
Crystallographic Data Collection Using a Multilayer Monochromator on an Undulator Beamline at the Shanghai Synchrotron Radiation Facility


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Performance of the DMM
2.2. Sample Preparation
2.3. Data Collection
2.4. Data Processing
2.5. Structure Determination
3. Results
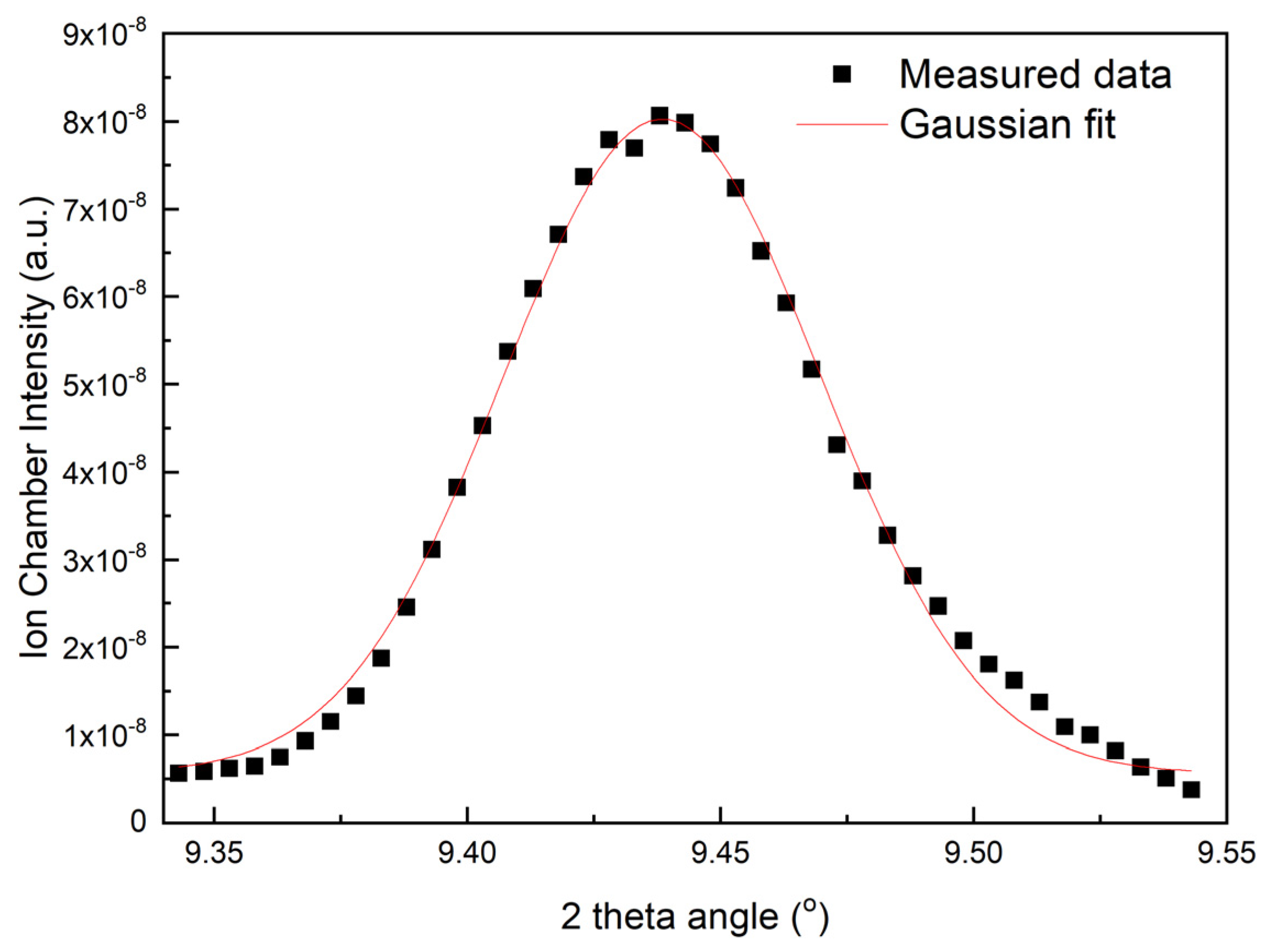
3.1. DMM and DCM Data Collection
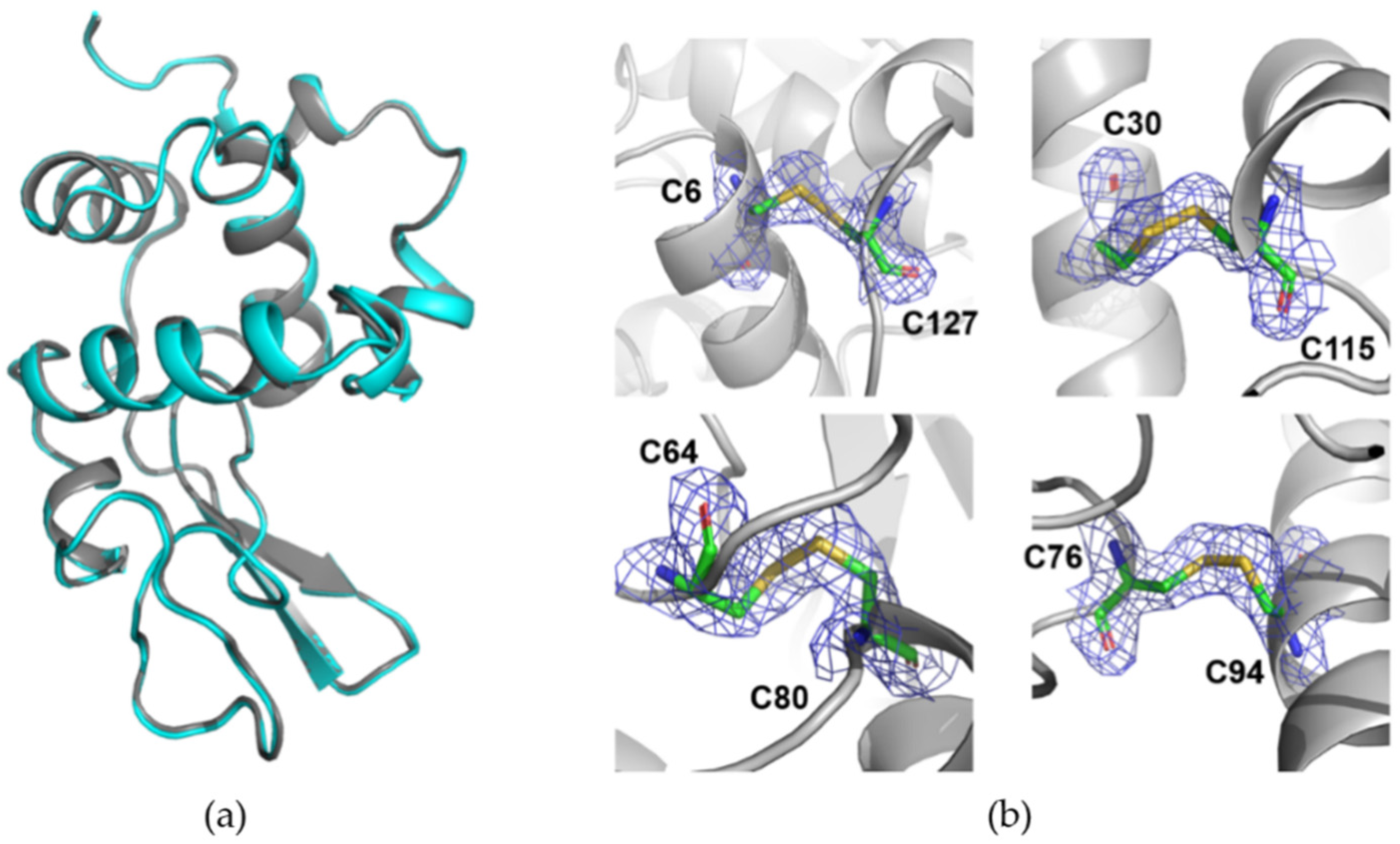
3.2. Room-Temperature Data Collection with DMM
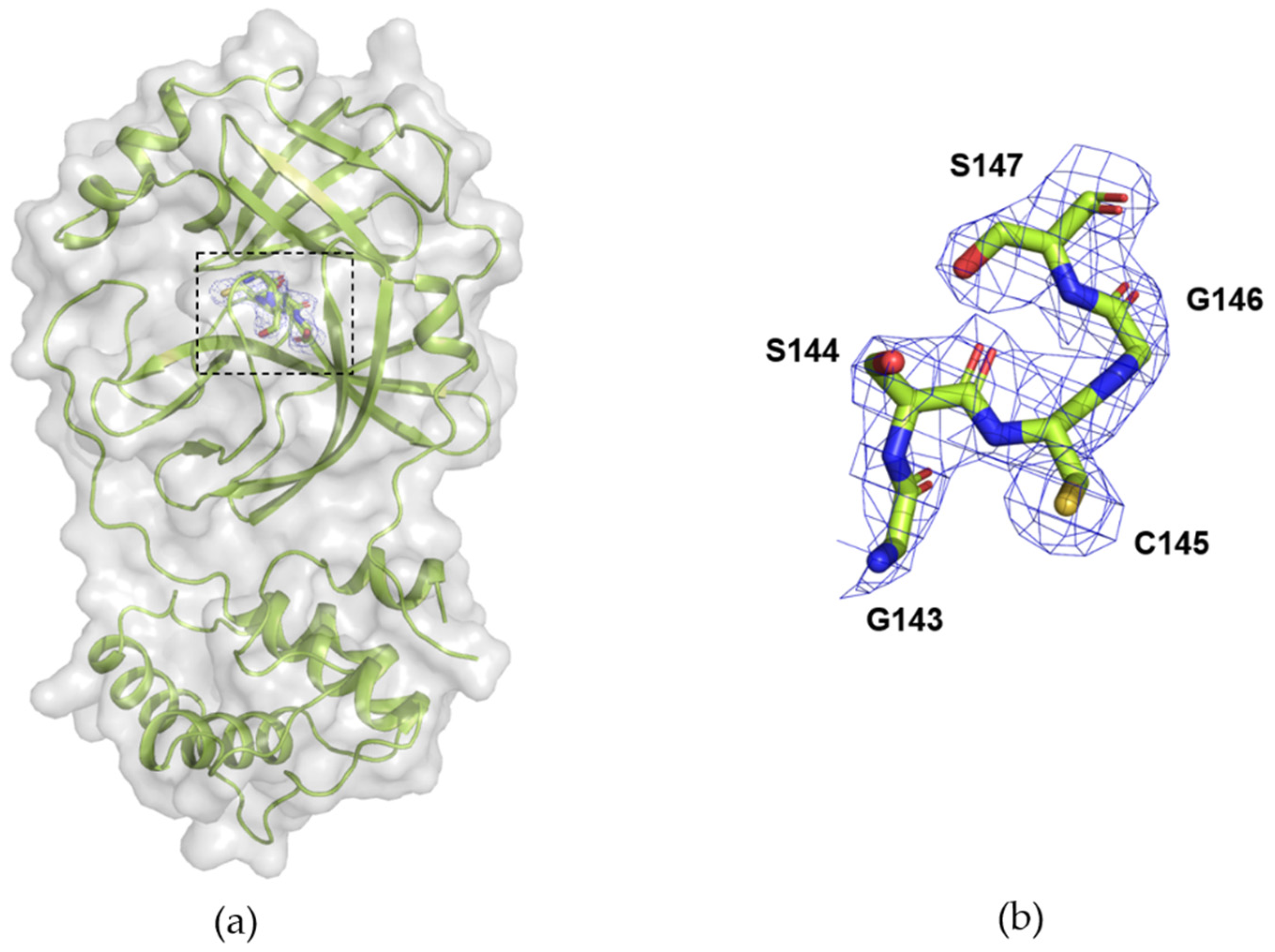
3.3. Data Collection of Mpro from SARS-CoV-2 with DMM
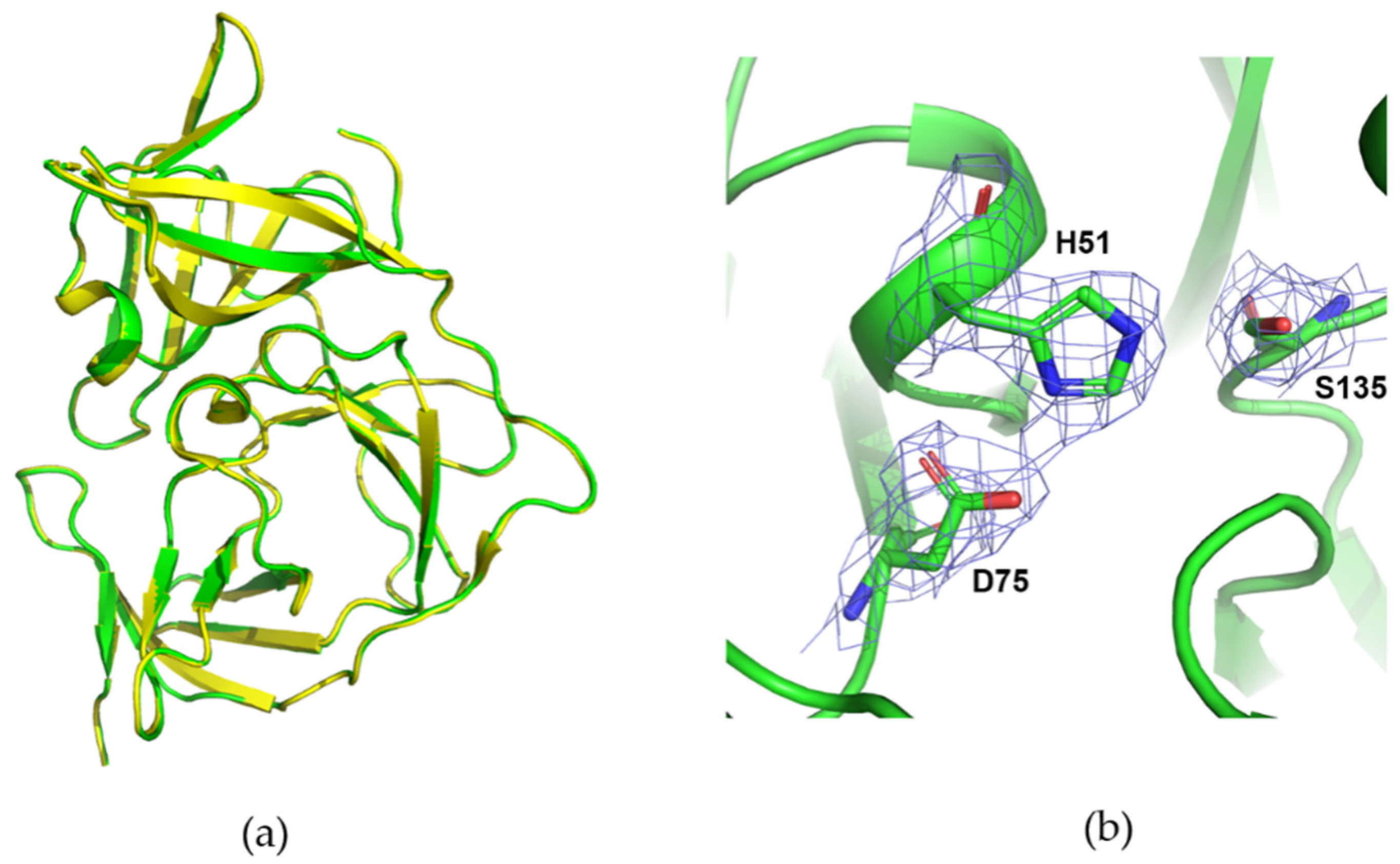
3.4. Data Collection of ZIKV NS2B/NS3 Protease with DMM
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Popov, A.M.; Dorovatovskii, P.V.; Mamichev, D.A.; Marchenkova, M.A.; Nikolaeva, A.Y. Development of a microfluidic chip for protein crystallization by the microbatch method. Crystallogr. Rep. 2019, 64, 282–286. [Google Scholar] [CrossRef]
- Liebschner, D.; Dauter, M.; Rosenbaum, G.; Dauter, Z. How good can our beamlines be? Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1430–1436. [Google Scholar] [CrossRef]
- Deacon, A.M.; Appleby, T.; Bilderback, D.H.; Ealick, S.E.; Fontes, E.; Thiel, D.J. Protein crystallography using a multilayer monochromator. J. Synchrotron Radiat. 1998, 5, 494–496. [Google Scholar] [CrossRef]
- Knapp, G.S.; Beno, M.A. A new method to do time-resolved, x-ray-diffraction studies—The rotating crystal laue method. Rev. Sci. Instrum. 1992, 63, 1032–1034. [Google Scholar] [CrossRef]
- Ren, Z.; Bourgeois, D.; Helliwell, J.R.; Moffat, K.; Srajer, V.; Stoddard, B.L. Laue crystallography: Coming of age. J. Synchrotron Radiat. 1999, 6, 891–917. [Google Scholar] [CrossRef]
