
Synthesis and Crystal Structures of Rhomb-Shaped Dimeric Pd(II) Complexes with Arylethynyl-Substituted 2,2′-Bipyridine through CH⋯π Interactions in the Crystalline States

Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis and Crystallization
2.3. Crystal Structure Determination
3. Results and Discussion
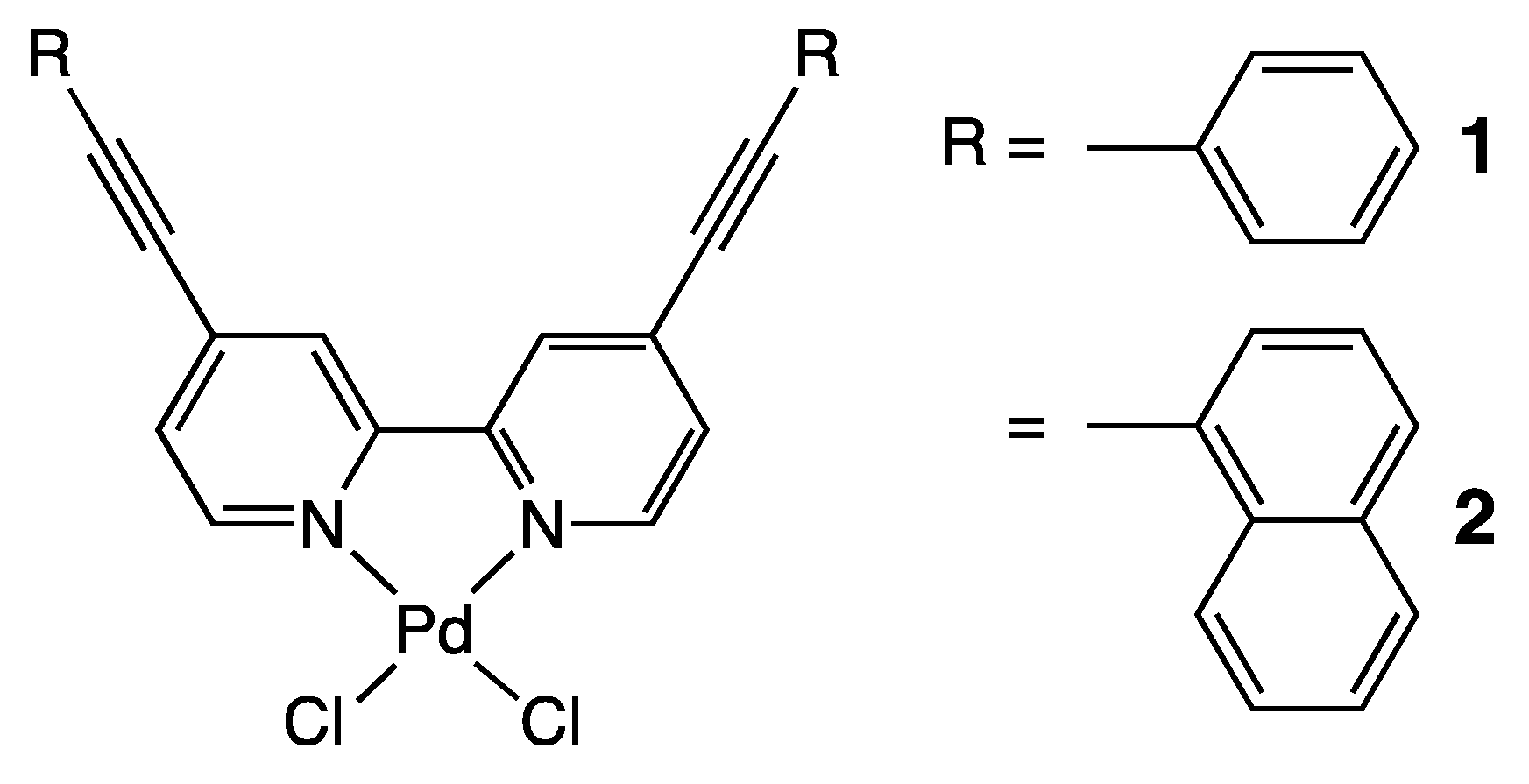
3.1. Preparations of 1 and 2
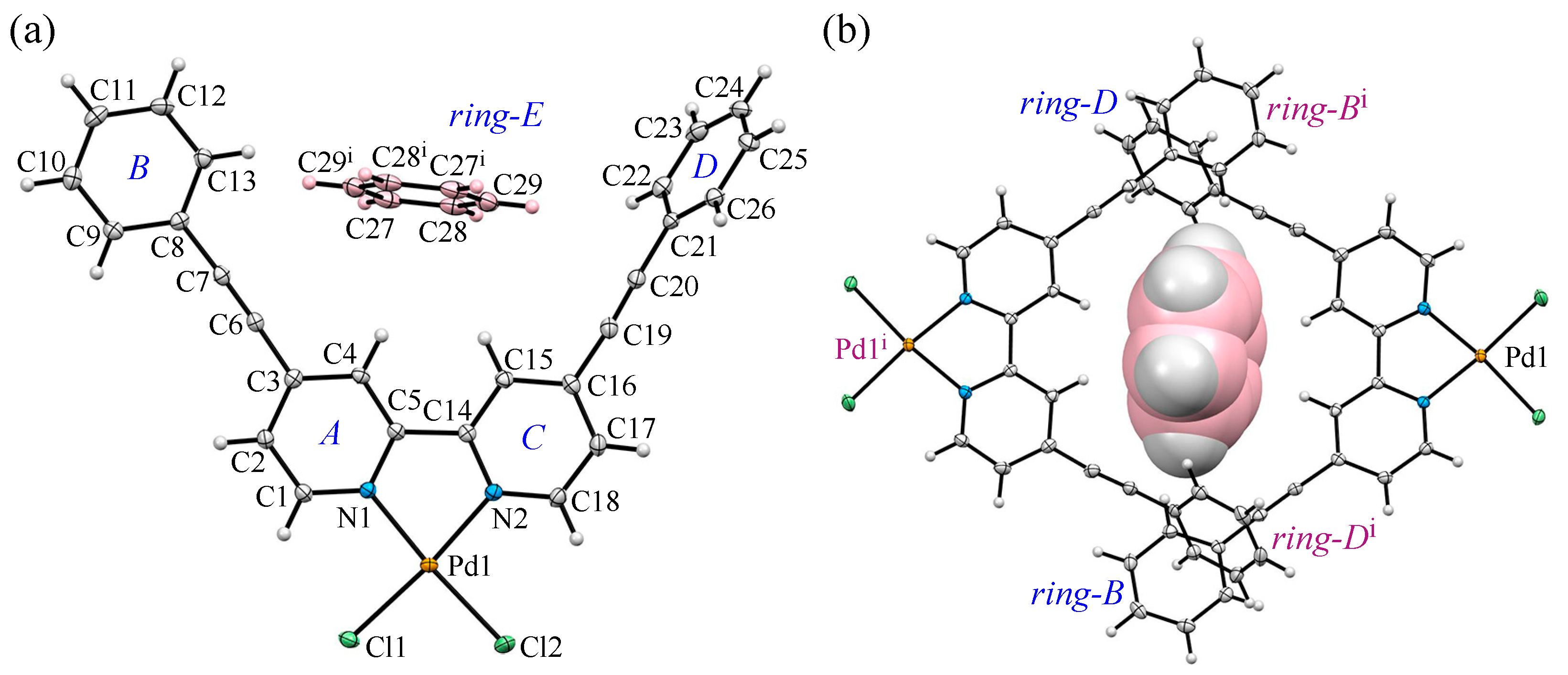
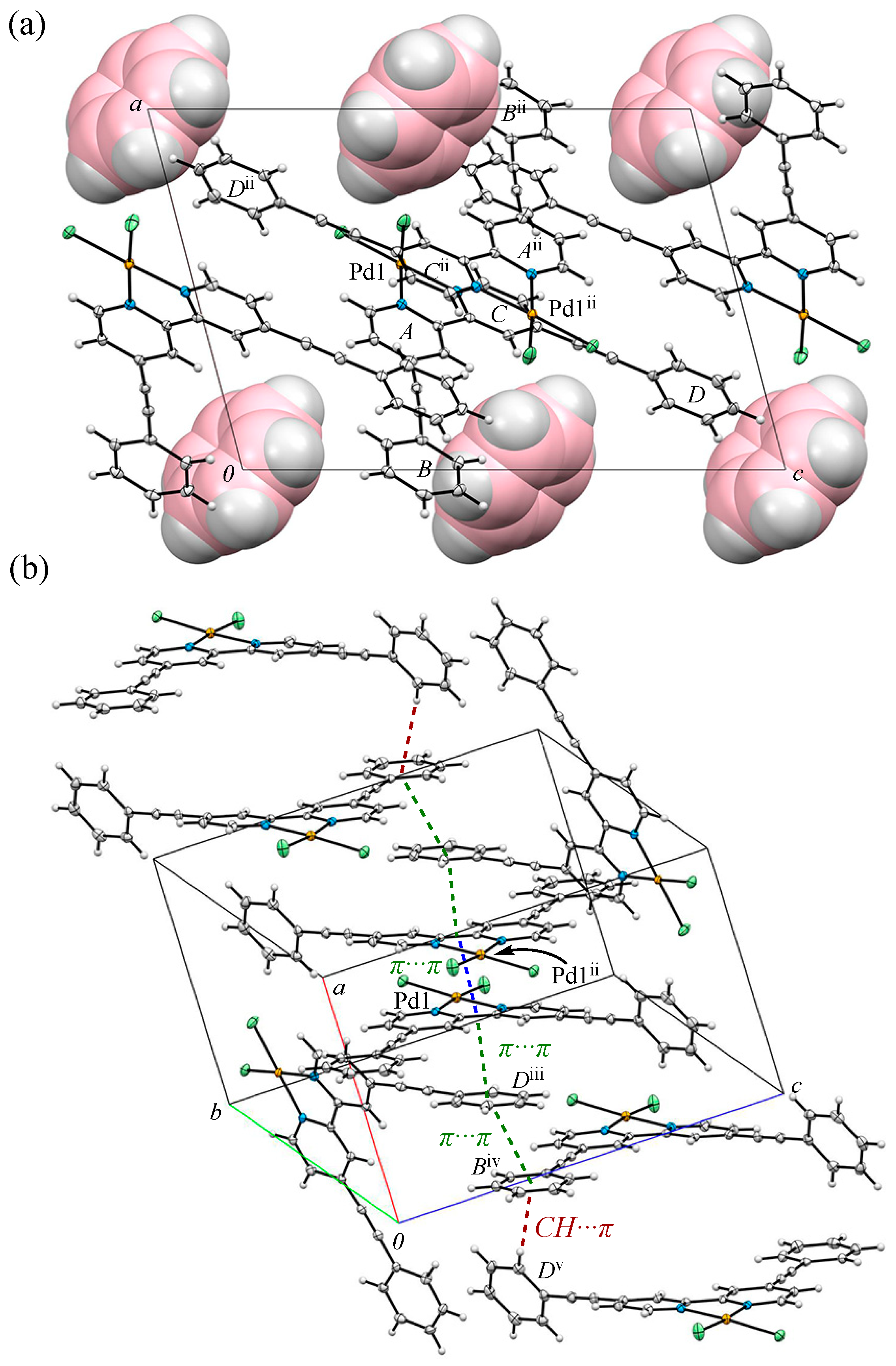
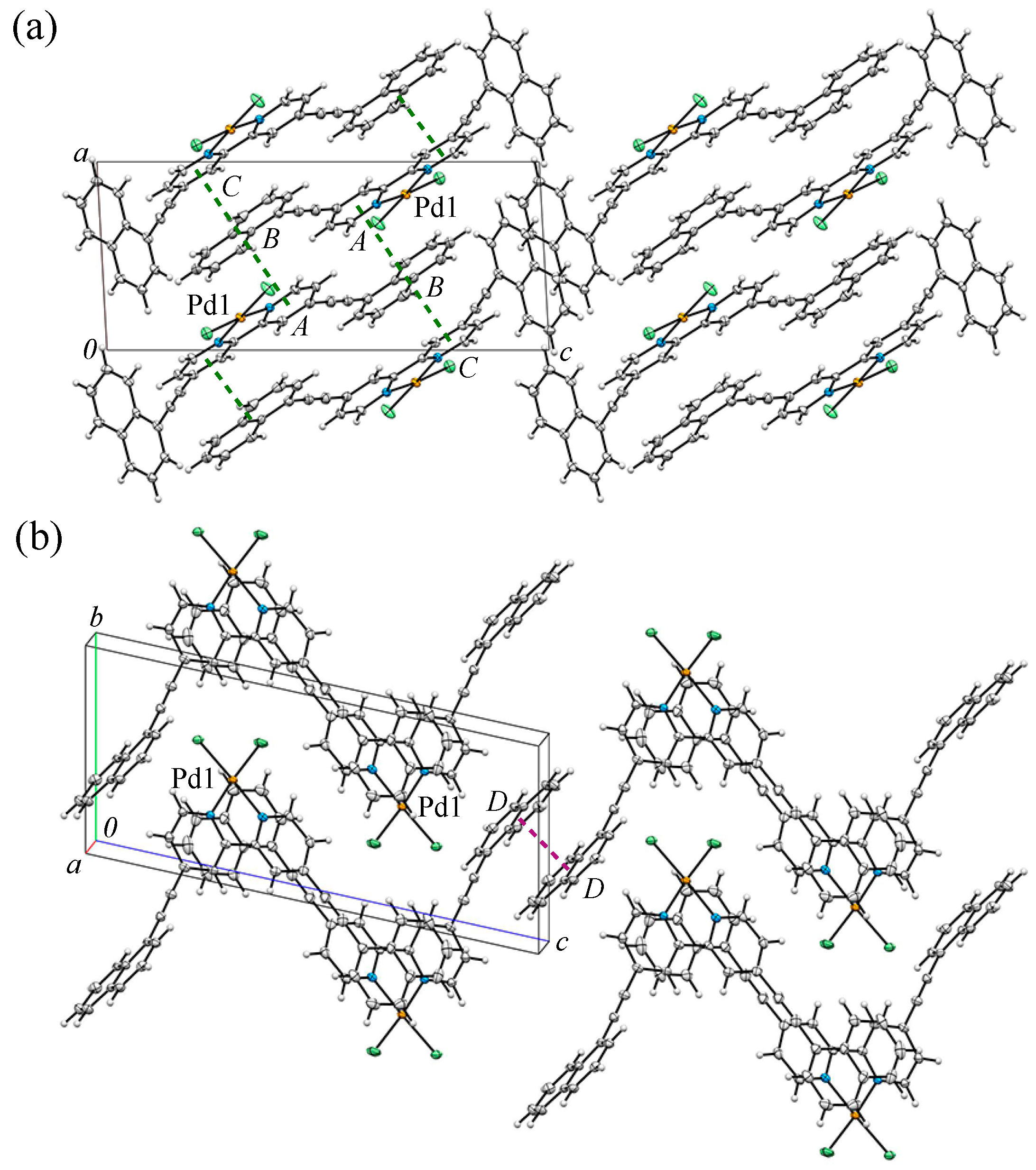
3.2. Crystal Structure and Intermolecular Interactions of 1•0.5C6H6
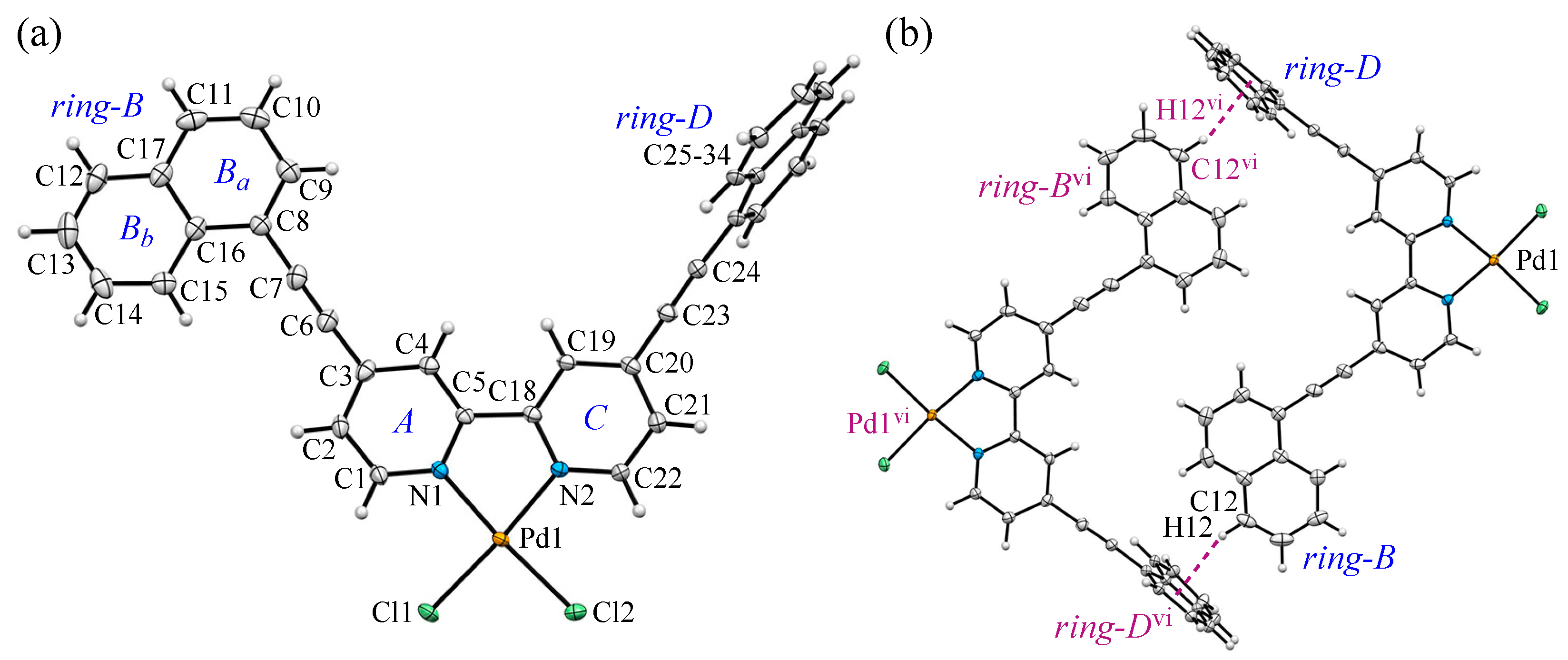
3.3. Crystal Structure and Intermolecular Interactions of 2
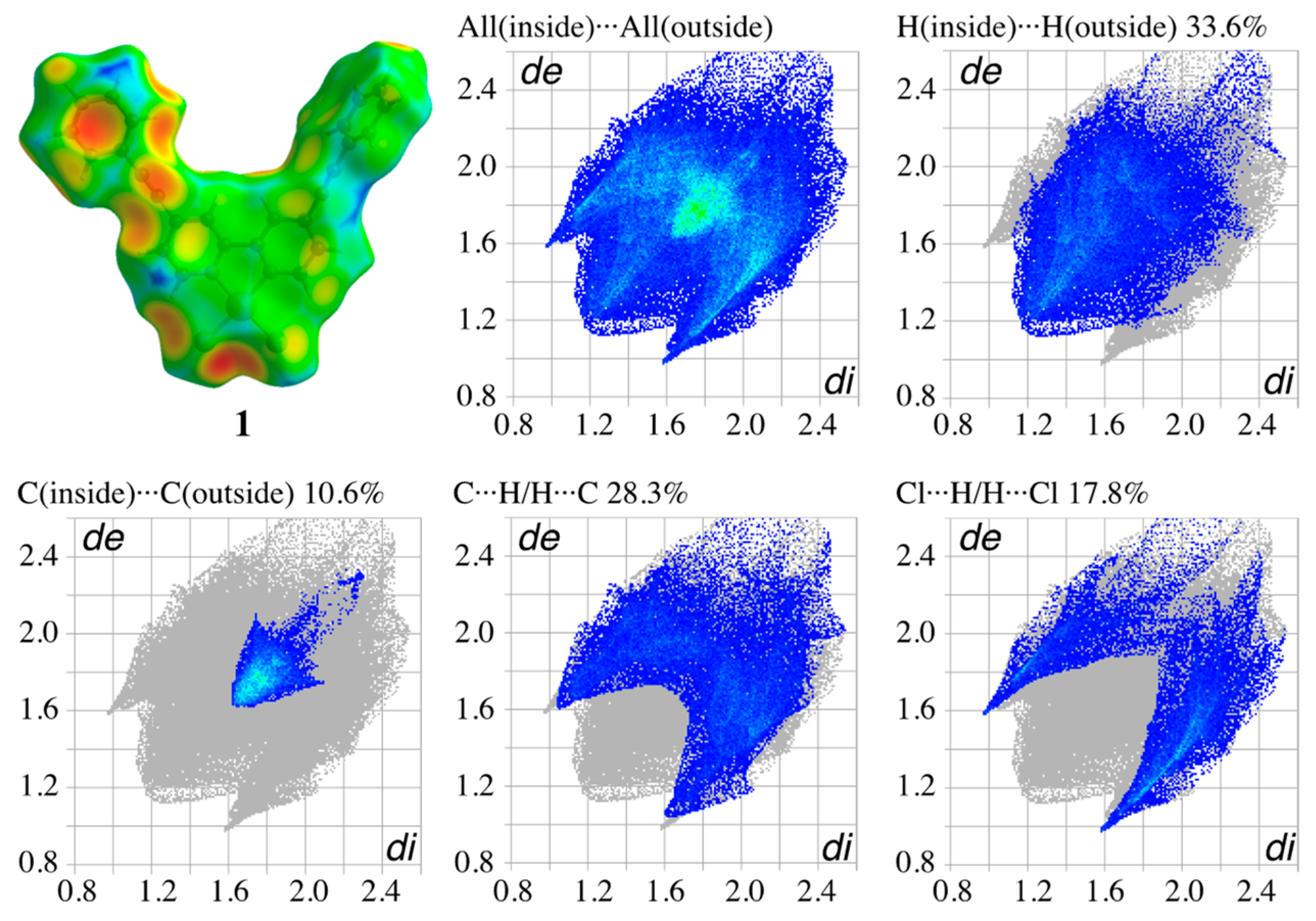
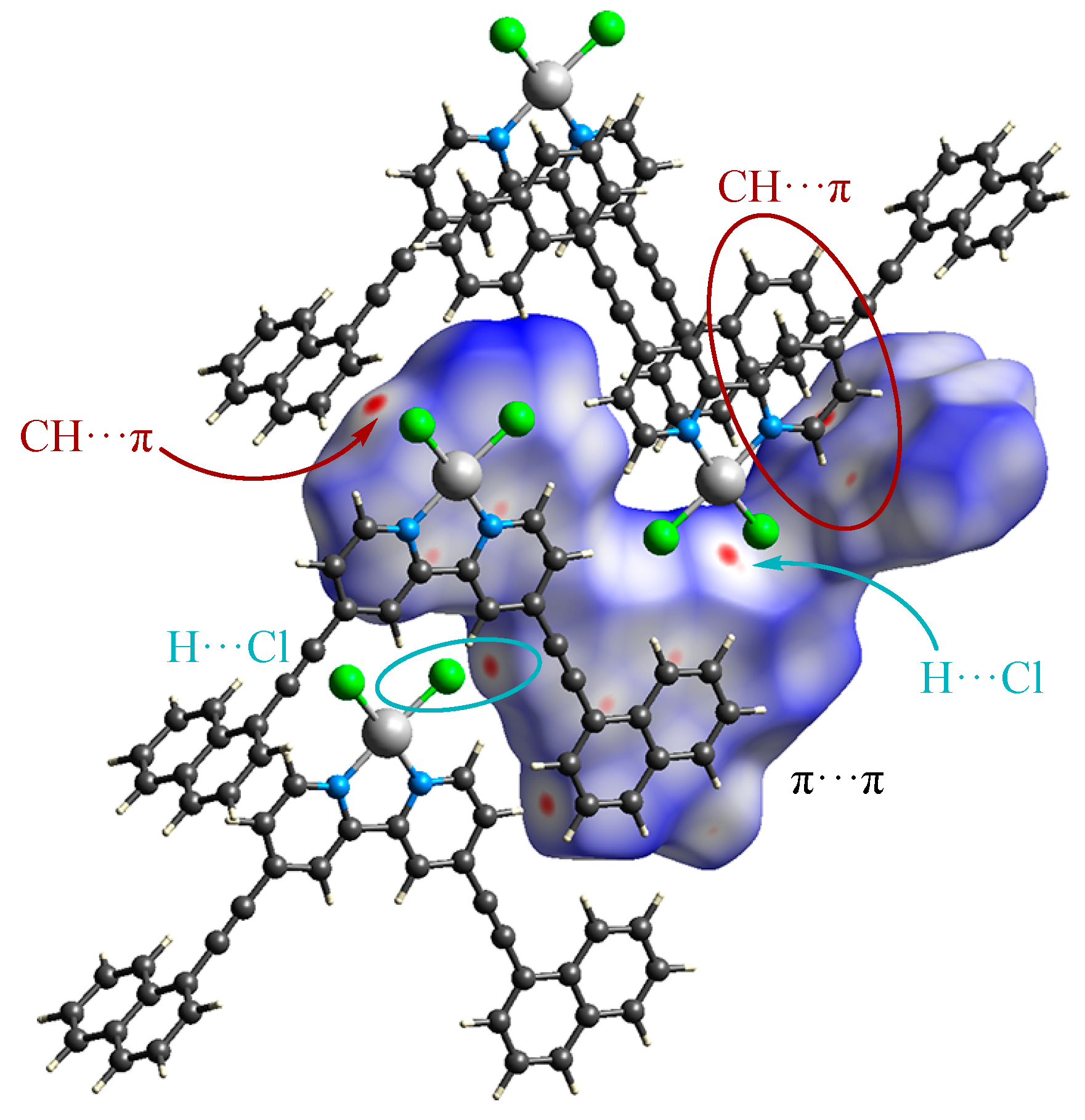
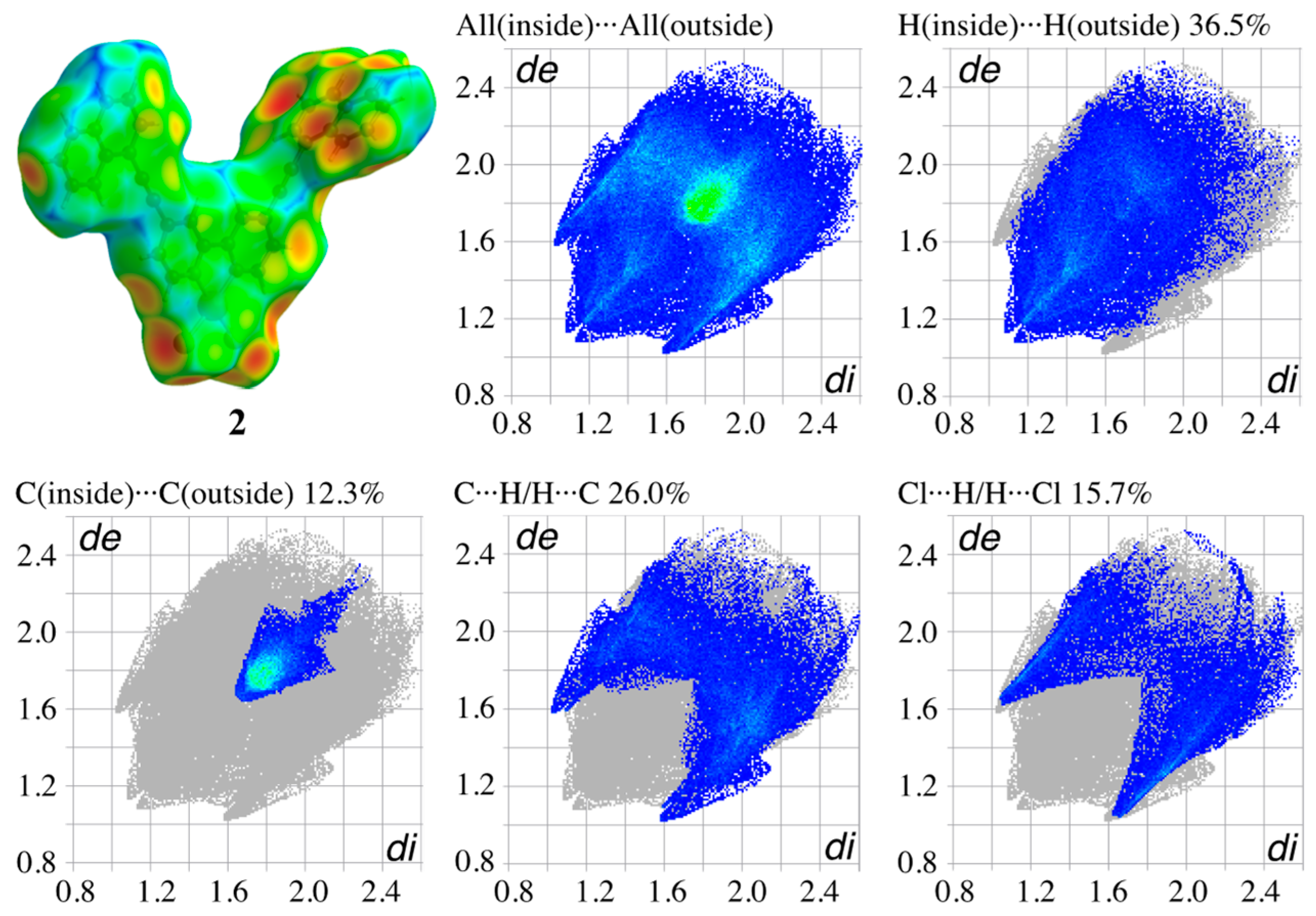
3.4. Hirshfeld Surface Analysis of the Structures
3.5. Density Functional Theory Calculations of the Structures
