Abstract
The three isomers of the tetraoxa[4]arene derivative, C24H16O4, which consist of two m-phenylenes and two phenylenes (meta 1, para 2, ortho 3), represent not only intriguing fundamental structures that induce molecular recognition toward non-porous adaptive crystals, but also attractive candidates for crystallographic polymorphism. In this study, we crystallized isomers 2 and 3, in comparison to isomer 1, in order to understand their stable orientations and the corresponding intermolecular interactions in the crystalline state. For example, m-phenylene derivative 1 exhibits polymorphism with both prismatic and block-shaped crystals. Therefore, we prepared p-phenylene derivative 2 and o-phenylene derivative 3, and their structures were fully characterized by SC-XRD, revealing two polymorphs of derivative 2, namely prismatic crystal 2-I and block-shaped crystal 2-II, along with changes to the crystal lattice parameters (2-Ia, 2-Ib, and 2-Ic) based on temperature dependence. In all of its crystal forms, derivative 2 adopts an O-shaped planar structure, where the p-phenylene units face each other. This suggests that the packing mode during the early stages of crystallization, rather than due to any remarkable changes in the molecular structure, directly affects the bulk crystal morphology. On the other hand, derivative 3 adopts a U-shaped vent structure and, to the best of our knowledge, does not form polymorphs. The Platon and Hirshfeld surface analyses indicated that the contributions to the crystal packing were C···C (av. 37.3% for 2-Ia, av. 38.2% for 2-II, and 18.7% for 3), C···H/H···C (av. 37.3% for 2-Ia, av. 38.2% for 2-II, and 18.7% for 3), and O···H/H···O (av. 17.8% for 2-Ia, av. 19.6% for 2-II, and 19.4% for 3), highlighting significant intermolecular CH···π interactions and pseudo-hydrogen bonding forms for derivative 2 and π···π interactions for derivative 3.
1. Introduction
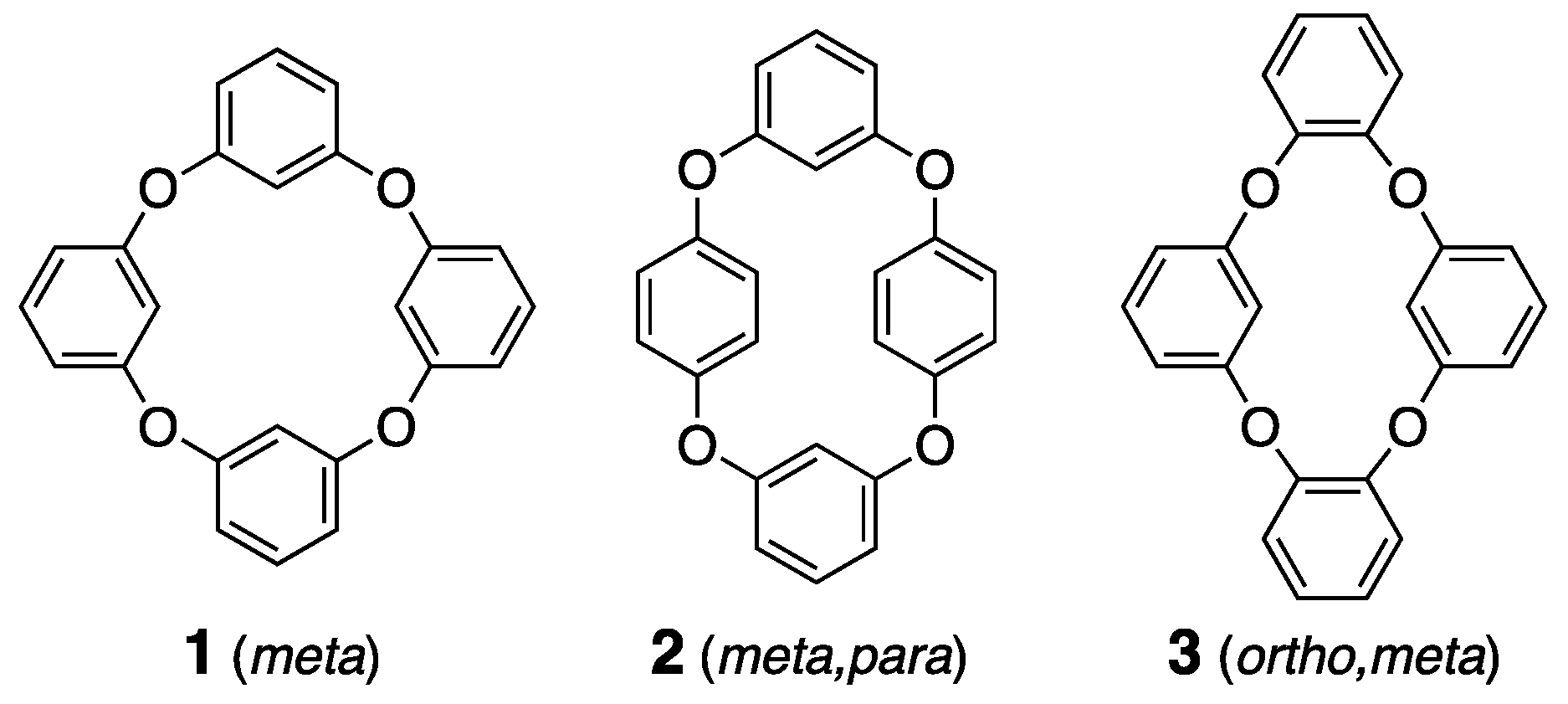
Calix[4]arene, composed of four aromatic rings linked by methylene groups (-CH2-) at the meta position, is a crucial material in host–guest chemistry, due to its unique structural features and versatile derivatization [1,2]. This compound has attracted considerable attention not only in terms of its synthetic and structural interest value, but also due to its potential applications in sensing and molecular recognition [3,4,5,6,7], which are vital for environmental monitoring and remediation. The ability of calix[4]arene to act as a receptor for various vapor and gas molecules make it an ideal candidate for developing sensors that detect pollutants in air and water. Cyclic compounds, like calix[4]arene, exhibit conformational stabilization through the macrocyclic effect, which is more pronounced than in acyclic structures of equivalent molecular weight, and show distinct spatial properties, both internally and externally [8,9,10]. The rational design of these internal spaces has facilitated molecular recognition in regard to both solution and solid states, contributing significantly to the development of precise analytical techniques and advanced separation processes [11]. Recent advancements in single-crystal X-ray analysis have revealed that even molecular crystals can incorporate molecules into their crystalline spaces, becoming reversible host materials [12,13]. This requires two design principles: higher melting points and moderate structural flexibility. Elevating the melting point of the host molecule enhances its structural stability and aids pre-treatment processes in regard to the elimination of impurities, like impurities in water, thereby improving their recyclability. Structural flexibility enhances selectivity by allowing adaptive conformational changes tailored to guest molecules [14,15,16,17,18,19,20,21,22,23,24]. Consequently, we have been focusing on the creation of host crystals using heavy-atom metal complexes [25,26]. In line with this approach, cyclic molecular crystals, even those composed of light atoms, are an intriguing subject, due to the potential for moderate increases in their melting points and structural flexibility.
Moreover, heterocalixarenes, where the bridging atoms are heteroatoms instead of carbon, have been understudied, due to synthetic challenges [27,28,29,30]. However, recent developments in various synthetic techniques have invigorated research into heterocalixarenes, which are expected to exhibit unique properties in terms of their size, conformational flexibility, and guest selectivity, due to the electronic alteration of the bridging heteroatoms [31,32,33,34]. Compounds like oxygen-bridged calix[4]arene analogues have sparked interest, due to their highly flexible orientation and the corresponding cavities that exhibit molecular recognition and separation properties, attributed to their macrocyclic structures. However, the corresponding isomers remain predominantly uncharacterized in crystallographic studies, with only a few structures defined [35,36,37]. The structural versatility of calixarene derivatives, which allows for various conformations, sets them apart from other host molecules and is advantageous for crystalline host structures. This flexibility increases the potential for forming both polymorphs and pseudo-polymorphs [38], making these derivatives particularly intriguing, as they exhibit diverse crystalline phases under different conditions, providing unique opportunities to study a wide range of solid-state behaviors. Three tetraoxacalix[4]arene derivatives, 1–3, are regarded as simple host frameworks, especially for non-polar adaptive crystals (Scheme 1), and the crystallization of compound 1, named 2,4,6,8-tetraoxa-1,3,5,7(1,3)-tetrabenzenacyclooctaphane, which shows polymorphs, such as prismatic (tetragonal, P-421c) and block (monoclinic, C2/c) crystals, has been reported [39,40]. This research prompted us to investigate the structural studies on derivatives 2 and 3.
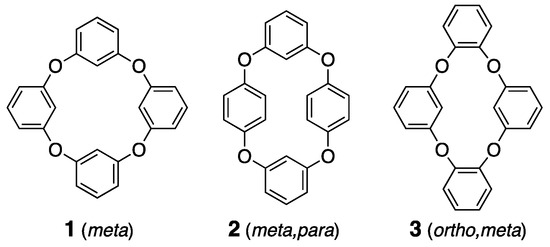
Scheme 1.
Molecular structures of derivatives 1–3.
2. Materials and Methods
2.1. General
All the chemicals were of reagent grade and were used without further purification. The 1H NMR spectral data were recorded on a JEOL ECS400 spectrometer (JEOL Ltd., Tokyo, Japan). The melting points were determined using Yanaco MP-500D melting point apparatus (Yanaco Technical Science Co., Kyoto, Japan). The mass data were obtained using the GC–MS QP2010 Plus system (Shimadzu Co., Kyoto, Japan).
2.2. Synthesis and Crystallization
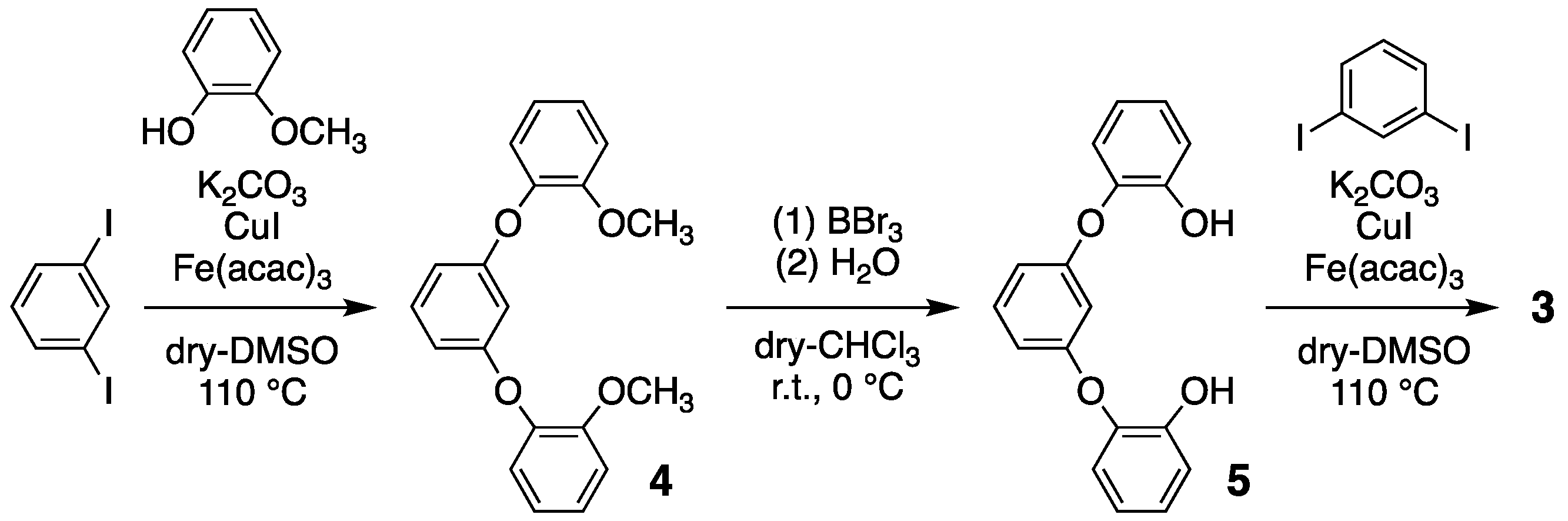
Compound 2 was prepared from resorcinol and 1,4-diiodobenzene in a single step, using a previously reported procedure [41]. In an inert gas atmosphere, resorcinol (1.315 g, 11.82 mmol), 1,4-diiodobenzene (3.967 g, 12.03 mmol), K2CO3 (6.735 g, 48.25 mmol), CuI (252.9 mg, 1.319 mmol), and Fe(acac)3 (447.2 mg, 1.242 mmol), were dissolved in dry DMSO (40 mL). The mixture was stirred and heated at 110 °C for 5 days. After cooling to room temperature, the reaction mixture was filtered and the filtrate was extracted with CH2Cl2. The organic layer was then washed with a 1 M sodium hydroxide solution, dried over sodium sulfate, and purified using flash column chromatography (CH2Cl2:hexane = 1:3) and gel permeation chromatography (GPC). The resulting colorless crystals, both block and prismatic in form, were obtained by evaporating the CHCl3 solution at room temperature (138 mg, 374 μmol, Rf = 0.49): m.p. > 190 °C; 1H NMR (400 MHz, CDCl3, TMS) of δ 7.30 (t, J = 8.0 Hz, 2H), 6.97 (s, 8H), 6.82 (d, J = 8.0 Hz, 4H), and 5.17 (s, 2H); 13C NMR (100 MHz, CDCl3) of 161.3, 151.8, 130.8, 124.2, 110.2, and 100.8; DI-MS of 368 m/z (M+).
