Electron traps are responsible for the photorefractive effect. The amount of antisite niobium decreases in LN:B crystals to a concentration of boron cations introduced into the crystal structure [14,17]. This amount is not too great; the concentration of boron in LN crystals is about 4 × 10−4 wt%. However, the photorefractive effect in such crystals is quite low. In fact, the photorefractive response in LN:B is at the same level as in the LN crystals doped with threshold concentrations of known non-photorefractive cations. The threshold concentration in these cases is quite high: 4.5 mol% for MgO, 6 mol% for ZnO, 1.5 mol% for Sc2O3 and In2O3, 4 mol% for HfO2, and 6 mol% for ZrO2 [42].
We have considered several possible cationic compositions of model clusters of LN:B(1) and LN:B(2) crystals. The following issues were taken into account when forming the clusters [15]: unit cell periods, Table 1; the type and coordinates of the considered defects (NbLi, NbV), Table 2; and interatomic distances Me-O, Table 3. The building of separate clusters for each studied crystal is the first step in the adjustment of real XRD data for calculations.
Figure 2 shows the structure of some clusters. Clusters 1.1, 2.1 (Figure 2a,c), 2.2, 3.1, and 3.2 model a fragment of the LN:B(1) crystal structure, and Clusters 1.2, 2.3 (Figure 2b,d), 2.4, 3.3, and 3.4 model a fragment of the LN:B(2) crystal structure using real XRD data (Table 1, Table 2 and Table 3, [15]). Clusters 1.1 and 1.2 demonstrate the ideal (characteristic of a stoichiometric crystal) alternation of Li, Nb, and V along the polar axis of the crystal, as shown in Figure 2a,b and Figure 3. Clusters 2.1 and 2.3 contain an antisite NbLi defect in layer I and a compensating [27,37] VNb in layer II, as can be seen in Figure 2c,d. Clusters 2.2 and 2.4, conversely, contain a NbLi defect in layer II and the VNb defect in layer I. The case of the implementation of different models is considered in Clusters 3.1–3.4. In this case, the model M3 of niobium vacancies [27,37] and the model M2 of filling empty octahedra [1,27] are combined. According to the empty octahedron model, some of the niobium cations fall into the vacant octahedron with the formation of NbV point defects. Clusters 3.1 and 3.3 contain the NbV defect in layer I, and the VNb in layer II. Clusters 3.2 and 3.4, conversely, contain the NbV defect in layer II, and the VNb defect in layer I.
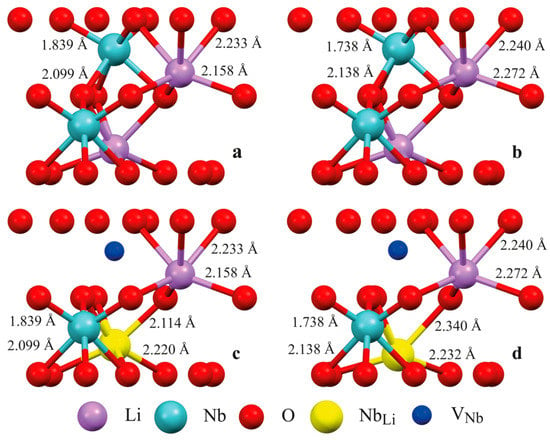
Figure 2.
Some configurations of clusters of LN:B(1) and LN:B(2) crystals built using real XRD data of these crystals: (a)—Cluster 1.1; (b)—Cluster 1.2; (c)—Cluster 2.1; and (d)—Cluster 2.3. The composition and structure of clusters are given in Table 4. Boron cation is supposed to occupy a face of a tetrahedron formed by the main octahedra (see Figure 3 for details).
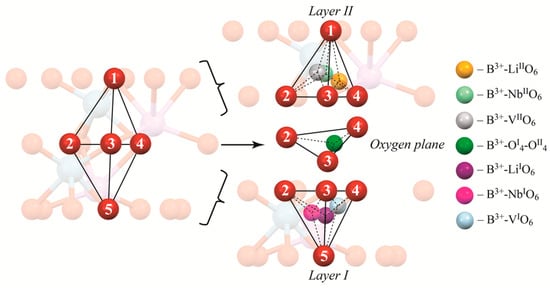
Figure 3.
Cluster 1.1 in more detail. Numbers show the number of oxygen atom to make the description of the exact tetrahedron face easier. Colored balls show possible positions of boron in tetrahedra faces.