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Archer, S.; Weinstein, J.A. Charge-separated excited states in platinum(II) chromophores: Photophysics, formation, stabilization and utilization in solar energy conversion. Coord. Chem. Rev. 2012, 256, 2530–2561. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, F.; Huang, C. Phosphorescent chemosensors based on heavy-metal complexes. Chem. Soc. Rev. 2010, 39, 3007–3030. [Google Scholar] [CrossRef]
- Balzani, V.; Bergamini, G.; Marchioni, F.; Ceroni, P. Ru(II)-bipyridine complexes in supramolecular systems, devices and machines. Coord. Chem. Rev. 2006, 250, 1254–1266. [Google Scholar] [CrossRef]
- Bhasikuttan, A.C.; Suzuki, M.; Nakashima, S.; Okada, T. Ultrafast fluorescence detection in tris(2,2′-bipyridine)ruthenium(II) complex in solution: Relaxation dynamics involving higher excited states. J. Am. Chem. Soc. 2002, 124, 8398–8405. [Google Scholar] [CrossRef]
- Palmans, R.; MacQueen, D.B.; Pierpont, C.G.; Frank, A.J. Synthesis and characterization of bis(2,2′-bipyridyl)platinum(I): A novel microtubular linear-chain complex. J. Am. Chem. Soc. 1996, 118, 12647–12653. [Google Scholar] [CrossRef]