Compound 3 was prepared in three steps, as per the previously reported protocols (Scheme 2) [41]; (1) guaiacol and 1,3-diiodobenzene were reacted with K2CO3, CuI, and Fe(acac)3, in order to yield white powder of 1,3-bis(3-methoxyphenoxy)benzene 4 (63% yield); (2) the methyl groups of 4 were removed in order to yield 1,3-bis(3-hydroxyphenoxy)benzene 5 using BBr3/H2O (95% yield); (3) finally, a cyclized reaction between 5 and 1,3-diiodobenzene was performed in order to yield colorless microcrystals of compound 3, with a 54% yield. Compound 3: m.p. 166–167 °C; 1H NMR (400 MHz, CDCl3, TMS) of δ 7.13 (br, 8H), 6.93 (br, 2H), 6.51 (br, 4H), and 5.39 (br, 2H); 13C NMR (100 MHz, CDCl3) 158.2, 146.6, 130.2, 126.4, 125.5, 110.5, and 103.0; DI-MS of 368 m/z (M+).
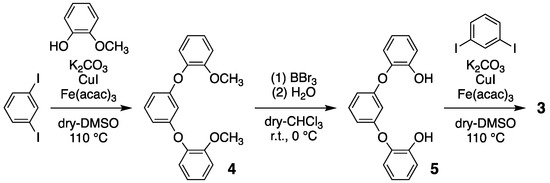
Scheme 2.
Three-step reaction involving compound 3.
2.3. Crystal Structure Determination
The single-crystal X-ray structure was determined using a Bruker D8 QUEST diffractometer (Bruker Japan Co., Yokohama, Japan), with a multilayered confocal X-ray mirror and MoKα radiation (λ = 0.71073 Å), generated at 50 kV and 1 mA. The crystals were coated with paraton-N and measured at 100~300 K. The crystal data for derivatives 2 and 3 (C24H16O4, m.w. 368.37) are summarized in Table 1 and Appendix A. For the compound, cell refinement and reduction were performed using the Bruker SAINT program (V8.40B), the determination of the structure solution was carried out using SHELXT [42], and the refinement was conducted using SHELXL [43]. Empirical adsorption corrections were applied using the SADABS program (2016/2). All the non-hydrogen atoms were refined anisotropically, unless otherwise stated, and the hydrogen atoms were constrained at idealized positions, with the C–H distances set at 0.95 Å for aromatic compounds, and Uiso(H) = 1.2Ueq(C). These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/structures/ (accessed on 23 November 2024).

Table 1.
Crystal data and structure refinement for derivatives 2 and 3.
3. Results and Discussion
3.1. Preparations of Derivatives 2 and 3
Compounds 2 and 3 were prepared through a coupling reaction between dihydroxybenene and diiodobenzene molecules. Both compounds were purified using column chromatography (silica, CHCl3) in order to yield a white powder and were crystallized from CHCl3 and/or CH2Cl2 solutions to produce colorless single crystals. For the crystallization of compound 2, prismatic (2-I) and block (2-II) crystals were obtained as major and minor products, respectively, in a CHCl3 solution, while only prismatic 2-I crystals were obtained from a CH2Cl2 solution. From a 1:1 CHCl3/CH2Cl2 solution, prismatic 2-I crystals were also mainly obtained, along with 2-II crystals, similar to the results from the CHCl3 solution, indicating that the major product was prismatic 2-I crystals, with minor 2-II crystals occasionally obtained from slow crystallization processes. Single crystallographic studies suggest that compound 2 exists as a polymorph, i.e., the major prismatic form 2-I (P21/n) and the minor block form 2-II (P21/c), similar to compound 1, which exhibits a major prismatic form 1-I (P-421c) and a minor block form 1-II (C2/c). The prismatic 2-I crystals provide the crystal structure of 2-Ia, which contains 1.5 molecules in the asymmetric unit at a low temperature (100 K). However, this crystal undergoes a reversible transformation into a more symmetrical packing structure, 2-Ic, at room temperature, where the asymmetric unit contains 0.5 molecules. During the packing change, an intermediate crystal state 2-Ib was also observed at 200 K, which contains two molecules in the asymmetric unit and is distinguishable in regard to 2-Ia and 2-Ic. While the cell parameters of 2-I show remarkable temperature dependence, the 2-II and 3 crystals show stable conformation with 2.5 and 1 molecules, respectively, in the asymmetric unit.
3.2. Crystal Structure and Intermolecular Interactions of 2-Ia
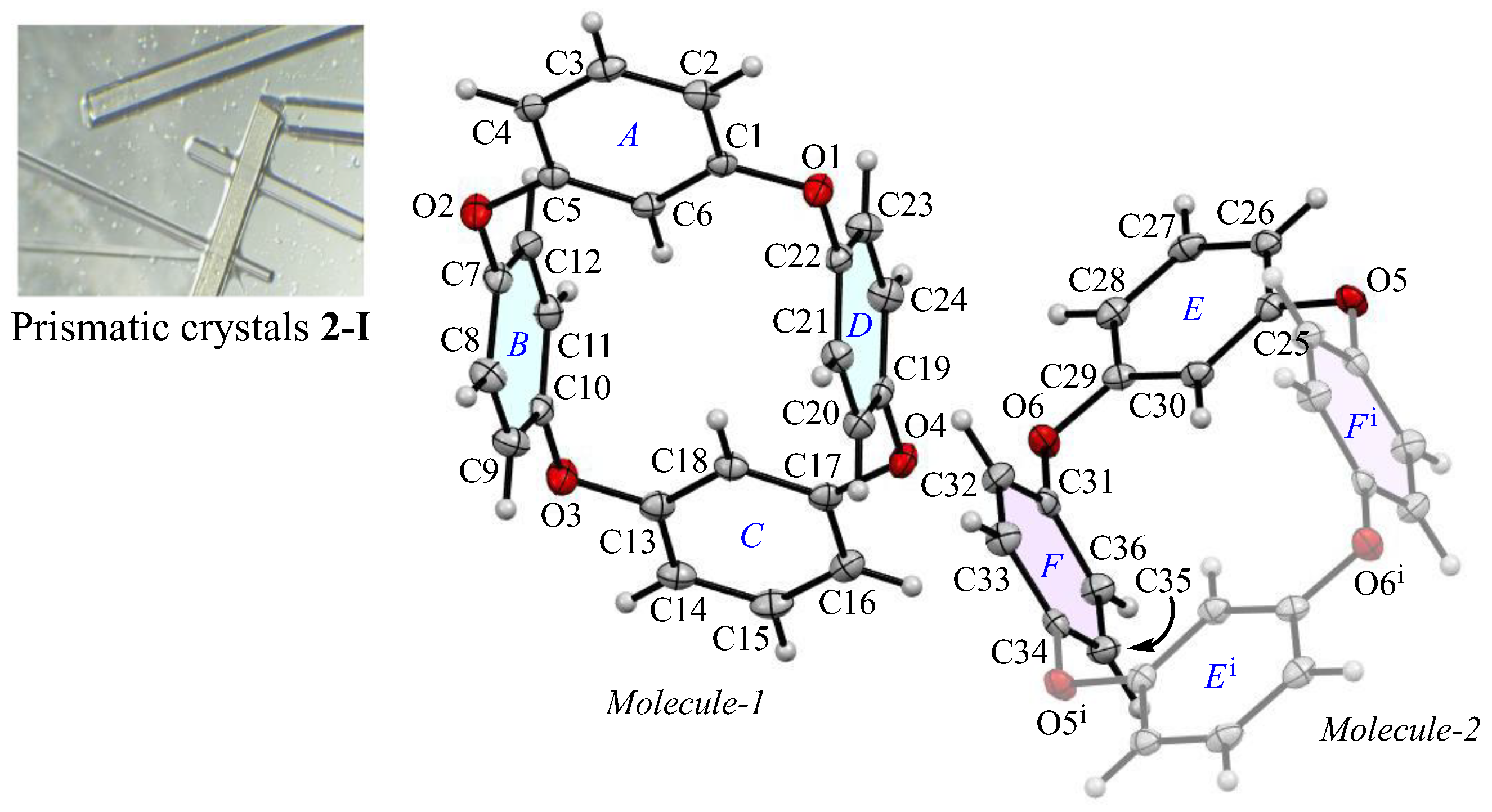
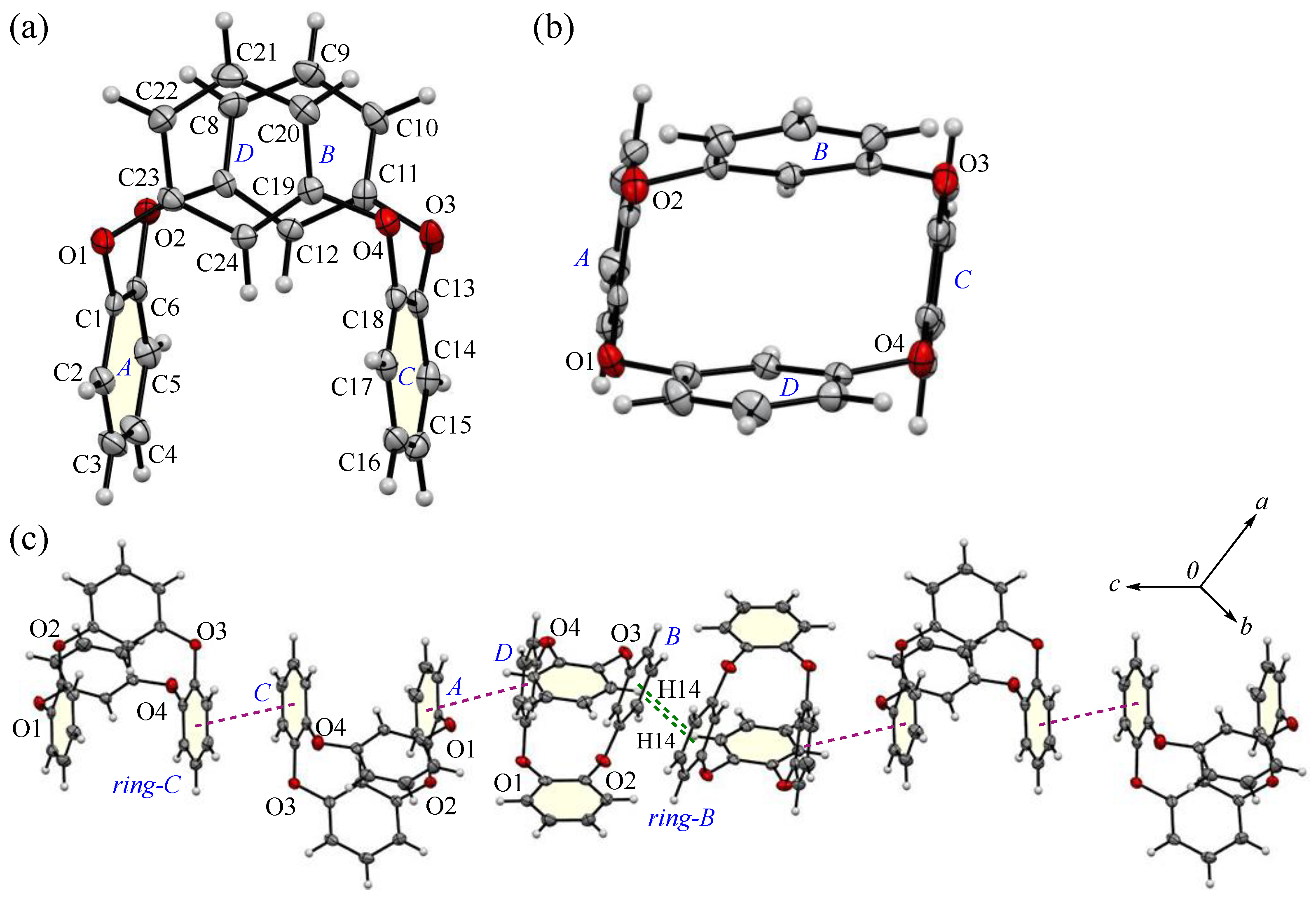
The appearance and ORTEP view, with the numbering schemes, of the prismatic crystal 2-Ia at 100 K are shown in Figure 1. In the crystal, the asymmetric unit contains a whole compound, Molecule-1, and one half of a compound, Molecule-2. In Molecule-1, two m-phenylene and two p-phenylene rings are alternately linked by oxygen atoms to form a cyclic structure. The average bond distance of C-O and the angle of C-O-C for six oxygen atoms (O1–O6) are 1.393 Å and 116.9°, respectively, showing single bonds and localized π-electrons. The two m-phenylene faces are nearly planar [the dihedral angle between rings-A and C is 1.26(16)°] across the plane of the ring framework, while the two p-phenylene faces are oriented face-to-face [the dihedral angle of rings-B and D is 4.05(15)°], rising perpendicular to the plane. The intramolecular distance between the centroids of two p-phenylene rings is 4.890(4) Å for Cg<ring-B>···Cg<ring-D>. For Molecule-2, the cyclic structure is centrosymmetric, with an intramolecular distance of 4.905(4) Å for Cg<ring-F>···Cg<ring-Fi>. The corresponding RMS deviation in the structural overlay of the ring frameworks, 24 carbon and 4 oxygen atoms, of Molecule-1 and Molecule-2, is only 0.045 Å.