The model calculations of the Coulomb energy of boron (EC, eV) interacting with the whole cluster are given in Table 4 and in Figure 4. We compare these calculations with the results of earlier model calculations given as Cluster 1 [14,17]. The earlier results were obtained based on the unit cell periods of a congruent crystal (a and c, 5.1483 and 13.8631 Å, respectively [1]). In those calculations, we have placed lithium and niobium cations in the corresponding octahedra, and structural defects have not been considered at all.
Table 4.
The Coulomb energy of boron (EC, eV) interacting with the whole cluster for all considered configurations of the Clusters.
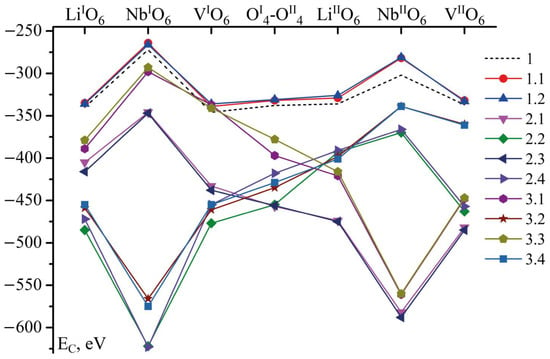
Figure 4.
EC is dependent on B3+ cation position in tetrahedra in all considered configurations of Clusters. Positions of boron on the bar above curves are given in Clusters 1, 1.1 and 1.2. In other Clusters, positions are appropriate to the cluster configuration but the sites correspond to the given ones.
Figure 3 shows Cluster 1.1 in more detail. First of all, it demonstrates layers I and II. In addition, it shows what the designations LiIO6, NbIO6, and VIIO6, etc., in Table 4 mean. Designations with I, such as LiIO6, NbIO6, etc., show the location of boron in lower tetrahedron faces (faces 235, 345, and 245 in Figure 3), designation with II (LiIIO6, NbIIO6, etc.) in upper tetrahedron faces (faces 123, 134, and 124 in Figure 3). The designation OI4-OII4 means that boron occupies a tetrahedral face in the oxygen plane in the middle of two tetrahedra (face 234 in Figure 3). As one can see, Figure 3 demonstrates the exact face boron occupies in a tetrahedron during calculations. Tetrahedral faces are simultaneously the faces of octahedra occupied by atom, defect or vacant. We have named the corresponding tetrahedral face by the atom, defect or vacancy in the octahedron than shares this face with the tetrahedron. Thus, the designation LiIO6 in Table 4 means that B3+ is placed into a tetrahedron face joint with the octahedron (O6) occupied by lithium in its own site (Li) located in the layer I (I); NbLiIIO6 into octahedron (O6), occupied by a niobium in lithium site defect (NbLi), located in layer II (II), etc. Designations of the tetrahedra faces occupied by boron, in Table 4, for other Clusters are similar to that shown, in Figure 3, for Cluster 1.1.
The dependence of EC on B3+ cation position in Clusters 1.1 and 1.2 (EC(1.1) and EC(1.2)) is similar to the dependence in Cluster 1 (EC(1)), as shown in Figure 4 and Table 4. However, EC(1.1) and EC(1.2) in each of the B3+ possible positions is 4–21 eV higher than the corresponding EC(1). Such a slight increase in EC(1.1) and EC(1.2) can be explained as follows. A congruent crystal (Cluster 1) grows from a congruent charge, i.e., the ratio of the main components in the melt and the crystal growing from it coincides (R = [Li]/[Nb] = 0.946). Obtaining stoichiometric (R = 1) and near-stoichiometric (0.946 < R < 1 [1,24]) crystals is a complex technological task; the complexity is caused by some features of the phase diagram of the Li2O-Nb2O5 system in the region of the LN phase [24,43,44]. The stoichiometric composition of the LN phase is at the edge of the homogeneity region, and a pronounced maximum is absent from the intersection of the solidus and liquidus lines. This leads to a partial dissociation of the compound. Therefore, even very weak doping of the melt with a chemically active element B3+ greatly changes the composition of the melt. This changes the type and concentration of reactive ionic complexes in the melt. The complexes determine the crystal structure during LN crystallization. The layer crystal-melt changes most seriously. This inevitably increases the energy of such a system. Therefore, the EC(1.1) and EC(1.2) energies are greater than EC(1). Thus, despite this increase in the energy, B3+ cations in Clusters 1.1 and 1.2 (Table 4 and Figure 4) will prefer to localize in the same tetrahedra faces as in Cluster 1: in lithium (LiIO6, LiIIO6), vacant (VIO6, VIIO6) octahedra, or in the middle oxygen plane (OI4-OII4).