- Houlding, V.H.; Miskowski, V.M. The effect of linear chain structure on the electronic structure of Pt(II) diimine complexes. Coord. Chem. Rev. 1991, 111, 145–152. [Google Scholar] [CrossRef]
- Herber, R.H.; Croft, M.; Coyer, M.J.; Bilash, B.; Sahiner, A. Origin of polychromism of cis square-planar platinum(II) complexes: Comparison of two forms of [Pt(2,2′-bpy)(Cl2)]. Inorg. Chem. 1994, 33, 2422–2426. [Google Scholar] [CrossRef]
- Valiente, R.; García-Lastra, J.M.; García-Fernández, P.; García-Revilla, S.; Wenger, O.S. Red-to-yellow pressure-induced phase transition in Pt(bpy)Cl2: Spectroscopic study supported by DFT calculations. Eur. J. Inorg. Chem. 2007, 36, 5735–5742. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Kondinski, A.; Monakhov, K.Y.; Koshevoy, I.O.; Grachova, E.V. Synthesis, photophysical properties and cation-binding studies of bipyridine-functionalized gold(I) complexes. Inorg. Chem. Front. 2018, 5, 160–171. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Kirina, Y.V.; Kukushkin, V.Y. Halogen bonding between metal centers and halocarbons. Chem. Commun. 2016, 52, 5565–5568. [Google Scholar] [CrossRef]
- Baykov, S.V.; Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Boyarskiy, V.P. Pt/Pd and I/Br isostructural exchange provides formation of C–I⋯Pd, C–Br⋯Pt, and C–Br⋯Pd metal-involving halogen bonding. Cryst. Growth Des. 2018, 18, 5973–5980. [Google Scholar] [CrossRef]
- Pang, X.; Wang, H.; Wang, W.; Jin, W.J. Phosphorescent π-hole⋯π bonding cocrystals of pyrene with halo-perfluorobenzenes (F, Cl, Br, I). Cryst. Growth Des. 2015, 15, 4938–4945. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Coord. Chem. Rev. 2017, 345, 209–228. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse arene sandwich structures based upon π-hole⋯[MII] (d8 M = Pt, Pd) interactions, where positively charged metal centers play the role of a nucleophile. Angew. Chem. Int. Ed. 2019, 58, 4164–4168. [Google Scholar] [CrossRef]