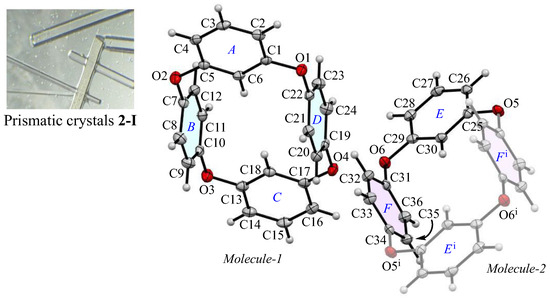
Figure 1.
The molecular structure of 2-Ia crystals at 100 K, showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 50% probability level. Symmetry code i: −x, −y, −z + 1.
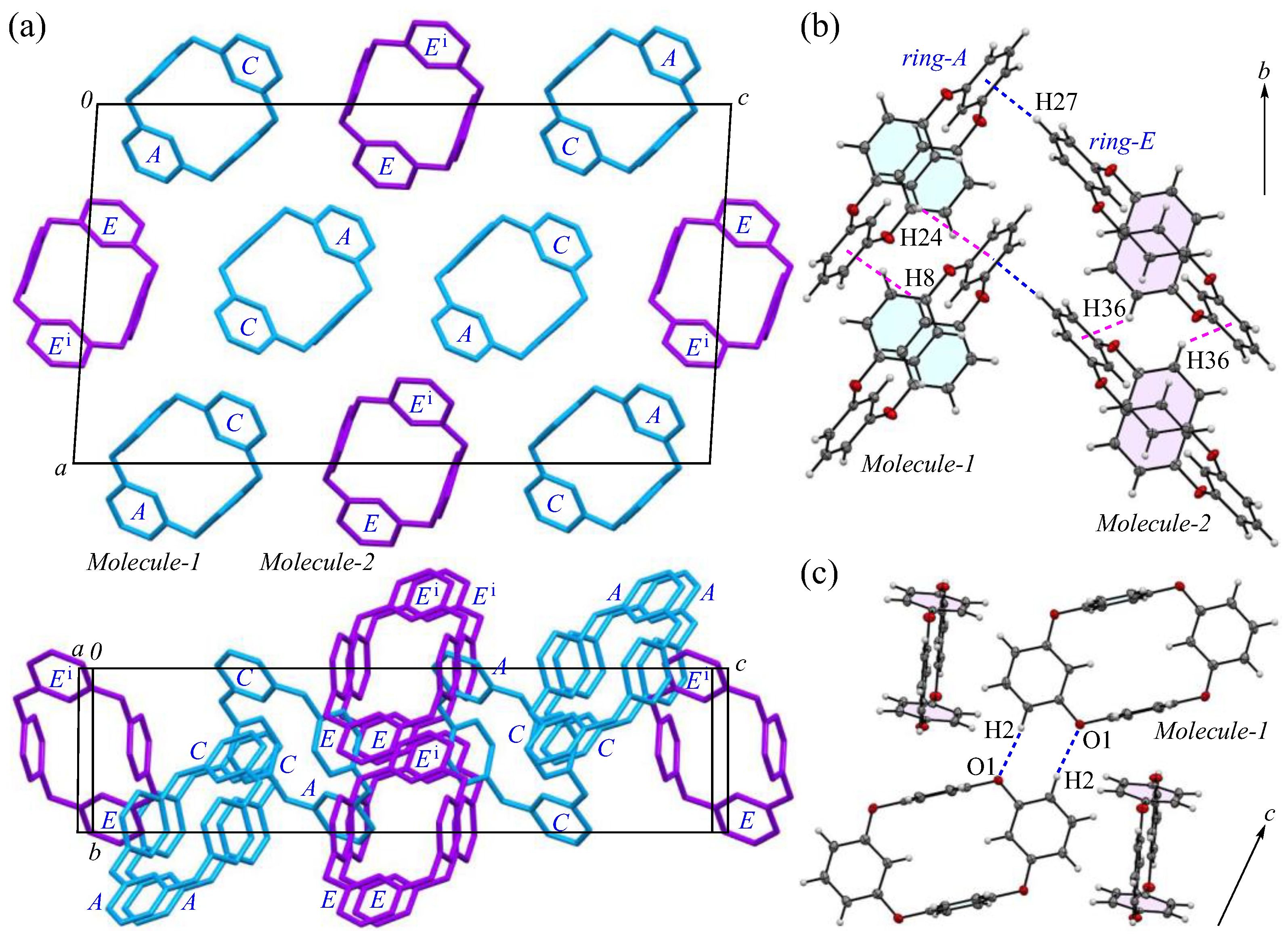
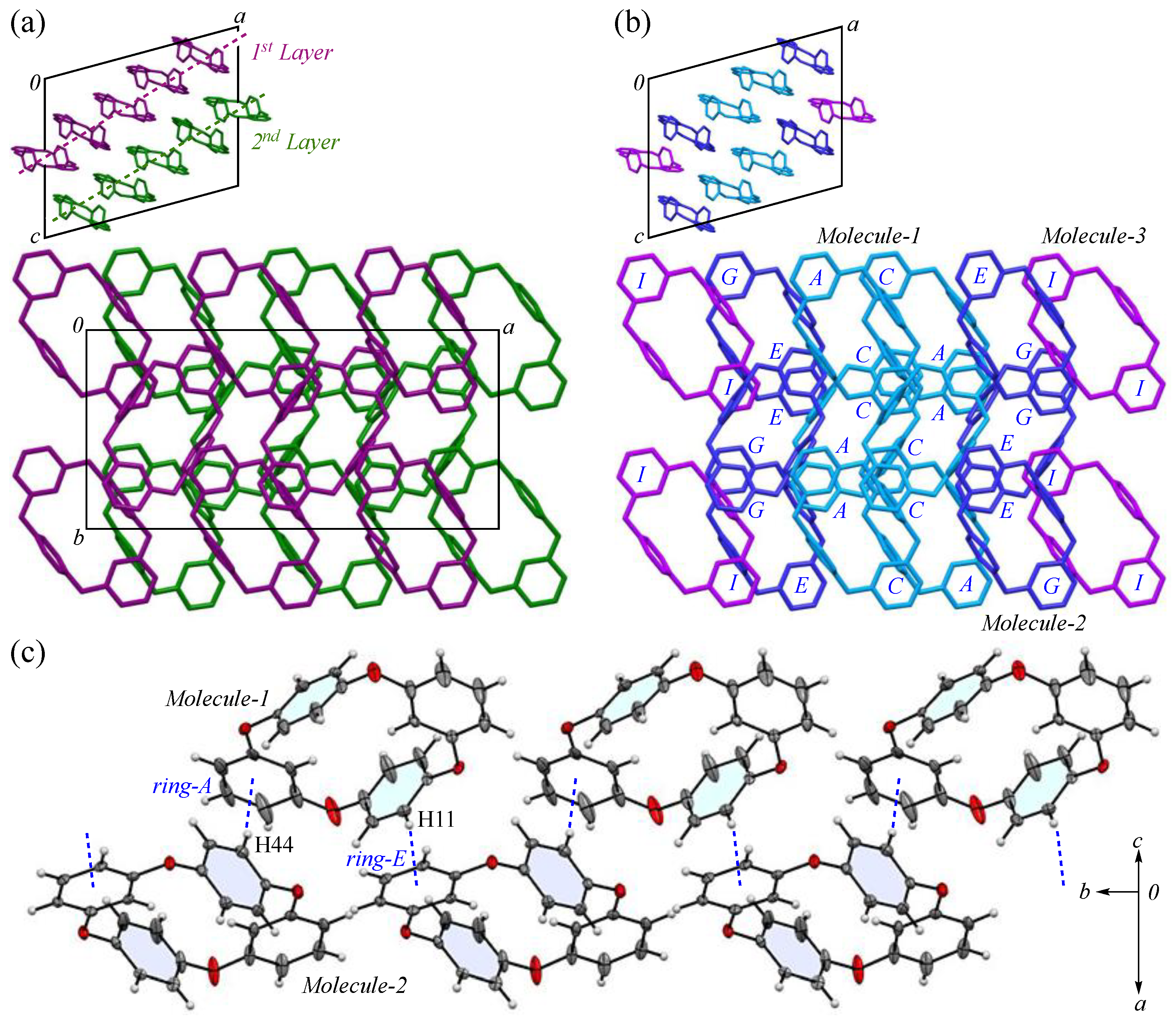
The packing structures of the 2-Ia crystals are shown in Figure 2, with the two crystallographically independent molecules; Molecule-1 and Molecule-2 are in blue and purple, respectively. As illustrated in Figure 2a, both molecules are arranged in a columnar alignment along the b-axis, positioned obliquely to the ac plane: the dihedral angles between the ac plane and the planes, defined by the four oxygen atoms in Molecule-1 and Molecule-2, are 47.93° and 48.61°, respectively, while the dihedral angle between the planes of Molecule-1 and Molecule-2 themselves is 12.89°. These two crystallographically nonequivalent molecules exhibit a distinct difference in their packing arrangement. For Molecules-1 and 2, which are aligned on the ac plane, particularly along the c-axis, the dihedral angle between the mirror–axes connecting the m-phenylene units in each molecule is 26.9°, indicating that the offset of Molecule-2 from Molecule-1 induces asymmetry in the crystal system, leading to an enlarged lattice parameter. Additionally, when viewed along the a-axis, the molecules adopt a zigzag arrangement, resulting in no π···π stacking [44] between the aromatic phenylene rings. The primary intermolecular interaction within the crystal is the CH···π interaction, with three interactions identified: C3-H3 of Molecule-1 [symmetry code: x, y, z] interacts with ring-E of the adjacent Molecule-2 [symmetry code: −x + 1, −y, −z + 1]; C27-H27 of Molecule-2 [symmetry code: x, y, z] interacts with ring-A of the neighboring Molecule-1 [symmetry code: −x + 1, −y + 1, −z + 1]; and C28-H28 of Molecule-2 [symmetry code: x, y, z] interacts with ring-D of the adjacent Molecule-1 [symmetry code: x, y, z]. The H···Cg(ring) distances for these interactions are 2.66, 2.51, and 2.80 Å, respectively. The shortest distance, observed for C27-H27 in ring-E, forms a strong CH···π interaction between ring-A and ring-E (Figure 2b), with ring-Ei on the opposite side establishing an equivalent CH···π interaction [45,46] with the adjacent ring-A, creating a zigzag molecular arrangement, which is often seen in planar aromatic compounds. Furthermore, weak CH···π interactions are also present along the molecular columns in the b-axis direction. Potential hydrogen bonds were also observed between C2-H2···O1 and C14-H14···O5, with intermolecular C···O distances of 3.444(5) and 3.483(5) Å, and C-H···A angles of 164° and 161°, respectively (Figure 2c).