- Englich, U.; Kazimirov, A.; Shen, Q.; Bilderback, D.H.; Gruner, S.M.; Hao, Q. Crystallographic data collection using a 0.22% bandwidth multilayer. J. Synchrotron Radiat. 2005, 12, 345–348. [Google Scholar] [CrossRef]
- Uesugi, K.; Hoshino, M.; Koyama, T.; Yamazaki, H.; Senba, Y.; Takeuchi, T.; Yumoto, H.; Ohashi, H.; Yamada, J.; Osaka, T.; et al. Demonstration of the x-ray imaging capabilities of the newly installed multilayer monochromator at spring-8 bl20b2. J. Phys. Conf. Ser. 2022, 2380, 012120. [Google Scholar] [CrossRef]
- Kim, Y.; Nam, K.H. Pink-beam serial synchrotron crystallography at pohang light source ii. Crystals 2022, 12, 1637. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Kovalevsky, A.; Coates, L. Room-temperature neutron and x-ray data collection of 3cl mprofrom SARS-CoV-2. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 483–487. [Google Scholar] [CrossRef]
- Schneps, C.M.; Ganguly, A.; Crane, B.R. Room-temperature serial synchrotron crystallography of drosophila cryptochrome. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 975–985. [Google Scholar] [CrossRef]
- Carrillo, M.; Mason, T.J.; Karpik, A.; Martiel, I.; Kepa, M.W.; McAuley, K.E.; Beale, J.H.; Padeste, C. Micro-structured polymer fixed targets for serial crystallography at synchrotrons and xfels. IUCrJ 2023, 10, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.R.; Mehrabi, P. Serial synchrotron crystallography for time-resolved structural biology. Curr. Opin. Struct. Biol. 2020, 65, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.S.; Yamada, Y.; Jakoncic, J.; McSweeney, S.; Sweet, R.M.; Skinner, J.; Foadi, J.; Fuchs, M.R.; Schneider, D.K.; Shi, W.; et al. Serial crystallography with multi-stage merging of thousands of images. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2022, 78, 281–288. [Google Scholar] [CrossRef]