The dynamics of the EC(2.1–3.4) strongly differ from those of EC(1), EC(1.1), and EC(1.2), as shown in Table 4 and Figure 4. EC in Clusters 2.1–3.4 is significantly smaller (sometimes more than two times) than that in Clusters 1, 1.1, and 1.2. Regular clusters without defects have EC values around −250–−350 eV, whilst some of EC values of clusters with a defect can reach −620 eV and even less. This fact means that boron is more likely to be embedded near a defect than in a regular structure. For boron, according to our calculations, this is energetically favorable. Later, we will prove that B is more likely to occupy a position near an empty octahedron, and not just regularly empty, but defectively empty (VNb). Thus, boron decreases the distortion of such an octahedron and strengthens the whole anion sublattice at a local level of separate clusters. Moreover, it has a charge +3, this decreases the charge tension caused by VNb defects, since VNb brings an extra -5 charge to the crystal lattice. The defects VNb are supposed to compensate for NbLi and NbV in the whole crystal but, locally, the defects strongly distort the electrical field of a crystal. The B3+ cation favorably incorporating near VNb5− decreases this local charge tension. Thus, B not only structures the LN melt upon doping [14], but also positively influences the local substructure of a doped LN crystal despite its small concentration. This is probably one of the reasons why boron-doped LN crystals are optical damage resistant.
The exceptions are energies of B localized in: NbIO6 position for Clusters 2.1 and 2.3; NbIO6 and NbVIO6 positions for Clusters 3.1 and 3.3; and NbIIO6 position for Clusters 3.2 and 3.4, as shown in Table 4 and Figure 4. EC of these exceptions are close to EC in Clusters 1, 1.1, and 1.2 in positions VIO6 and VIIO6. As we have already mentioned above, these positions of B cation are the most probable ones in Clusters 1, 1.1, and 1.2, since their Coulomb energy is lower than energies of other positions of boron, Figure 4.
The minimum EC in Clusters 2.1 and 2.3 corresponds to the boron position VNbIIO6: −582 and −588 eV, respectively. The minimum EC in Clusters 2.2 and 2.4 also corresponds to the position of boron in the tetrahedral face joint with the niobium vacancy octahedron, but this time is located in layer I, VNbIO6: −622 and −623 eV, respectively, as shown in Table 4 and Figure 4.
Cluster pairs built on the XRD data of different crystals (LN:B(1) and LN:B(2)), but with the same defect in one layer (I or II), have very similar EC values in similar positions; the difference between them is only 13 eV or even less, as shown in Table 4. We speak about Cluster pairs 2.1 and 2.3, 2.2 and 2.4, 3.1 and 3.3, and 3.2 and 3.4. Sometimes the energy minimum belongs to LN:B(1) (Clusters 2.1, 2.2, 3.1, and 3.2), and in other cases to LN:B(2) (Clusters 2.3, 2.4, 3.3, and 3.4). However, a greater difference in these pairs is determined only in several points: VIO6 EC(2.4) − EC(2.2) = 22 eV; OI4-OII4 EC(2.4) − EC(2.2) = 37 eV; and OI4-OII4 EC(3.3) − EC(3.1) = 19 eV. Clusters with lower energies (2.2 and 3.1) belong to LN:B(1) crystal. Hence, homogeneous doping forms a certain mutual arrangement of the structural units of the cationic and anionic sublattices of the LN crystal. With this arrangement, the minimal interaction of the Coulomb energy of trace amounts of boron cations with the crystal structure is deeper in LN:B(1) than in LN:B(2).
EC(3.1) and EC(3.3) are minimum when the B3+ cation is localized in the VNbIIO6 position, −561 and −560 eV, respectively, as shown in Table 4. In Clusters 3.2 and 3.4 EC is minimum when the B3+ cation is localized in the VNbIO6 position, −566 and −575 eV, respectively, as shown in Table 4. Thus, boron will favorably occupy a position near a vacant niobium octahedron (VNbIO6 and VNbIIO6) while incorporating into a defect-containing cluster.
Despite the symmetry of defect positioning in oxygen layers I and II for each crystal and each defect type (Cluster 2.1 has a NbLi defect in layer I, Cluster 2.2–in layer II, all other configuration is the same, both clusters refer to one crystal LN:B(1)), the most energetically favorable boron positions are not symmetric in similar clusters. Thus, Coulomb energy is sensitive to the mutual location of defects along the polar axis. This is true, at least, in LN clusters as small as those used in our study.
Figure 4 and Table 4 show that EC depends on boron localization in the faces of vacant tetrahedral voids, the type of structural defects, and the doping technology. The most interesting dependence is on the doping technology. Thus, we have to compare real LN:B(1) and LN:B(2) crystals in terms of the EC. However, Table 4 demonstrates boron localization energy only in separate clusters, not in the whole crystal. Real crystals contain a lot of described clusters. We have developed a method that allows us to evaluate the probability of boron occupying all sites. The method includes the second adjustment of real XRD data into our model.