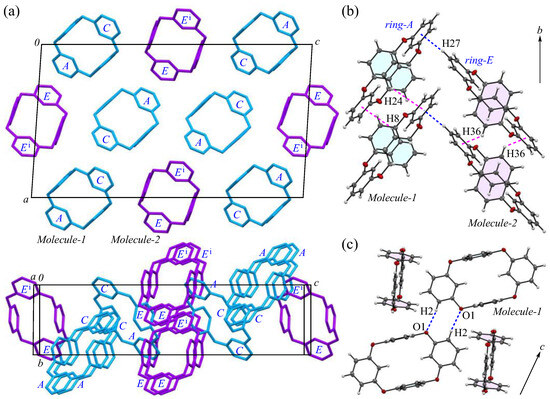
Figure 2.
Components of the crystal structure of 2-Ia: (a) top and side views of the molecular packing along the b and c-axes, respectively, with Molecule-1 in blue and Molecule-2 in purple; (b) columnar stacking along the b-axis; and (c) weak dipole interactions along the c-axis. Color scheme: C, gray; H, white; O, red.
3.3. Crystal Structure and Intermolecular Interactions of 2-II
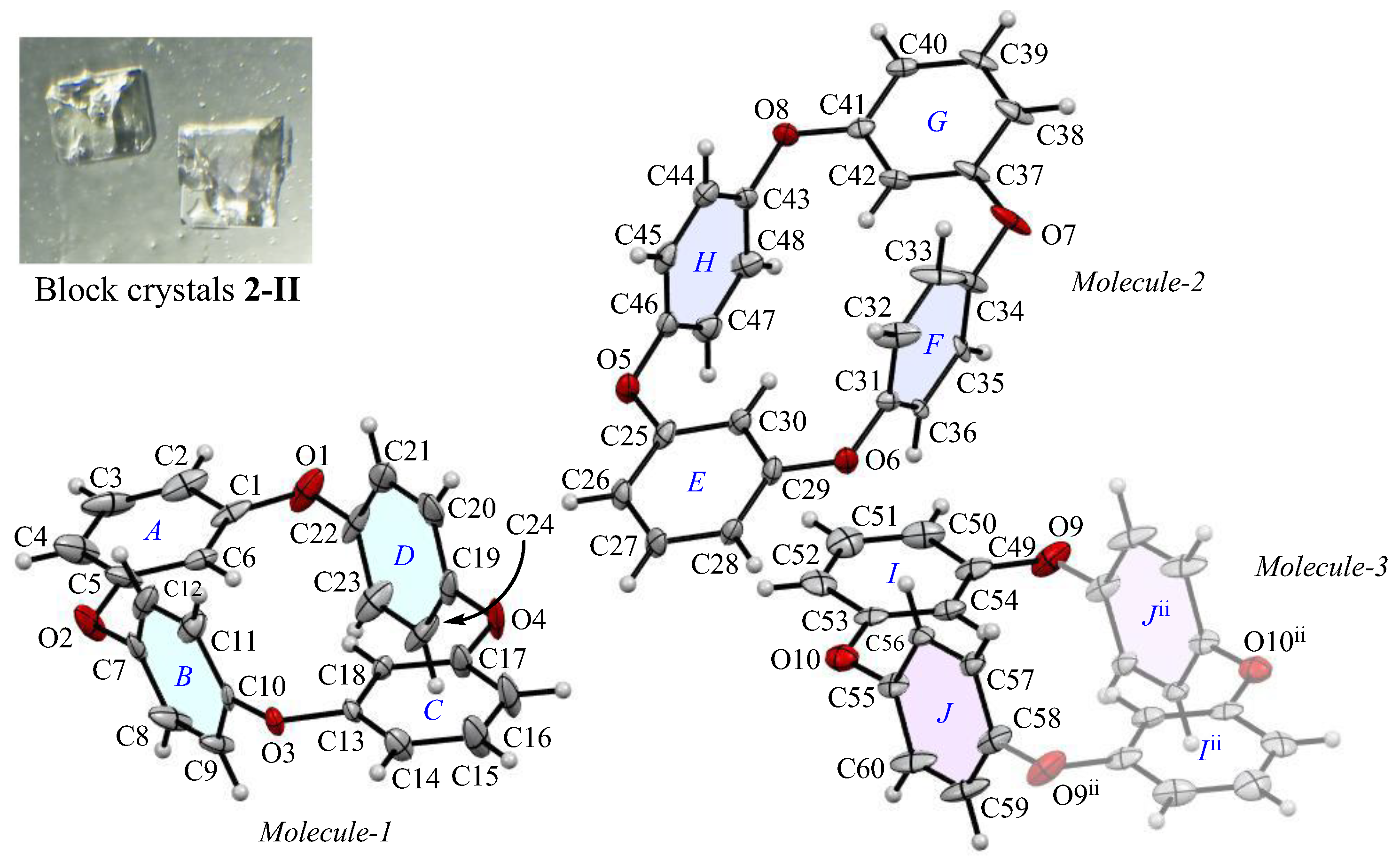
The appearance and ORTEP view, with the numbering schemes, of block crystal 2-II are shown in Figure 3. The crystal forms are minor, which are occasionally obtained from a CHCl3 solution. In the 2-II crystal, the asymmetric unit contains two types of whole compounds (Molecules-1 and 2) and one half compound (Molecules-3). The average bond distance of C-O and angle of C-O-C for six oxygen atoms (O1–O10) are 1.392 Å and 117.0°, respectively, showing similar structural and electronic configurations to 2-Ia crystals. The two m-phenylene faces are almost flat [the dihedral angles are 9.07(2)° of rings-A and C in Molecules-1, 5.25(2)° of rings-E and G in Molecules-2, and 0° of rings-I and Iii in Molecules-3] across the plane of the ring framework, and the corresponding two p-phenylene faces are oriented face-to-face [the dihedral angles are 12.6(2)° of rings-B and D, 6.4(2)° of rings-F and H, 0° of rings-J and Jii], and the intramolecular distances between the centroids of two p-phenylene rings are 4.880(3) Å for Cg<ring-B>···Cg<ring-D>, 4.869(3) Å for Cg<ring-F>···Cg<ring-H>, and 4.880 Å for Cg<ring-J>···Cg<ring-Jii>. The corresponding RMS deviation in the structural overlay of 24 carbon and 4 oxygen atoms of Molecule-1 and Molecule-2 is 0.166 Å, that of Molecule-2 and Molecule-3 is 0.084 Å, and that of Molecule-3 and Molecule-1 is 0.131 Å, indicating that the difference in superposition between the molecular structures present in the crystallographic space is greater for 2-II crystals than for 2–1a crystals (RMS = 0.045 Å). In terms of the crystal form, both 2-Ia and 2-II crystals adopt the same O-shaped planar structure, where the p-phenylene units face each other. This suggests that the packing mode during the early stages of crystallization, rather than due to any remarkable changes in the molecular structure, directly affects the bulk crystal morphology.
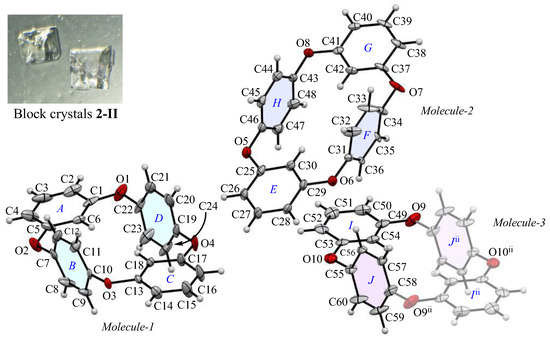
Figure 3.
The molecular structure of 2-II crystals at 100 K, showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 50% probability level. Symmetry code ii: −x + 2, −y, −z + 1.
The packing structure of 2-II crystals is shown in Figure 4. The large image provides a view along the c-axis, where the cyclic structure is displayed face on, while the small image shows the view along the b-axis, showing the layered arrangement formed by these cyclic molecules. In Figure 4a, each layer extending along the ab plane is color coded in green and purple. Within each layer, the cyclic molecules are aligned with a half-overlap along the b-axis and these rows form a herringbone-like pattern that constructs the (2 0 1) plane. Consequently, when viewed along the c-axis, the packing arrangement differs from the columnar structure in crystal 2-Ia, filling the space in a distinctive way; however, the density of 2-II crystals, estimated by crystal structure analysis, is 1.306 Mg m−3, indicating a less compact structure than 2-Ia crystals, which have a density of 1.329 Mg m−3.
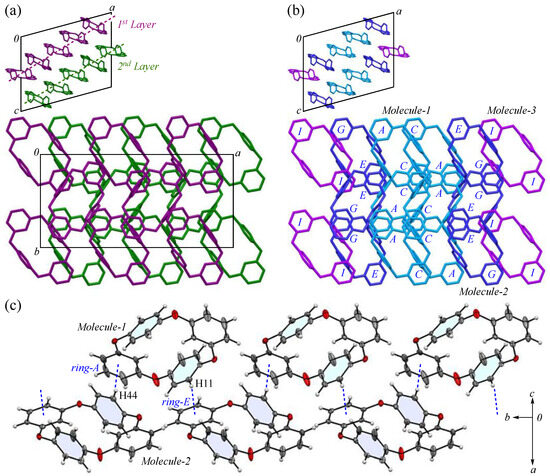
Figure 4.
Components of the 2-II crystal structure: (a) layer view; (b) three types of molecules viewed from the b-axis, with the first layer in maroon, the second layer in green, Molecule-1 in blue, Molecule-2 in navy Molecule-3 in purple; (c) zigzag arrangement along the b-axis. Color scheme: C, gray; H, white; O, red.
In Figure 4b, the packing is color coded by the three crystallographically independent molecules defined in Figure 3, revealing that the orientation and alignment of each molecule are nearly identical crystallographically. Using PLATON analysis (ver. 90522) [47], we examined whether any π···π stacking interactions exist between the aromatic phenylene rings. However, the shortest ring interaction, observed between the centroids of rings-H and J, was 4.645(2) Å, suggesting no significant interaction. The primary intermolecular interactions within the crystals are CH···π interactions: C11-H11 of Molecule-1 interacts with ring-E of Molecule-2 [symmetry code: −x + 1, −y, −z + 1]; C23-H23 of Molecule-1 interacts with ring-C of Molecule-1 [symmetry code: −x + 1, y + 1/2, −z + 1/2]; C36-H36 of Molecule-2 interacts with ring-I of Molecule-3 [symmetry code: x, y, z]; C38-H38 of Molecule-2 interacts with ring-F of Molecule-2 [symmetry code: −x + 2, y + 1/2, −z + 3/2]; C44-H44 of Molecule-2 interacts with ring-A of Molecule-1 [symmetry code: −x + 1, −y + 1, −z + 1]; and C57-H57 of Molecule-3 interacts with ring-G of Molecule-2 [symmetry code: x, y − 1, z], and the corresponding H···Cg(ring) distances for these interactions are 2.49, 2.59, 2.56, 2.85, 2.47, and 2.58 Å, respectively. In particular, the CH···π interactions between rings-A and E are thought to contribute to the stability of the packing (Figure 4c). Potential hydrogen bonds were also observed between C15-H15···O5, C27-H27···O4, and C56-H56···O6, with C···O distances of 3.353(5), 3.378(5), and 3.363 Å, and C-H···A angles of 149°, 143°, and 166°, respectively. The increase in the number of interactions is considered to be due to the reduced symmetry of the molecule, which leads to more interactions being counted, rather than an actual increase in the number of interactions.