- Hough, M.A.; Owen, R.L. Serial synchrotron and xfel crystallography for studies of metalloprotein catalysis. Curr. Opin. Struct. Biol. 2021, 71, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.L.; Panepucci, E.; Kaminski, J.W.; Aumonier, S.; Huang, C.-Y.; Eris, D.; Buntschu, D.; Meier, N.; Glettig, W.; McAuley, K.E.; et al. Sdu—Software for high-throughput automated data collection at the swiss light source. J. Synchrotron Radiat. 2023, 30, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Maveyraud, L.; Mourey, L. Protein x-ray crystallography and drug discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, J.W.; Vera, L.; Stegmann, D.P.; Vering, J.; Eris, D.; Smith, K.M.L.; Huang, C.Y.; Meier, N.; Steuber, J.; Wang, M.; et al. Fast fragment- and compound-screening pipeline at the swiss light source. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.S.; Wollenhaupt, J.; Correy, G.J.; Fraser, J.S.; Heine, A.; Klebe, G.; Krojer, T.; Thunnissen, M.; Pearce, N.M. Of problems and opportunities-how to treat and how to not treat crystallographic fragment screening data. Protein Sci. 2022, 31, e4391. [Google Scholar] [CrossRef]
- Xu, Q.; Kong, H.-T.; Liu, K.; Zhou, H.; Zhang, K.-H.; Wang, W.-W.; Li, M.-J.; Pan, Q.-Y.; Wang, X.-Y.; Wang, Y.-Z.; et al. The biosafety level-2 macromolecular crystallography beamline (bl10u2) at the shanghai synchrotron radiation facility. Nucl. Sci. Tech. 2023, 34, 202. [Google Scholar] [CrossRef]