In order to evaluate real crystals data we have to consider site population factors for intrinsic cations (G(Li), G(Nb)) and defects (G(NbLi), G(NbV)), as shown in Table 2 [15]. We also have to take into consideration the fraction of vacant octahedra not occupied by NbV. We are thus led to the second step of real XRD data adjustment in our calculations.
The main volume of the LN:B(1) and LN:B(2) crystal is the alteration of cations, vacancies, and defect corresponding to the perfect Clusters 1.1 and 1.2, respectively. This perfect structure contains some small amounts of defect clusters of different configurations scattered in the bulk of the crystal. So, if we want to calculate the contribution of each type of cluster to the whole real crystal, we have to take population factors into consideration.
The formula for the calculation of the real contribution of energy of each boron position () to the whole crystal for Clusters 1.1 and 1.2 is as follows (Cluster 1.1 is given as an example):
where is the Coulomb interaction energy of a boron cation located in the LiIO6 face of a tetrahedron of Cluster 1.1, is a corresponding value from Table 4, and K1.1 is a coefficient taking into account the site population factors of the main metal cations and defects in the structure of the considered Cluster 1.1 (see Table 2 for data for crystal LN:B(1)). The coefficient is calculated using the formula:
where G(LiLi) is the population factor of Li atoms in lithium sites, G(NbNb) is the population factor of Nb atoms in niobium sites, and G(VV) is the amount of strictly vacant octahedra in vacant sites calculated using the formula:
where G (NbV) is the population factor of Nb atoms occupying vacant octahedra.
Strictly speaking, we cannot discuss vacancy ‘population’, since these are octahedra free of any cations; they are not populated. However, LN structure vacancies are crucial; we must take their amount into consideration.
Calculations of and Ki for all possible boron positions (in LiIO6, NbIO6, VIO6, OI4-OII4, LiIIO6, NbIIO6, and VIIO6 faces of tetrahedra), made for Clusters 1.1 and 1.2 according to Formulas (3)–(5), are given in Table 5. In the case of Cluster 1.2, corresponding data were taken from Table 2.
Table 5.
Calculation of the Coulomb interaction energy of a boron cation located in the corresponding face of a tetrahedron of studied Clusters (), considering site population factors for intrinsic cations (Li, Nb), defects (NbLi, NbV), amount of vacant octahedra in vacant and niobium sites, and values of coefficients Ki calculated according to Formulas (4), (7) and (10).
The calculations for Clusters 2.1–2.4 were similar, but they included data for corresponding defect (NbLi) and compensated for niobium vacancy (VNb). The formula for Cluster 2.1 is given as an example; Clusters 2.2–2.4 were calculated correspondingly:
where is the Coulomb interaction energy of a boron cation located in the NbLiIO6 face of a tetrahedron of Cluster 2.1, is a corresponding value from Table 4, and K2.1−2 is a coefficient calculated using the formula:
where G(NbLi) is the population factor of Nb atoms occupying lithium octahedra, and G(VNb) is the amount of vacant niobium octahedra calculated as:
The coefficient K2.1−2 is used because the structure of Clusters 2.1 and 2.2 is different only in the mutual location of defects; all other data (the amount of defects in the cluster, the site population factor for them) are the same. The coefficients are identical for Clusters 2.3 and 2.4 (K2.3−4), 3.1 and 3.2 (K3.1−2), and 3.3 and 3.4 (K3.3−4). The coefficients and calculations according to the Formula (7) are given in Table 5.
Clusters 3.1–3.4 contain a defect NbV, which should be taken into consideration in calculations. The Coulomb energy calculation in Cluster 3.1 is given as an example; all other calculations for Clusters 3.2–3.4 were made similarly, but with the according data from Table 2 and Table 4:
where is the Coulomb interaction energy of a boron cation located in the LiIO6 face of a tetrahedron of Cluster 3.1, is a corresponding value from Table 4, and coefficient K3.1−2 was calculated using the formula:
where G(i) is the corresponding population factor for the corresponding crystal (LN:B(1) or LN:B(2)). The results of the calculations are given in Table 5.
We have obtained and summed them for each studied crystal in each position:
Thus, we have summed all numbers in the columns of Table 5 for each LN:B crystal. The results are given in Figure 5.