3.4. Thermal and Comparison Studies of Crystal Structures of 2-I
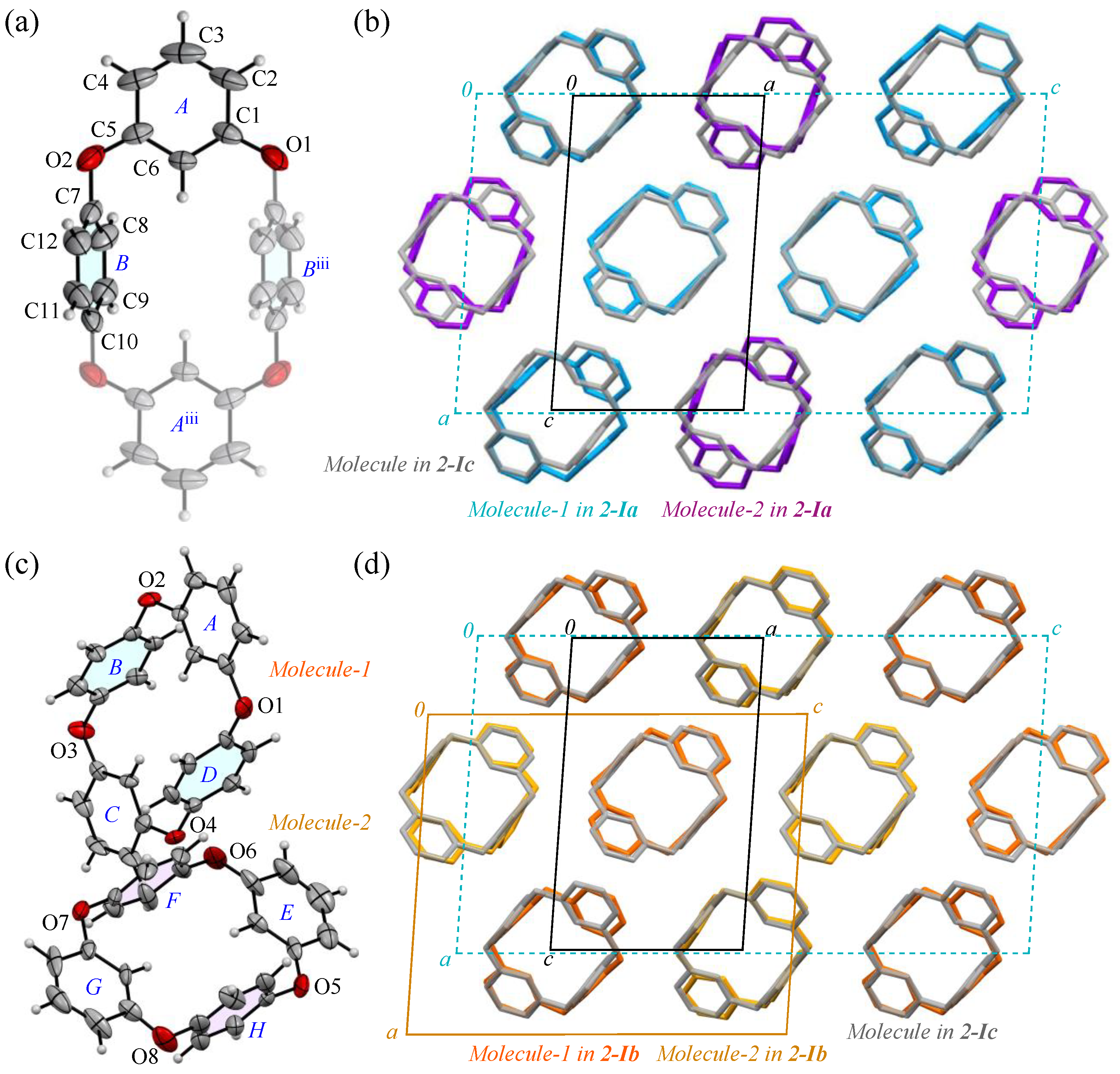
The prismatic crystal 2-I, contrary to its seemingly uniform formation, exhibited temperature-dependent reflection pattern disturbances during the crystal structure analysis. To verify the stability of the 2:1 molecular arrangement (Molecules-1 and 2) within the crystal, as shown in Figure 2a, we examined the crystal’s temperature-dependent behavior. Typically, organic crystals composed of low-weight atoms are cooled to suppress the electron density fluctuations. Accordingly, we cooled the crystal to a low temperature of 100 K, at which point we observed the unit cell of the 2-Ia crystals, with a volume of 2781(5) Å3. Near room temperature, the unit cell volume decreased significantly to 940.2(1) Å3, approximately one-third of its value at 100 K. This suggests that, as the temperature increases and molecular vibrations occur, Molecule-1 and Molecule-2 become indistinguishable due to averaging (Table 2). The reduction in the unit cell volume is accompanied by a decrease in density, making it comparable to that of 2-II crystals. When we actually retrieved the prismatic crystal and measured it at room temperature, the number of reflections decreased and a highly symmetrical crystal containing 0.5 molecules of compound 2 per unit cell was obtained as 2-Ic. The ORTEP view, with the numbering schemes, of 2-Ic crystals at 302 K is shown in Figure 5a. The average bond distance of C-O and angle of C-O-C for two oxygen atoms (O1 and O2) are 1.388 Å and 117.2°, respectively, and the two p-phenylene faces are oriented face-to-face. The intramolecular distance between the centroids of two p-phenylene rings is 4.8546(15) Å for Cg<ring-B>···Cg<ring-Biii>. The packing structure (Figure 5b) indicates that the molecules were aligned in the ac plane and the crystal lattice became one-third smaller by aligning in the same direction. The RMS deviation in the three structural overlays of Molecule-1 and Molecule-2 in the 2-Ia crystal and the molecule in the 2-Ic crystal is small, 0.054 Å.
Table 2.
Unit cell parameters and volumes for 2-I crystals at 100–300 K.
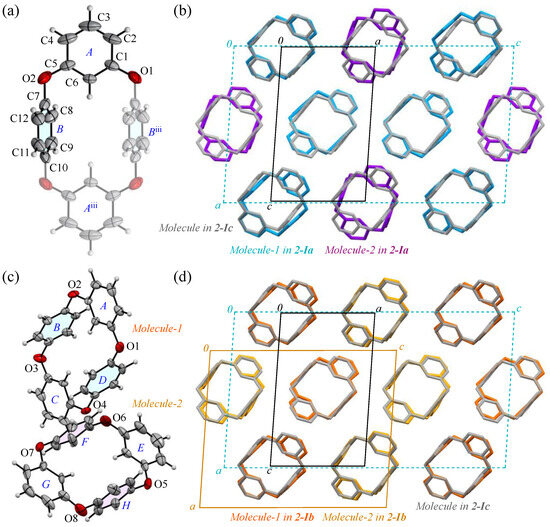
Figure 5.
(a) The molecular structure of 2-Ic crystals at 302 K, showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 30% probability level. Symmetry code iii: −x + 1, −y + 1, −z + 1. (b) Packing structures of 2-Ia crystals (blue and purple) and 2-Ic crystals (gray). (c) The molecular structure of 2-Ib crystals at 200 K, showing a partial atom-labeling scheme. Displacement ellipsoids are drawn at the 30% probability level. (d) Packing structures of 2-Ib crystals (orange and yellow) and 2-Ic crystals (gray). Color scheme: C, gray; H, white; O, red.
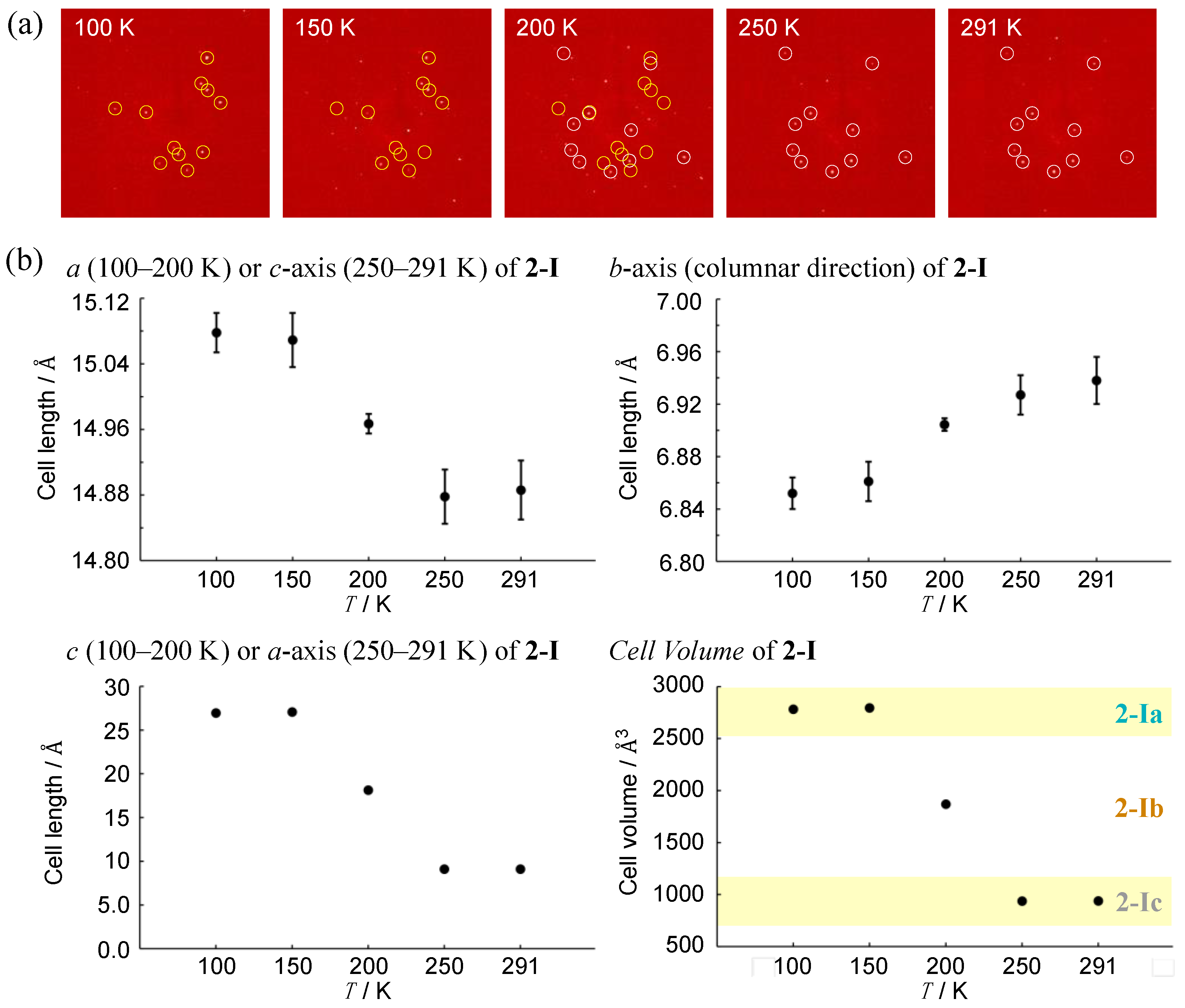
To investigate a possible phase transition between 2-Ia and 2-Ic crystals, an approximate determination of their respective transition temperatures was considered necessary. Several experiments were conducted, leading to the discovery of a metastable intermediate state, 2-Ib. In this crystal system, the unit cell volume is precisely between the volumes of the two other phases (with Z also being an intermediate value) and two crystallographically distinct molecules were identified in the asymmetric unit. The structure (Figure 5c) and packing diagram with the unit cell (Figure 5d), indeed, suggest an intermediate phase between the two crystal systems. However, due to the structural similarity, the possibility of a mixed phase cannot be ruled out. To investigate further, the diffraction patterns and lattice parameters were measured at intervals of 50 K and the results were compared (Figure 6). At the intermediate temperature of 200 K, a unique lattice in the 2-Ib crystal was observed that could not be explained by a simple overlap of the diffraction patterns from the phases at 100 K and 302 K (291 K when measured on the same crystal). Plotting the values with three times the standard uncertainty as the estimated error, revealed a clear distinction in the lattice parameters. Attempts to analyze the diffraction pattern at this temperature using the lattice parameters of either 2-Ia or 2-Ic crystals did not yield sufficient results, with the 2-Ib crystal providing the best fit for refinement and no detectable errors in the lattice settings. Consequently, we concluded that the 2-Ib crystal represents an original crystal structure, indicating a three-stage change in packing structure. The crystal density of 2-Ib is 1.309 Mg m−3, similar to 2-Ic crystals at 302 K and the polymorph 2-II. Additionally, attempts to refine the 100 K data with 2-Ic parameters or the 300 K data with 2-Ia parameters confirmed that the refinement did not converge. Temperature measurements were also conducted on the polymorph 2-II, but no significant changes were observed. Due to the relatively loose packing suggested by the density of 2-II crystals, even at 100 K, it is likely that temperature changes have minimal impact on 2-II, whereas the packing structure of 2-Ia crystals at 100 K represents the densest structure in this system.