- Kusakawa, T.; Goto, T.; Hori, A. Supramolecular association of M2+⋯π induced by different electrostatic properties using naphthyl substituted β-diketonate complexes (metal = Cu, Pd, Pt). CrystEngComm. 2020, 20, 3090–3094. [Google Scholar] [CrossRef]
- Ikumura, Y.; Habuka, Y.; Sakai, S.; Shinohara, T.; Yuge, H.; Rzeznicka, I.I.; Hori, A. Enhanced and heteromolecular guest encapsulation in nonporous crystals of a perfluorinated triketonato dinuclear copper complex. Chem. Eur. J. 2020, 26, 5051–5060. [Google Scholar] [CrossRef]
- Hori, A.; Takatani, S.; Miyamoto, T.K.; Hasegawa, M. Luminescence from π–π stacked bipyridines through arene–perfluoroarene interactions. CrystEngComm 2009, 11, 567–569. [Google Scholar] [CrossRef]
- Williams, J.H. How Molecules Build Solids. In Crystal Engineering; Morgan & Claypool Publishers: Kentfield, CA, USA, 2017. [Google Scholar]
- Hori, A. The Importance of P-Interactions in Crystal Engineering; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 163–185. [Google Scholar]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π hydrogen bonds in organic and organometallic chemistry. CrystEngComm 2009, 11, 1757–1788. [Google Scholar] [CrossRef]
- Zhao, Z.; Yin, X.; Peng, T.; Wang, N.; Wang, S. Triarylboron/triarylamine-functionalized 2,2′-bipyridine ligands and their copper(I) complexes. Inorg. Chem. 2020, 59, 7426–7434. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Guo, Z.; Zhu, Q.; Liu, S.; Zhang, J. The two-stepwise luminescent switching properties of triple-stimuli-responsive platinum(II) complexes bearing 4,4′-bis (2-phenylethynyl)-2,2′-bipyridine ligand. Dye. Pigment. 2023, 217, 111406. [Google Scholar] [CrossRef]
- Hunter, C.A. Meldola Lecture. The role of aromatic interactions in molecular recognition. Chem. Soc. Rev. 1994, 23, 101–109. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira Reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Zhu, Q.; Liao, L.; Cheng, G.; Yang, W.; Deng, Y.; Yang, D. Palladium-catalyzed sonogashira coupling reaction of 2-amino-3-bromopyridines with terminal alkynes. Mod. Res. Catal. 