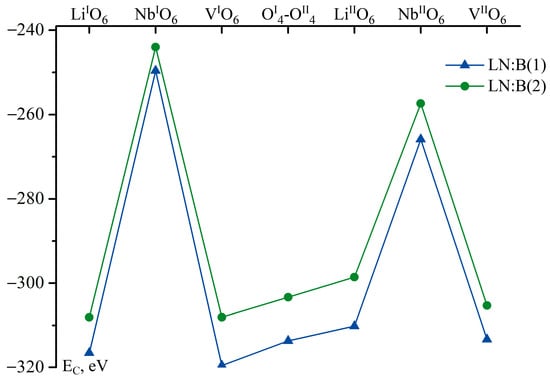
Figure 5.
Sum Coulomb interaction energies of a boron cation located in the corresponding face of a tetrahedron of studied Clusters (EC) considering site population factors for intrinsic cations (Li, Nb), defects (NbLi, NbV), and amount of vacant octahedra in vacant and niobium sites. Blue curve shows data for LN:B(1), green curve for LN:B(2).
Figure 5 shows that the localization of B3+ cations in LN:B(1) and (2) crystals (0.02 and 0.547 mol% B2O3 in the charge) is most probable in the three positions in the faces of vacant tetrahedral voids. The first position is the localization of B3+ cations in the faces of vacant tetrahedral voids, which are similar to lithium ones (LiIO6, LiIIO6); the second position is the localization of B3+ cations in the faces of vacant tetrahedral voids, which have vacant (VIO6, VIIO6) octahedra; and the third position is the localization of B3+ cations on the oxygen plane, which is common for adjacent tetrahedra (OI4-OII4). The obtained data are in a good agreement with the results of references [14,17]. The absolute energy minimum corresponds to the position of the B3+ cation in the VIO6 face of the vacant tetrahedral void of the LN:B(1) crystal obtained via homogeneous doping, as shown in Figure 5.
Thus, the technology of homogeneous doping, compared to the technology of direct solid-phase doping, leads to a general decrease in the energy of the Coulomb interaction of the B3+ cation with the crystal fragment surrounding it (Figure 5), regardless of its localization in the considered Clusters 1.1–3.4, as can be seen in Figure 4 and Figure 5, Table 4 and Table 5. The obtained data are in a good agreement with the results of XRD analysis presented in reference [15]. The R = [Li]/[Nb] value for the LN:B(1) crystal calculated on the basis of site population factors (Table 2 [15]) is 0.985. It is close to the value R of a stoichiometric crystal. The interatomic distances <Me-O> in the clusters of the main motif (NbO6, LiO6), and in the clusters of the sublattice of point defects (NbLiO6) of the LN:B(1) crystal, are closer to the interatomic distances of the NSLN(HTTSSG 5.5 wt% K2O) (Table 3) compared to those for the LN:B(2) crystal [15].
4. Conclusions
LN:B crystals (0.02 (homogeneous doping) and 0.547 (solid-phase doping) mol% B2O3 in the charge) served as examples for the double-adjustment calculations of LN clusters based on real XRD data. The study showed that the technology of homogeneous doping, compared to the technology of direct solid-phase doping, helps to obtain more pure LN:B crystals. They are closer in stoichiometry to a nominally pure stoichiometric LN crystal. The values of the site population factors G of lithium and niobium cations and interatomic distances (<Me-O>, Å) in clusters of the main motif (NbO6, LiO6) and secondary structure (NbLiO6) of LN:B(1) crystals are closer to the listed parameters of a near-stoichiometric crystal NSLN(HTTSSG 5.5 wt% K2O) in comparison to LN:B(2) [15]. Earlier, laser conoscopy [15] confirmed that the LN:B(1) is the more optically uniform crystal compared to LN:B(2). The results of calculating the energy of the Coulomb interaction of the B3+ cation with fragments of the structure of LN:B crystals, constructed on the basis of real XRD data, took into account point structural defects (NbLi, NbV, and VNb). The results of the double-adjustment calculation are consistent with the results of references [14,15,17] and the XRD data [15]. This fact indicates that the most probable localization of the B3+ cation in the faces of vacant tetrahedral voids of a defect-containing cluster is near a vacant niobium octahedron (VNbIO6 and VNbIIO6). The homogeneous doping technology reduces the energy of the Coulomb interaction of the boron cation with the LN crystal fragment surrounding it, regardless of its localization in the considered clusters. The absolute energy minimum corresponds to the position of the boron cation in the face of the VIO6 vacant tetrahedral void of the LN:B(1) crystal.
Coulomb energy is sensitive to the mutual location of defects along the polar axis in a LN cluster.
Boron is more likely to be embedded near a defect than in a regular structure. Considering charges of boron and defects towards the crystal lattice, this means that this dopant positively influences the local substructure of doped LN crystals.