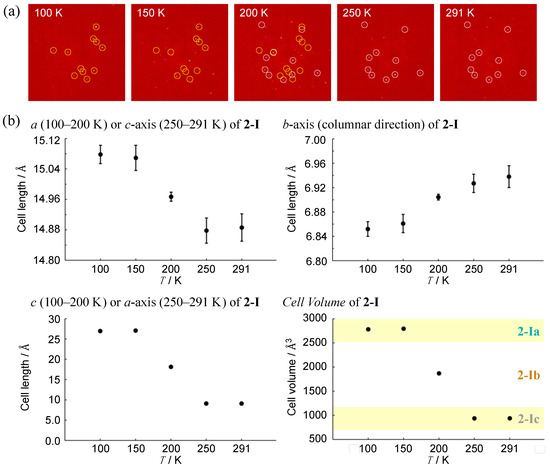
Figure 6.
Temperature dependence in terms of (a) selected diffraction patterns for the same measurement plane (yellow and white circles indicate 2-Ia and 2-Ic, respectively) and (b) lattice constants of 2-I crystals. Data were obtained from the same single crystal by gradually increasing the temperature from 100 K. Error bars represent three times the standard uncertainty of the structural analysis results.
Compound 1, composed of four m-phenylene groups, exhibited molecular symmetry in terms of C2 or S4 in the 1,3-alternate structure [44,45], which determined the packing structure and, consequently, the external appearance of the crystal. However, in the molecule composed of m- and p-phenylene groups, the rigidity of the p-phenylene groups takes precedence, resulting in minimal variation in the molecular structure. Repeated crystallization experiments suggest that the ellipsoidal molecule 2, when aligned in the same direction, produces a stable structure in terms of 2-I. This anisotropy in the molecular arrangement promotes columnar crystal growth, while the block-shaped 2-II crystals are thought to precipitate only when a kinetically favored herringbone-type crystal nucleus forms. Such a discrete aromatic cyclic molecular polymorphism is an intriguing example, as it affects molecular recognition in the solid state.
3.5. Hirshfeld Surface Analysis of the Structures
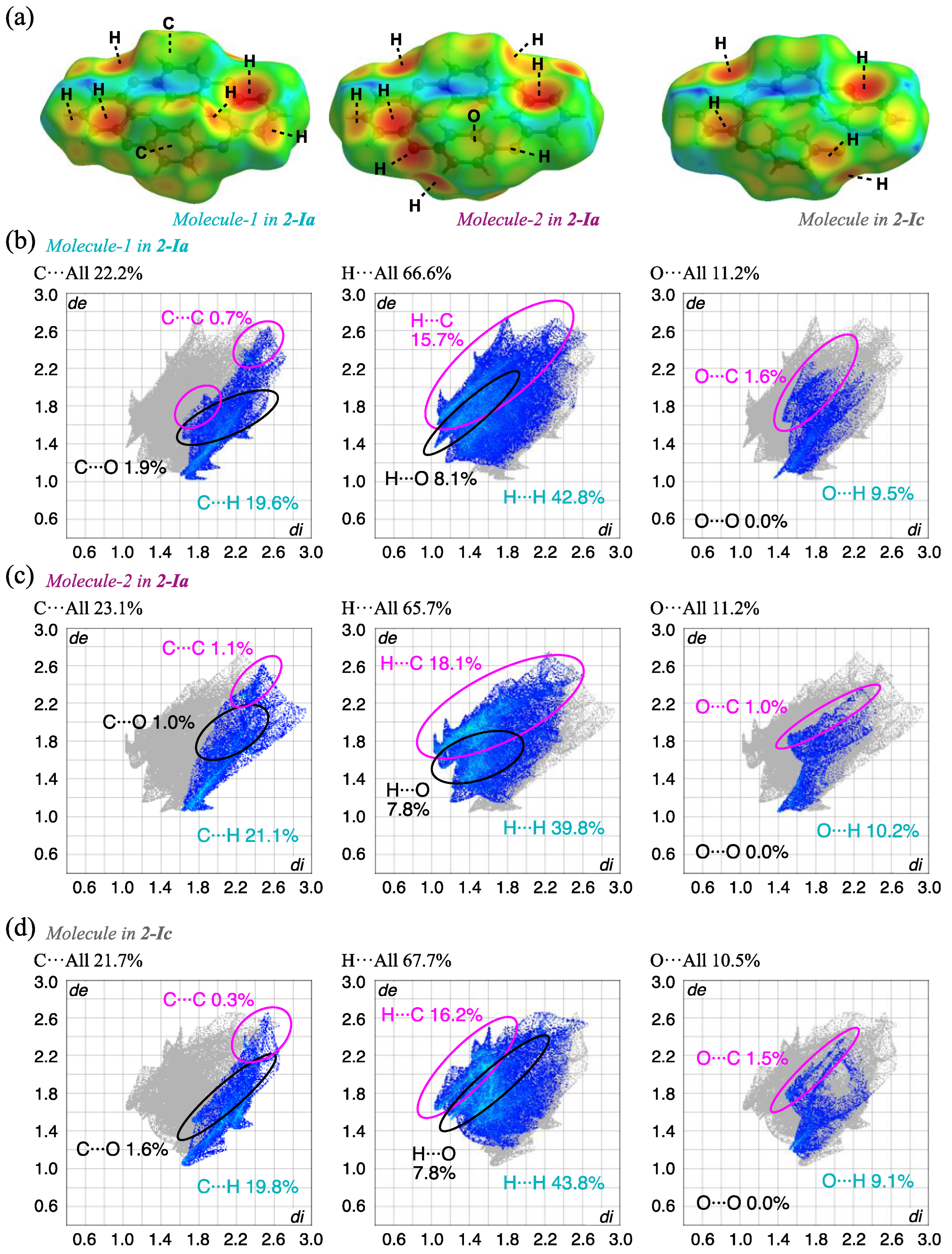
To understand the detailed intermolecular interactions, HS analysis [48,49] of each complex was carried out using Crystal Explorer 17.5 [50]. The results for the 2-Ia and 2-Ic crystals mapped with de (the distance from a point on the surface to the nearest nucleus outside the surface) and the corresponding fingerprint plots are shown in Figure 7 and Table 3. The red spots indicate short intermolecular distances of the molecules, indicating the existence of notable intermolecular interactions; in this case, electrostatic CH···π is mainly observed between the compounds and the corresponding fingerprint plots show the contribution of H(inside)···C(outside), with 15.7% and 18.1% for Molecule-1 and Molecule-2, respectively, in 2-Ia crystals. The CH···π interaction in 2-Ic crystals at a higher temperature (302 K) is 16.2%, which is slightly lower than the average value of 16.9% for 2-Ia crystals. The increase in temperature weakens this interaction, as evidenced by the slight loss of sharpness in the fingerprint plot, indicating that these specific interactions weaken as the temperature increases. The contribution of the pseudo-hydrogen bond, O···H, also decreases from 9.5% and 10.2% in 2-Ia crystals to 9.1% in 2-Ic crystals, with a simultaneous loss of sharpness. The contribution of C···C, which suggests π···π stacking that is electrostatic and considered to contribute to the overall packing structure, decreases from 0.7% and 1.1% in 2-Ia crystals to 0.3% in 2-Ic crystals, with an increase in the intermolecular distance, consistent with the above discussion. As these interactions diminish, the relative contribution of surface hydrogen increases, although it is unlikely to influence the crystal surface stabilization: H···H interactions are 42.8% and 39.8% for 2-Ia crystals and 43.8% for 2-Ic crystals. In other words, the thermal dependence of the prismatic 2-I crystal once again shows that the increase in temperature loosens the overall packing structure, rather than inducing molecular structure changes, and contributes to enhanced symmetry.
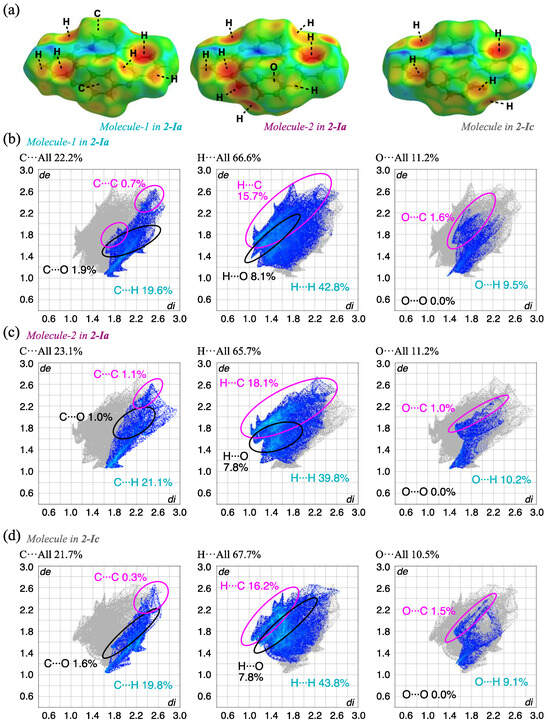
Figure 7.
HS analysis of (a) the three molecules in 2-Ia and 2-Ic crystals mapped with de and the corresponding fingerprint plots, showing the contributions of intermolecular interactions with surrounding atoms; (b) Molecule-1 in 2-Ia; (c) Molecule-2 in 2-Ia; and (d) Molecule in 2-Ic.
Table 3.
The percentage contributions of intermolecular interactions for each molecule in crystals 2-Ia, 2-Ib, 2-Ic, 2-II, and 3.
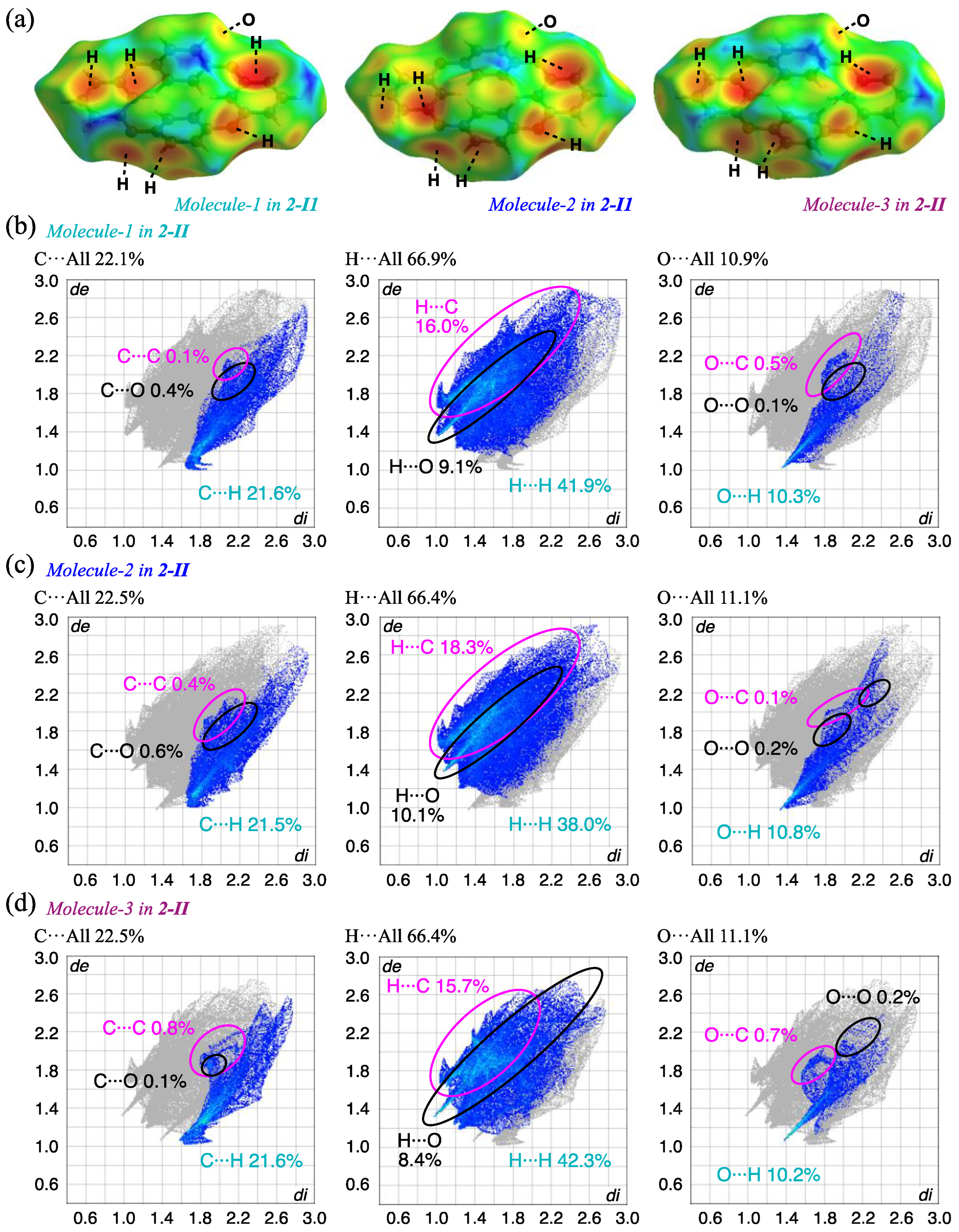
The HS analysis for 2-II crystal shows similar results to those of the 2-Ia crystal at the same temperature (Figure 8). However, the fingerprint plots of the 2-II crystal indicate a greater spatial distribution of the interactions, with only a minor quantitative impact. The increased contribution of C···C interactions, clustered around 2 Å in both de and di, was observed. The proportion of C···H/H···C, suggesting CH···π interactions, also increased: at the same temperature, the contributions of C···H/H···C interactions in 2-Ia crystals were 35.3% (Molecule-1) and 39.2% (Molecule-2), with an average of 37.3%, while in 2-II crystals, the contributions were 37.5% (Molecule-1), 39.8% (Molecule-2), and 37.3% (Molecule-3), with an average of 38.2%. Additionally, a decrease in the contribution of O···C interactions and an increase in pseudo-hydrogen bonding of O···H were observed: the contributions of O···H/H···O interactions in 2-Ia crystals were 17.6% for Molecule-1 and 18.0% for Molecule-2, averaging 17.8%, whereas in 2-II crystals, the values were 19.4% for Molecule-1, 20.9% for Molecule-2, and 18.6% for Molecule-3, with an average of 19.6%. This suggests that while the molecules are not in tight contact, the specific interactions are enhanced.