- Liu, K.; Zhou, H.; Xu, Q.; Kong, H.-T.; Zhang, K.-H.; Wang, W.-W.; Li, M.-J.; Wang, Z.-J.; Pan, Q.-Y.; Wang, X.-Y.; et al. Bl02u1: The relocated macromolecular crystallography beamline at the shanghai synchrotron radiation facility. Nucl. Sci. Tech. 2023, 34, 193. [Google Scholar] [CrossRef]
- Sun, B.; Wang, Y.Z.; Liu, K.; Wang, Q.S.; He, J.H. Design of new sub-micron protein crystallography beamline at ssrf. In Proceedings of the 13th International Conference on Synchrotron Radiation Instrumentation (SRI2018), Taipei, Taiwan, 11–16 June 2018; Volume 2054. [Google Scholar]
- Hill, C.P.; Johnston, N.L.; Cohen, R.E. Crystal-structure of a ubiquitin-dependent degradation substrate—A 3-disulfide form of lysozyme. Proc. Natl. Acad. Sci. USA 1993, 90, 4136–4140. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Li, H.; Nie, T.; Jian, X.; Yu, C.; Li, J.; Su, H.; Zhang, X.; Li, S.; Yang, X.; et al. Discovery and mechanism study of SARS-CoV-2 3c-like protease inhibitors with a new reactive group. J. Med. Chem. 2023, 66, 12266–12283. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Cheng, F.; Zhang, J.; Su, H.; Hu, H.; Zou, Y.; Li, M.; Xu, Y. Structure-based design of a novel inhibitor of the zika virus ns2b/ns3 protease. Bioorg. Chem. 2022, 128, 106109. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Struct. Biol. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr. Sect. D Struct. Biol. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Thai, Q.M.; Pham, M.Q.; Minh, P.T.H.; Phung, H.T.T. Machine learning combines atomistic simulations to predict SARS-CoV-2 mpro inhibitors from natural compounds. Mol. Divers. 2023, 564, 111709. [Google Scholar] [CrossRef]