This study proved that the developed method of calculation can be used to confirm the real physical properties of boron-doped LN crystals and even predict them.
Author Contributions
Conceptualization, A.K., R.T. and D.M.; methodology, R.T., A.K. and S.M.; software, R.T.; validation, I.B. and I.E.; formal analysis, O.T. and N.T.; investigation, R.T., A.K., O.T., N.T., I.B., I.E. and D.M.; resources, M.P. and S.M.; data curation, R.T. and A.K.; writing—original draft preparation, R.T., A.K., N.S. and D.M.; writing—review and editing, R.T., N.S. and D.M.; visualization, R.T.; supervision, R.T.; project administration, M.P.; funding acquisition, N.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Ministry of Science and Higher Education Russian Federation scientific topic 0186-2022-0002 (registration FMEZ-2022-0016).
Data Availability Statement
The raw data required to reproduce these findings are available from authors A.K., R.T. and D.M. upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Prokhorov, A.M.; Kuz’minov, Y.S. Physics and Chemistry of Crystalline Lithium Niobate; Adam Hilger: New York, NY, USA, 1990; p. 377. [Google Scholar]
- Kitamura, K.; Yamamoto, J.K.; Iyi, N.; Kimura, S.; Hayashi, T. Stoichiometric LiNbO3 single crystal growth by double crucible Czochralski method using automatic powder supply system. J. Cryst. Growth 1992, 116, 327–330. [Google Scholar] [CrossRef]
- Zheng, Y.; Shi, E.; Lu, Z.; Cui, S.; Wang, S.; Zhong, W. A novel technique to grow stoichiometric lithium niobate single crystal. J. Cryst. Growth 2005, 275, e895–e898. [Google Scholar] [CrossRef]
- Palatnikov, M.N.; Shcherbina, O.B.; Sandler, V.A.; Sidorov, N.V. Study of a stoichiometric lithium tantalate crystal obtained by VTE (Vapor transport equilibration) processing. Perspect. Mater. 2011, 2, 659–664. (In Russian) [Google Scholar]
- Lengyel, K.; Peter, A.; Kovacs, L.; Corradi, G.; Palfalvi, L.; Hebling, J.; Unferdorben, M.; Dravecz, G.; Hajdara, I.; Szaller, Z.; et al. Growth, defect structure, and THz application of stoichiometric lithium niobate. Appl. Phys. Rev. 2015, 2, 040601. [Google Scholar] [CrossRef]
- Polgar, K.; Peter, A.; Kovacs, L.; Corradi, G.; Szaller, Z. Growth of stoichiometric LiNbO3 single crystals by top seeded solution growth method. J. Cryst. Growth 1997, 177, 211–216. [Google Scholar] [CrossRef]
- Shi, Q.; Nozawa, J.; Uda, S. Effect of interface electric field on partitioning during the growth of conventional and true congruent-melting LiNbO3 crystals. J. Cryst. Growth 2020, 549, 125864. [Google Scholar] [CrossRef]
- Yakhnevych, U.; Vasylechko, L.; Hurskyj, S.; Buryy, O.; Sugak, D.; Zhydachevskyy, Y.; Sydorchuk, V.; Lakhnik, A.; Syvorotka, I.; Suchocki, A.; et al. A1 Piezoelectric High-Temperature Sensors I (Special Session). In SMSI 2021—Sensors and Instrumentation; 2021; pp. 45–46. Available online: https://www.ama-science.org/proceedings/details/3934 (accessed on 6 March 2023).