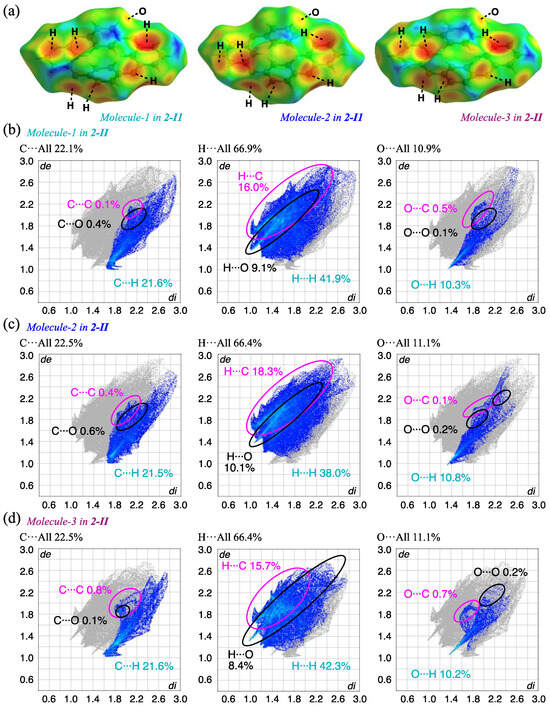
Figure 8.
HS analysis of (a) the three molecules in 2-II crystals mapped with de and the corresponding fingerprint plots, showing the contributions of intermolecular interactions with surrounding atoms; (b) Molecule-1; (c) Molecule-2; and (d) Molecule-3 in 2-II.
3.6. Crystal Structure and Intermolecular Interactions of 3 Crystals
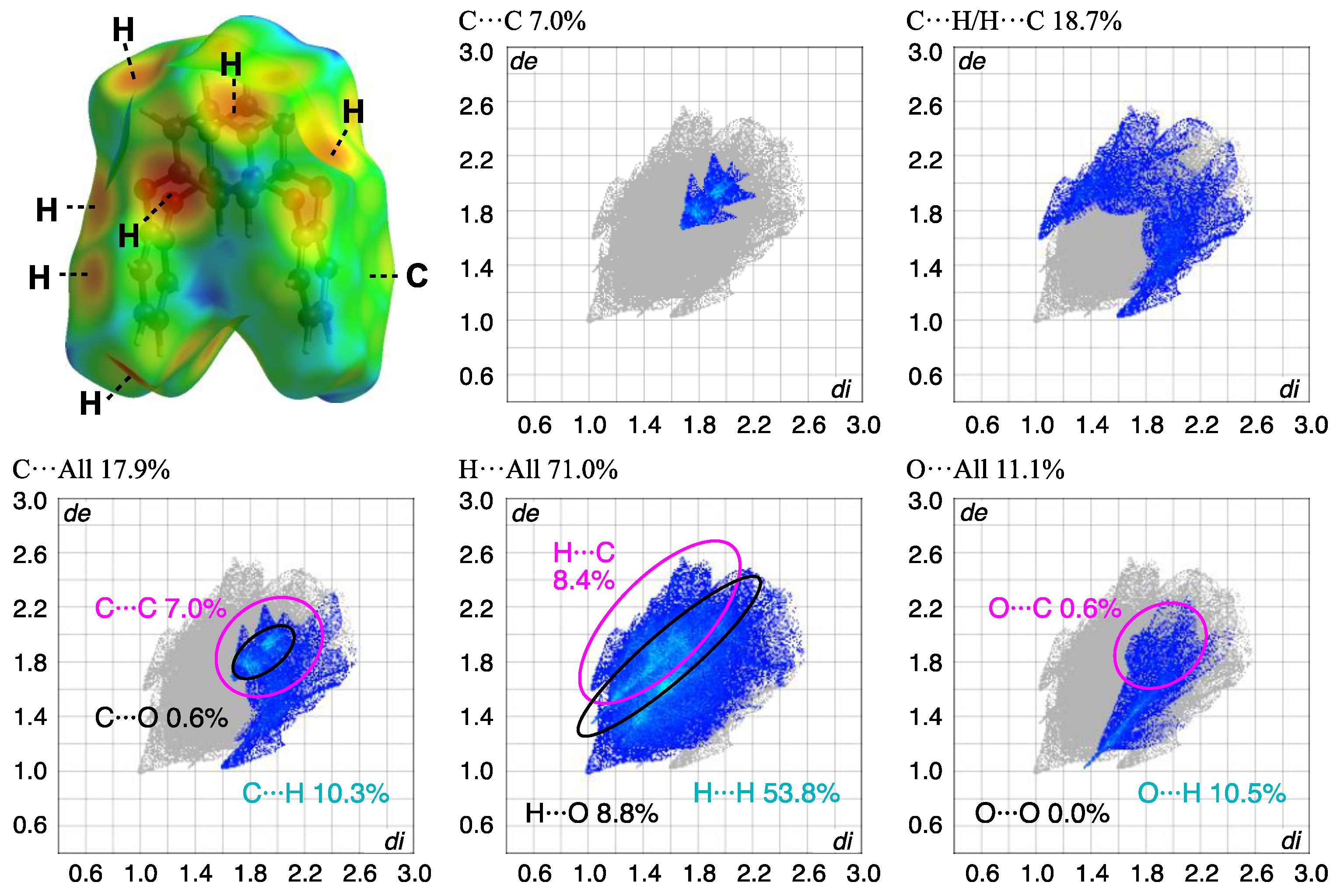
The block crystal in 3 was only one obtained, to the best of our knowledge. A whole compound was found in the asymmetric unit, and the side and top views of the molecular structures and the packing structure of 3 crystals are as shown in Figure 9. Compound 3 adopts a U-shaped vent structure (Figure 9a), and the average bond distance of C-O and the angle of C-O-C for six oxygen atoms (O1–O6) are 1.393 Å and 116.7°, respectively, similar to those observed in compound 2. The two o-phenylene faces are oriented face-to-face; the dihedral angle and the intramolecular distance between rings-A and C are 4.69(9)° and 4.9027(17) Å, respectively, and the two m-phenylene faces are also oriented face-to-face; the dihedral angle and the intramolecular distance between rings-B and D are 30.14(10)° and 3.6946(15) Å, respectively (Figure 9b). The primary intermolecular interactions within the crystal are π···π interactions: the intermolecular distances between the centroids of two phenylene rings are 3.8389(15) Å for Cg<ring-A>···Cg<ring-D>, 3.6946(15) Å for Cg<ring-B>···Cg<ring-D>, and 3.5457(15) Å for Cg<ring-C>···Cg<ring-C>. The shortest intermolecular stacking between ring-C and the adjacent ring-C is shown in Figure 9c, which is further linked through the stacking between rings-A and D. The CH···π interaction is also observed between C14-H14 and ring-B of the adjacent molecule, complementarily, and the corresponding H···Cg<ring-B> distance is 2.75 Å, aligned with the c-axis to form a linear arrangement. A potential hydrogen bond was also observed between C2-H2···O2, with a C···O distance of 3.446(3) Å and a C-H···A angle of 163°. Notable π-π stacking was visualized through HS analysis (Figure 10 and Table 3). In crystal 2, the contribution of C···C interactions was less than 1%, while in compound 3, it reached 7%, indicating a concentrated distribution typical of π-π stacking. This suggests the presence of more than two types of stacking, based on the electron density distribution. Additionally, a comparison with this plot confirms the minimal influence of π-π stacking in crystal 2. The overall plot of compound 3 shows a contraction towards shorter interaction distances compared to compound 2, indicating stronger interactions. This is well correlated with the higher crystal density of compound 3, showing a value greater than that of 2-Ia crystals: 1.350 Mg m−3 (3) > 1.329 Mg m−3 (2-Ia). The contribution of C-H/H-C interactions was lower in compound 3, which correlates well with the decrease in CH···π interactions. It can be inferred that this crystal does not form polymorphs, because the most stable structure is a supramolecular assembly, utilizing its bent molecular structure and surface area.
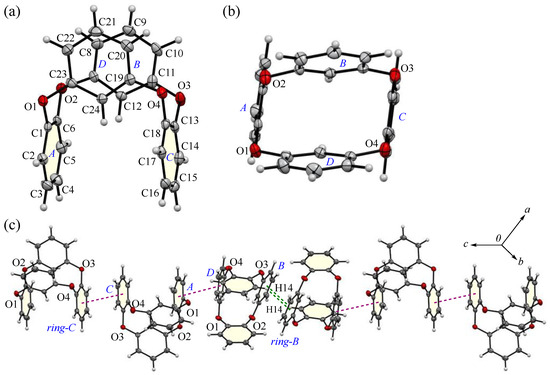
Figure 9.
(a) Side and (b) top views of molecular structure of compound 3 at 100 K, showing the atom-labeling scheme, and (c) a part of the packing structure of compound 3. Displacement ellipsoids are drawn at the 50% probability level. Color scheme: C, gray; H, white; O, red.
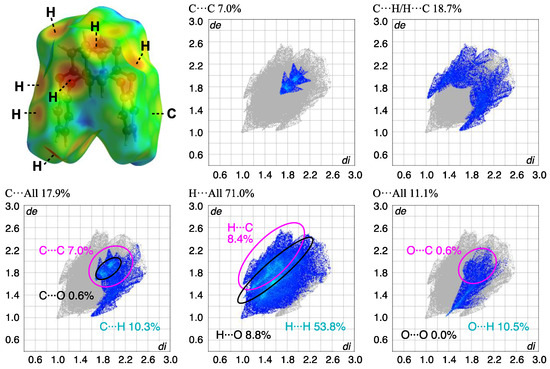
Figure 10.
HS analysis of the molecule mapped with de and the corresponding fingerprint plots showing the contributions of intermolecular interactions with surrounding atoms in compound 3.
4. Conclusions
In conclusion, we conducted an in-depth investigation of the crystallographic and thermal properties of tetraoxacalix[4]arene derivatives 2 and 3, highlighting distinct polymorphic behaviors and temperature-dependent structural changes. Compound 2 exhibited two polymorphs: a prismatic form (2-I) and a block form (2-II). Detailed phase analysis revealed that 2-I polymorphs underwent a three-stage phase transition, spanning low-temperature (2-Ia), intermediate (2-Ib), and high-temperature (2-Ic) forms, demonstrating unique thermal adaptability in terms of the packing structure. The Hirshfeld surface analysis identified significant CH···π interactions and pseudo-hydrogen bonding in 2-Ia crystals, which decreased at higher temperatures, resulting in a relaxed packing structure and increasing the overall symmetry, without altering the molecular framework. These findings suggest that crystallization conditions and thermal changes are pivotal in determining the polymorphism and stability of these structures, offering a promising strategy for designing adaptive host materials. The stability and structural flexibility observed here provide a basis for applications that require selective molecular recognition and environmental responsiveness. Future research will focus on utilizing the inner and outer π planes of these aromatic cyclic molecular crystals, aiming to develop non-porous adaptive crystals (NACs) [12] and enhanced molecular recognition capabilities, particularly in regard to external field applications.