2017, 6, 121–133. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 17.5; University of Western Australia: Perth, Australia, 2017; Available online: http://hirshfeldsuface.net (accessed on 19 February 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1•0.5C6H6 | 2 | |
|---|---|---|
| Chemical formula | C29H19Cl2N2Pd | C34H20Cl2N2Pd |
| Formula weight | 572.76 | 633.82 |
| Crystal system | monoclinic | triclinic |
| Space group | P21/c | P-1 |
| a [Å] | 11.950(4) | 8.019(3) |
| b [Å] | 11.726(4) | 8.623(4) |
| c [Å] | 17.388(6) | 19.261(8) |
| α [°] | 90 | 102.519(5) |
| β [°] | 104.727(3) | 92.193(5) |
| γ [°] | 90 | 93.798(5) |
| V [Å3] | 2356.4(13) | 1295.5(9) |
| Z | 4 | 2 |
| Dc [Mg m−3] | 1.614 | 1.625 |
| μ [mm−1] | 1.04 | 0.95 |
| F(000) | 1148 | 636 |
| Rint | 0.0264 | 0.0341 |
| GOF | 1.038 | 1.039 |
| R [(I) > 2σ(I)] | 0.0212 | 0.0321 |
| wR (Fo2) | 0.0541 | 0.0730 |
| CCDC No. | 2031735 | 2031736 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hori, A.; Ichisugi, R.; Azegami, D.; Toyama, N.; Yuge, H. Synthesis and Crystal Structures of Rhomb-Shaped Dimeric Pd(II) Complexes with Arylethynyl-Substituted 2,2′-Bipyridine through CH⋯π Interactions in the Crystalline States. Crystals 2024, 14, 255. https://doi.org/10.3390/cryst14030255
Hori A, Ichisugi R, Azegami D, Toyama N, Yuge H. Synthesis and Crystal Structures of Rhomb-Shaped Dimeric Pd(II) Complexes with Arylethynyl-Substituted 2,2′-Bipyridine through CH⋯π Interactions in the Crystalline States. Crystals. 2024; 14(3):255. https://doi.org/10.3390/cryst14030255
Chicago/Turabian StyleHori, Akiko, Reo Ichisugi, Daiki Azegami, Naoki Toyama, and Hidetaka Yuge. 2024. "Synthesis and Crystal Structures of Rhomb-Shaped Dimeric Pd(II) Complexes with Arylethynyl-Substituted 2,2′-Bipyridine through CH⋯π Interactions in the Crystalline States" Crystals 14, no. 3: 255. https://doi.org/10.3390/cryst14030255