- Shi, Q.; Uda, S. Non-steady-state crystal growth of LiNbO3 in the presence of an interface electric field. J. Cryst. Growth 2021, 566–567, 126161. [Google Scholar] [CrossRef]
- Ma, C.; Liu, K.; Ma, C.; Liu, Y.; Xu, Y.; Yu, S. Absorption enhancement in visible range from Fano resonant silicon nanoparticle arrays embedded in single crystal Mg:Er:LiNbO3 synthesized by direct ion implantation. Nanotechnology 2022, 33, 375201. [Google Scholar] [CrossRef]
- Yan, X.; Tian, T.; Wang, M.; Shen, H.; Zhou, D.; Zhang, Y.; Xu, J. High Homogeneity of Magnesium Doped LiNbO3 Crystals Grown by Bridgman Method. Crystals 2020, 10, 71. [Google Scholar] [CrossRef]
- Yang, J.; Lai, M.; Shang, J.; Li, Q.; Zhang, L.; Sun, J. Defect structure of near-stoichiometric Mg-doped LiNbO3 crystals prepared by different method. J. Cryst. Growth 2022, 580, 126478. [Google Scholar] [CrossRef]
- Liu, J.; Liu, A.; Chen, Y.; Tu, X.; Zheng, Y. Growth and optical properties of Pr-Mg co-doped LiNbO3 crystal using Bridgman method. Phys. B Condens. Matter 2022, 624, 413419. [Google Scholar] [CrossRef]
- Sidorov, N.V.; Teplyakova, N.A.; Makarova, O.V.; Palatnikov, M.N.; Titov, R.A.; Manukovskaya, D.V.; Birukova, I.V. Boron influence on defect structure and properties of lithium niobate crystals. Crystals 2021, 11, 458. [Google Scholar] [CrossRef]
- Palatnikov, M.N.; Sidorov, N.V.; Kadetova, A.V.; Titov, R.A.; Biryukova, I.V.; Makarova, O.V.; Manukovskaya, D.V.; Teplyakova, N.A.; Efremov, I.N. Growing, structure and optical properties of LiNbO3:B crystals, a material for laser radiation transformation. Materials 2023, 16, 732. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Sidorov, N.V.; Titov, R.A.; Voskresenskiy, V.M.; Palatnikov, M.N. Localization of B3+ cations in the LiNbO3 crystal structure and its effect on the crystal properties. J. Struct. Chem. 2021, 62, 221–229. [Google Scholar] [CrossRef]
- Sidorov, N.V.; Serebryakov, Y.A. The structural orderings and photorefraction in lithium niobate admixed crystals. Ferroelectrics 1994, 160, 101–105. [Google Scholar] [CrossRef]
- Palatnikov, M.N.; Biryukova, I.V.; Makarova, O.V.; Efremov, V.V.; Kravchenko, O.E.; Kalinnikov, V.T. Preparation and properties of lithium niobate crystals grown from melts of congruent composition doped with boron. Proceedings KSC RAS. Chem. Mater. Sci. 2015, 5, 434–438. (In Russian) [Google Scholar]
- Masloboeva, S.M.; Efremov, I.N.; Biryukova, I.V.; Palatnikov, M.N. Growth and characterization of a boron-doped lithium niobate single crystal. Inorg. Mater. 2020, 56, 1147–1152. [Google Scholar] [CrossRef]
- Shur, V.Y.; Akhmatkhanov, A.R.; Baturin, I.S. Micro- and nano-domain engineering in lithium niobate. Appl. Phys. Rev. 2015, 2, 040604. [Google Scholar] [CrossRef]
- Kemlin, V.; Jegouso, D.; Debray, J.; Boursier, E.; Segonds, P.; Boulanger, B.; Ishizuki, H.; Taira, T.; Mennerat, G.; Melkonian, J.-M.; et al. Dual-wavelength source from 5% MgO:PPLN cylinders for the characterization of nonlinear infrared crystals. Opt. Express 2013, 21, 28886–28891. [Google Scholar] [CrossRef]
- Murray, R.T.; Runcorn, T.H.; Guha, S.; Taylor, J.R. High average power parametric wavelength conversion at 3.31–3.48 μm in MgO:PPLN. Opt. Express 2017, 25, 6421–6430. [Google Scholar] [CrossRef] [PubMed]
- Volk, T.; Wohlecke, M. Lithium Niobate. Defects, Photorefraction and Ferroelectric Switching; Springer: Berlin, Germany, 2008; p. 250. [Google Scholar]
- Sidorov, N.V.; Teplyakova, N.A.; Palatnikov, M.N. Influence of the method of doping on uniformity and optical properties of LiNbO3:Mg crystals. Phys. Chem. Asp. Study Clust. Nanostr. Nanomater. 2021, 13, 383–391. (In Russian) [Google Scholar]
- Fulei, W.; Dehui, S.; Qilu, L.; Yukun, S.; Feng, Z.; Weijia, Z.; Yuanhua, S.; Dongzhou, W.; Hong, L. Growth of large size near-stoichiometric lithium niobate single crystals with low coercive field for manufacturing high quality periodically poled lithium niobate. Opt. Mater. 2022, 125, 112058. [Google Scholar] [CrossRef]