Author Contributions
Conceptualization, writing—original draft preparation, formal analysis, data curation, investigation, Y.I. and T.K.; validation, writing—original draft preparation, writing—review and editing, funding acquisition, methodology, supervision, A.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Grant-in-Aid for Scientific Research B (no. 23K21122) of JSPS KAKENHI.
Data Availability Statement
The crystallographic data have been deposited at the CCDC; additional data are available in Appendix A.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Appendix A
The crystallographic data have been deposited with the Cambridge Crystallographic Data Centre and the deposition CCDC numbers are 2396584-2396588. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/ structures/ (accessed on 26 November 2024).
References
- Gutsche, C.D. Calixarenes. Acc. Chem. Res. 1983, 16, 161–170. [Google Scholar] [CrossRef]
- Ikeda, A.; Shinakai, S. Novel cavity design using calix[n]arene skeletons: Toward molecular recognition and metal binding. Chem. Rev. 1997, 97, 1713–1734. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Isabelle, L. Design principles of fluorescent molecular sensors for cation recognition. Coord. Chem. Rev. 2000, 205, 3–40. [Google Scholar] [CrossRef]
- Dalgarno, S.J.; Thallapally, P.K.; Barbour, L.J.; Atwood, J.L. Engineering void space in organic van der Waals crystals: Calixarenes lead the way. Chem. Soc. Rev. 2007, 36, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Yamagishi, T.; Nakamoto, Y. Pillar-shaped macrocyclic hosts pillar[n]arenes: New key players for supramolecular chemistry. Chem. Rev. 2016, 116, 7937–8002. [Google Scholar] [CrossRef] [PubMed]
- Isabelle, L.; Bernard, V. Calixarene-based fluorescent molecular sensors for toxic metals. Eur. J. Inorg. Chem. 2009, 24, 3525–3535. [Google Scholar] [CrossRef]
- Zadmard, R.; Akbarzadeha, A.; Jalalia, M.R. Highly functionalized calix[4]arenes via multicomponent reactions: Synthesis and recognition properties. RSC Adv. 2019, 9, 19596–19605. [Google Scholar] [CrossRef]
- Han, X.-N.; Han, Y.; Chen, C.-F. Recent advances in the synthesis and applications of macrocyclic arenes. Chem. Soc. Rev. 2023, 52, 3265–3298. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-P.; Wang, X.; Yao, H.; Jiang, W. Naphthotubes: Macrocyclic hosts with a biomimetic cavity feature. Acc. Chem. Res. 2020, 53, 198–208. [Google Scholar] [CrossRef]
- Tian, H.-W.; Liu, Y.-C.; Guo, D.-S. Assembling features of calixarene-based amphiphiles and supra-amphiphiles. Mater. Chem. Front. 2020, 4, 46–98. [Google Scholar] [CrossRef]
- Lumetta, G.J.; Rogers, R.D.; Gopalan, A.S. Calixarenes for Separations; ACS: Washington, WA, USA, 2000; pp. 107–312. [Google Scholar]
- Jie, K.; Zhou, Y.; Li, E.; Huang, F. Nonporous adaptive crystals of pillararenes. Acc. Chem. Res. 2018, 51, 2064–2072. [Google Scholar] [CrossRef]
- Weissbuch, I.; Addadi, L.; Lahav, M.; Leiserowit, L. Molecular recognition at crystal interfaces. Science 1991, 253, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Gutsche, C.D.; Kye, C.-N. Calixarenes. 22. Synthesis, properties, and metal complexation of aminocalixarenes. J. Am. Chem. Soc. 1988, 110, 6153–6162. [Google Scholar] [CrossRef] [PubMed]
- Ohto, K.; Yano, M.; Inoue, K.; Nagasaki, T.; Goto, M.; Nakashio, F.; Shinkai, S. Effect of coexisting alkaline metal ions on the extraction selectivity of lanthanido ions with calixarene carboxylate derivatives. Polyhedron 1997, 16, 1655–1661. [Google Scholar] [CrossRef]
- Danil de Namor, A.F.; Chahine, S.; Kowalska, D.; Costellano, E.E.; Piro, O.E. Selective interaction of lower rim calix[4]arene derivatives and bivalent cations in solution. Crystallographic evidence of the versatile behavior of acetonitrile in lead(II) and cadmium(II) complexes. J. Am. Chem. Soc. 2002, 124, 12824–12836. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.; Tanaka, M.; Ando, H.; Iwamoto, H.; Kimura, K. Chromened t-butylcalix[4]arenes: Cooperation effect of chromene and calixarene moieties on photochromism and metal-ion binding ability. Tetrahedron 2003, 59, 4135–4142. [Google Scholar] [CrossRef]
- Ogoshi, T.; Kanai, S.; Fujinami, S.; Yamagishi, T.; Nakamoto, Y. para-Bridged symmetrical pillar[5]arenes: Their Lewis acid catalyzed synthesis and host–guest property. J. Am. Chem. Soc. 2008, 130, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Hongo, T.; Matsuo, N.; Konishi, M.; Kawaguchi, T.; Ihara, H. Hemisphere-shaped calixarenes and their analogs: Synthesis, structure, and chiral recognition ability. Tetrahedron 2011, 67, 4716–4722. [Google Scholar] [CrossRef]
- Aragay, G.; Hernández, D.; Verdejo, B.; Escudero-Adán, E.C.; Martínez, M.; Ballester, P. Quantification of CH-π interactions using calix[4]pyrrole receptors as model systems. Molecules 2015, 20, 16672–16686. [Google Scholar] [CrossRef]
- Romero, J.; Barberá, J.; Blesa, M.-J.; Concellón, A.; Romero, P.; Serrano, J.L.; Marcos, M. Liquid crystal organization of calix[4]arene-appended schiff bases and recognition towards Zn2+. ChemistrySelect 2017, 2, 101–109. [Google Scholar] [CrossRef]
- He, L.; Wang, S.-C.; Lin, L.-T.; Cai, J.-Y.; Li, L.; Tu, T.-H.; Chan, Y.-T. Multicomponent metallo-supramolecular nanocapsules assembled from calix[4]resorcinarene-based terpyridine ligands. J. Am. Chem. Soc. 2020, 142, 7134–7144. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sasaki, T.; Tonosaki, A.; Hattori, T. Mechanistic consideration for the selective inclusion of disubstituted benzene isomers with p-tert-butylcalix[4]arene crystals. Cryst. Growth Des. 2021, 21, 5006–5016. [Google Scholar] [CrossRef]
- Li, M.; Shao, L.; Liu, Z.; Liu, R.; Stoikov, I.I.; Khashab, N.M.; Hua, B.; Huang, F. Cis−trans and length-selective molecular discrimination of halogenated organic compounds by a crystalline hybrid macrocyclic arene. ACS Appl. Mater. Interfaces 2024, 16, 6614–6622. [Google Scholar] [CrossRef] [PubMed]
- Ikumura, Y.; Habuka, Y.; Sakai, S.; Shinohara, T.; Yuge, H.; Rzeznicka, I.I.; Hori, A. Enhanced and heteromolecular guest encapsulation in nonporous crystals of a perfluorinated triketonato dinuclear copper complex. Chem. Eur. J. 2020, 26, 5051–5060. [Google Scholar] [CrossRef]
- Habuka, Y.; Usui, H.; Okawa, M.; Shinohara, T.; Yuge, H.; Mao, Y.; Gong, J.; Richards, G.J.; Hori, A. Selective recognition and reversible encapsulation of tetrameric alcohol clusters via hydrogen bonds using a perfluorinated dinuclear nickel(II) complex. CrystEngComm 2024, 26, 3014–3020. [Google Scholar] [CrossRef]
- Smith, G.W. Crystal Structure of a nitrogen isostere of pentacyclo-octacosadodecaene. Nature 1963, 198, 879. [Google Scholar] [CrossRef]
- Maesa, W.; Dehaen, W. Oxacalix[n](het)arenes. Chem. Soc. Rev. 2008, 37, 2393–2402. [Google Scholar] [CrossRef]
- Kumagai, H.; Hasegawa, M.; Miyanari, S.; Sugawa, Y.; Sato, Y.; Hori, T.; Ueda, S.; Kamiyama, H.; Miyano, S. Facile synthesis of p-tert-butylthiacalix[4]arene by the reaction of p-tert-butylphenol with elemental sulfur in the presence of a base. Tetrahedron Lett. 1997, 38, 3971–3972. [Google Scholar] [CrossRef]
- Katz, J.L.; Feldman, M.B.; Conry, R.R. Synthesis of functionalized oxacalix[4]arenes. Org. Lett. 2005, 7, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-X. Nitrogen and oxygen bridged calixaromatics: Synthesis, structure, functionalization, and molecular recognition. Acc. Chem. Res. 2012, 45, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Lee, Y.O.; Bhalla, V.; Kumar, M.; Kim, J.S. Recent developments of thiacalixarene based molecular motifs. Chem. Soc. Rev. 2014, 43, 4824–4870. [Google Scholar] [CrossRef] [PubMed]
- Canard, G.; Edzang, J.A.; Chen, Z.; Chessé, M.; Elhabiri, M.; Giorgi, M.; Siri, O. 1,3-Alternate tetraamido-azacalix[4]arenes as selective anion receptors. Chem. Eur. J. 2016, 22, 5756–5766. [Google Scholar] [CrossRef]
- Takemura, H. Synthesis of azacalixarenes and development of their properties. Molecules 2021, 26, 4885. [Google Scholar] [CrossRef] [PubMed]
- Kleyn, A.; Jacobs, T.; Barbour, L.J. Solid-state structural studies of oxacalix[2]arene[2]naphthalene as a molecular tweezer. CrystEngComm 2011, 13, 3175–3180. [Google Scholar] [CrossRef]
- Petersen, R.J.; Rozeboom, B.J.; Oburn, S.M.; Blythe, N.J.; Rathje, T.L.; Luna, J.A.; Kibby, S.K.; O'Brien, E.A.; Rohr, K.G.; Carpenter, J.R.; et al. Cambiarenes: Single-step synthesis and selective zwitterion binding of a clip-shaped macrocycle with a redox-active Core. Chem. Eur. J. 2019, 26, 1928–1930. [Google Scholar] [CrossRef] [PubMed]
- Wan, K.; Gao, S.-C.; Fang, X.; Xu, M.-Y.; Yang, Y.; Xue, M. Oxacalix[4]arene-bridged pillar[5]arene dimers: Syntheses, planar chirality and construction of chiral rotaxanes. Chem. Commun. 2020, 56, 10155–10158. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Zhang, S.; Wang, L.; Tao, X. Recent advances in polymorph discovery methods of organic crystals. Cryst. Growth Des. 2023, 23, 637–654. [Google Scholar] [CrossRef]
- Peterson, A.; Kaabel, S.; Kahn, I.; Pehk, T.; Aav, R.; Adamson, J. Unsubstituted Oxacalix[n]arenes (n=4 and 8): A conformational study in solution and solid state and interaction studies with aromatic guests. ChemistrySelect 2018, 3, 9091–9095. [Google Scholar] [CrossRef]
- Hori, A.; Betsugi, E.; Ikumura, Y.; Yoza, K. Polymorphic crystals of oxacalix[4]arene with 1,3-alternate conformations of S4 and C2 symmetry. Acta Cryst. 2019, C75, 265–270. [Google Scholar] [CrossRef]
- Zhou, Q.; Su, L.; Jiang, T.; Zhang, B.; Chen, R.; Jiang, H.; Ye, Y.; Zhu, M.; Han, D.; Shen, J.; et al. Copper/iron-catalyzed Ullmann coupling of diiodo- and dibromoarenes and diphenols for the synthesis of aryl ether macrocycles. Tetrahedron 2014, 70, 1125–1132. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The nature and applications of π–π interactions: A Perspective. Cryst. Growth Des. 2019, 19, 523–528. [Google Scholar] [CrossRef]
- Nishio, M.; Hirota, M. CH/π interaction: Implications in organic chemistry. Tetrahedron 1989, 45, 7201–7245. [Google Scholar] [CrossRef]
- Sun, J.; Tian, Z.-Y.; Liu, J.; Wan, C.; Dai, C.; Liu, Z.; Xing, Y.; Wu, Y.; Hou, Z.; Han, W.; et al. Intramolecular CH⋯π attraction mediated conformational polymorphism of constrained helical peptides. Chem. Sci. 2024, 15, 14264–14272. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17.5; University of Western Australia: Perth, Australia, 2017; Available online: http://hirshfeldsuface.net (accessed on 10 November 2024).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).