- Duan, Y.K.; Wang, H.F.; Yuan, Z.H.; Yang, H.T. Structural biology of SARS-CoV-2 mpro and drug discovery. Curr. Opin. Struct. Biol. 2023, 82, 102667. [Google Scholar] [CrossRef]
- Jin, Z.M.; Du, X.Y.; Xu, Y.C.; Deng, Y.Q.; Liu, M.Q.; Zhao, Y.; Zhang, B.; Li, X.F.; Zhang, L.K.; Peng, C.; et al. Structure of mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From sars to mers: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Ghasemlou, A.; Uskokovic, V.; Sefidbakht, Y. Exploration of potential inhibitors for SARS-CoV-2 mpro considering its mutants via structure-based drug design, molecular docking, md simulations, mm/pbsa, and dft calculations. Biotechnol. Appl. Biochem. 2023, 70, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Aplin, C.; Milano, S.K.; Zielinski, K.A.; Pollack, L.; Cerione, R.A. Evolving experimental techniques for structure-based drug design. J. Phys. Chem. B 2022, 126, 6599–6607. [Google Scholar] [CrossRef]
- Cellini, A.; Wahlgren, W.Y.; Henry, L.; Pandey, S.; Ghosh, S.; Castillon, L.; Claesson, E.; Takala, H.; Kubel, J.; Nimmrich, A.; et al. The three-dimensional structure of drosophila melanogaster (6-4) photolyase at room temperature. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Yabukarski, F.; Doukov, T.; Mokhtari, D.A.; Du, S.; Herschlag, D. Evaluating the impact of x-ray damage on conformational heterogeneity in room-temperature (277 k) and cryo-cooled protein crystals. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 945–963. [Google Scholar] [CrossRef]