- Zotov, N.; Boysen, H.; Frey, F.; Metzger, T.; Born, E. Cation substitution models of congruent LiNbO3, investigated by X-ray and neutron powder diffraction. J. Phys. Chem. Solid. 1994, 55, 145–152. [Google Scholar] [CrossRef]
- Wang, W.W.; Zheng, D.H.; Hu, M.Y.; Saeed, S.; Liu, H.D.; Kong, Y.F.; Zhang, L.X.; Xu, J.J. Effect of defects on spontaneous polarization in pure and doped LiNbO3: First-principles calculations. Materials 2019, 12, 100. [Google Scholar] [CrossRef]
- Cochard, C.; Guennou, M.; Spielmann, T.; van Hoof, N.; Halpin, A.; Granzow, T. Effect of optical damage resistant dopants on the dielectric properties of LiNbO3: Insight from broadband impedance spectroscopy and Raman scattering. J. Appl. Phys. 2018, 123, 154105. [Google Scholar] [CrossRef]
- Boukhtouta, M.; Megdoud, Y.; Benlamari, S.; Meradji, H.; Chouahda, Z.; Ahmed, R.; Ghemid, S.; Abu-Jafar, M.; Syrotyuk, S.; Rai, D.P.; et al. Predictions on structural, electronic, optical and thermal properties of lithium niobate via first-principle computations. Phyl. Mag. 2020, 100, 1150–1171. [Google Scholar] [CrossRef]
- Schmidt, F.; Kozub, A.L.; Biktagirov, T.; Eigner, C.; Silberhorn, C.; Schindlmayr, A.; Schmidt, W.G.; Gerstmann, U. Free and defect-bound (bi)polarons in LiNbO3: Atomic structure and spectroscopic signatures from ab initio calculations. Phys. Rev. Res. 2020, 2, 043002. [Google Scholar] [CrossRef]
- Schmidt, F.; Kozub, A.L.; Gerstmann, U.; Schmidt, W.G.; Schindlmayr, A. Electron polarons in lithium niobate: Charge localization, lattice deformation, and optical response. Crystals 2021, 11, 542. [Google Scholar] [CrossRef]
- Donnerberg, H.J.; Tomlinson, S.M.; Catlows, C.R.A. Defects in LiNbO3. II. Computer simulation. J. Phys. Chem. Solid. 1991, 52, 201–210. [Google Scholar] [CrossRef]
- Messerschmidt, S.; Krampf, A.; Vittadello, L.; Imlau, M.; Nörenberg, T.; Eng, L.M.; Emin, D. Small-polaron hopping and low-temperature (45–225 K) photo-induced transient absorption in magnesium-doped lithium niobate. Crystals 2020, 10, 809. [Google Scholar] [CrossRef]
- Vittadello, L.; Guilbert, L.; Fedorenko, S.; Bazzan, M. Polaron trapping and migration in iron-doped lithium niobate. Crystals 2021, 11, 302. [Google Scholar] [CrossRef]
- Yatsenko, A.V.; Evdokimov, S.V.; Palatnikov, M.N.; Sidorov, N.V. Features of the electrical properties of lithium niobate crystals grown from a melt containing K2O flux. Phys. Solid State 2019, 61, 1211–1217. [Google Scholar] [CrossRef]
- Sánchez-Dena, O.; Villalobos-Mendoza, S.D.; Farías, R.; Fierro-Ruiz, C.D. Lithium niobate single crystals and powders reviewed—Part II. Crystals 2020, 10, 990. [Google Scholar] [CrossRef]
- Abrahams, S.C.; Marsh, P. Defect structure dependence on composition in lithium niobate. Acta Crystallogr. B 1986, 42, 61–68. [Google Scholar] [CrossRef]
- Abrahams, S.C.; Reddy, J.M.; Bernstein, J.L. Ferroelectric lithium niobate. 3. Single crystal X-ray diffraction study at 24° C. J. Phys. Chem. Solid. 1966, 27, 997–1012. [Google Scholar] [CrossRef]
- Abrahams, S.C.; Levinstein, H.J.; Reddy, J.M. Ferroelectric lithium niobate. 5. Polycrystal X-ray diffraction study between 24 and 1200 °C. J. Phys. Chem. Solid. 1966, 27, 1019–1026. [Google Scholar] [CrossRef]
- Can, H.; Shichao, W.; Ning, Y. Subsolidus phase relations and the crystallization region of LiNbO3 in the system Li2O–B2O3–Nb2O5. J. Alloys Compd. 2010, 502, 211–214. [Google Scholar] [CrossRef]
- Kling, A.; Marques, J.G. Unveiling the defect structure of lithium niobate with nuclear methods. Crystals 2021, 11, 501. [Google Scholar] [CrossRef]
- Svaasand, L.O.; Eriksrud, M.; Nakken, G.; Grande, A.P. Solid-solution range of LiNbO3. J. Cryst. Growth 1974, 22, 230–232. [Google Scholar] [CrossRef]
- Azuma, Y.; Uda, S. Electric current induced compositional variation in LiNbO3 fiber crystal grown by a micro-pulling down method. J. Cryst. Growth 2007, 306, 217–224. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).