- Thorne, R.E. Determining biomolecular structures near room temperature using x-ray crystallography: Concepts, methods and future optimization. Acta Crystallogr. Sect. D Struct. Biol. 2023, 79, 78–94. [Google Scholar] [CrossRef]
- Hendrickson, W.A. Facing the phase problem. IUCrJ 2023, 10, 521–543. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 100K-DCM-Lys | 100K-DMM-Lys | RT-DMM-Lys | 100K-DMM-Mpro | 100K-DMM-NS2B/NS3 | |
|---|---|---|---|---|---|
| Resolution range (Å) | 38.58–1.90 (1.94–1.90) | 39.13–1.57 (1.59–1.57) | 39.41–2.10 (2.16–2.10) | 48.21–2.52 (2.62–2.52) | 45.76–2.70 (2.83–2.70) |
| Space group | P43212 | P43212 | P43212 | C121 | P43212 |
| Unit cell parameters | |||||
| a, b, c (Å) | 77.16, 77.16, 36.85 | 78.26, 78.26, 36.81 | 78.82, 78.82, 37.70 | 45.42, 53.42, 113.91 | 59.74, 59.74, 213.53 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 101, 90 | 90, 90, 90 |
| Multiplicity | 16.9 (9.8) | 18.8 (18.2) | 24.8 (24.8) | 6.4 (6.3) | 24.4 (23.9) |
| Completeness (%) | 99.9 (98.7) | 99.90 (97.1) | 100.0 (100.0) | 98.4 (96.3) | 99.4 (95.6) |
| 〈I/σ(I)〉 | 16.6 (1.8) | 30.90 (5.2) | 20.2 (6.6) | 4.7 (0.8) | 16.1 (6.2) |
| Wilson B factors (Å2) | 32.30 | 17.77 | 13.98 | 51.06 | 31.18 |
| Unique reflections | 9250 (587) | 16,608 (788) | 7407 (585) | 9012 (963) | 11,424 (1419) |
| 0.28 (0.07) | 0.15 (0.03) | 0.29 (0.08) | 0.79 (0.13) | 0.22 (0.07) | |
| CC1/2 | 0.99 (0.87) | 1.00 (0.94) | 0.99 (0.93) | 0.85 (0.43) | 0.99 (0.96) |
| Reflections used in refinement | 9216 (878) | 16,564 (1599) | 7376 (707) | 8887 (758) | 11,348 (1032) |
| 0.20 | 0.19 | 0.19 | 0.25 | 0.18 | |
| 0.25 | 0.21 | 0.24 | 0.30 | 0.25 | |
| Protein residues | 129 | 129 | 129 | 300 | 387 |
| RMS bonds (Å) | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| RMS angles (°) | 1.07 | 1.11 | 0.97 | 0.46 | 0.97 |
| Ramachandran plot (%) | |||||
| Favored | 97.64 | 99.21 | 99.21 | 96.64 | 97.56 |
| Allowed | 2.36 | 0.79 | 0.79 | 3.02 | 2.17 |
| Outliers | 0.00 | 0.00 | 0.00 | 0.34 | 0.27 |
| Overall B factors (Å2) | 41.06 | 23.62 | 24.88 | 57.15 | 35.34 |
| PDB ID | 8Y9W | 8YA1 | 8YA4 | 8YA5 | 8Y9V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Xu, Q.; Wang, W.; Liang, M.; Yu, L.; Li, M.; Zhu, Z.; Huang, L.; Li, Q.; Yu, F.; et al. Crystallographic Data Collection Using a Multilayer Monochromator on an Undulator Beamline at the Shanghai Synchrotron Radiation Facility. Crystals 2024, 14, 199. https://doi.org/10.3390/cryst14020199
Zhang C, Xu Q, Wang W, Liang M, Yu L, Li M, Zhu Z, Huang L, Li Q, Yu F, et al. Crystallographic Data Collection Using a Multilayer Monochromator on an Undulator Beamline at the Shanghai Synchrotron Radiation Facility. Crystals. 2024; 14(2):199. https://doi.org/10.3390/cryst14020199
Chicago/Turabian StyleZhang, Chenyu, Qin Xu, Weiwei Wang, Miao Liang, Li Yu, Minjun Li, Zhimin Zhu, Liqing Huang, Qianhui Li, Feng Yu, and et al. 2024. "Crystallographic Data Collection Using a Multilayer Monochromator on an Undulator Beamline at the Shanghai Synchrotron Radiation Facility" Crystals 14, no. 2: 199. https://doi.org/10.3390/cryst14020199
APA StyleZhang, C., Xu, Q., Wang, W., Liang, M., Yu, L., Li, M., Zhu, Z., Huang, L., Li, Q., Yu, F., Wang, Y., Zhou, H., & Wang, Q. (2024). Crystallographic Data Collection Using a Multilayer Monochromator on an Undulator Beamline at the Shanghai Synchrotron Radiation Facility. Crystals, 14(2), 199. https://doi.org/10.3390/cryst14020199