
Structural Analysis of 3,5-Bistrifluoromethylhydrocinnamic Acid


Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Crystallization
2.2. Single-Crystal X-ray Diffraction
2.3. Hirshfeld Surface and Pairwise Interaction Energy Analyses
2.4. Density Functional Theory Calculations
3. Results and Discussion
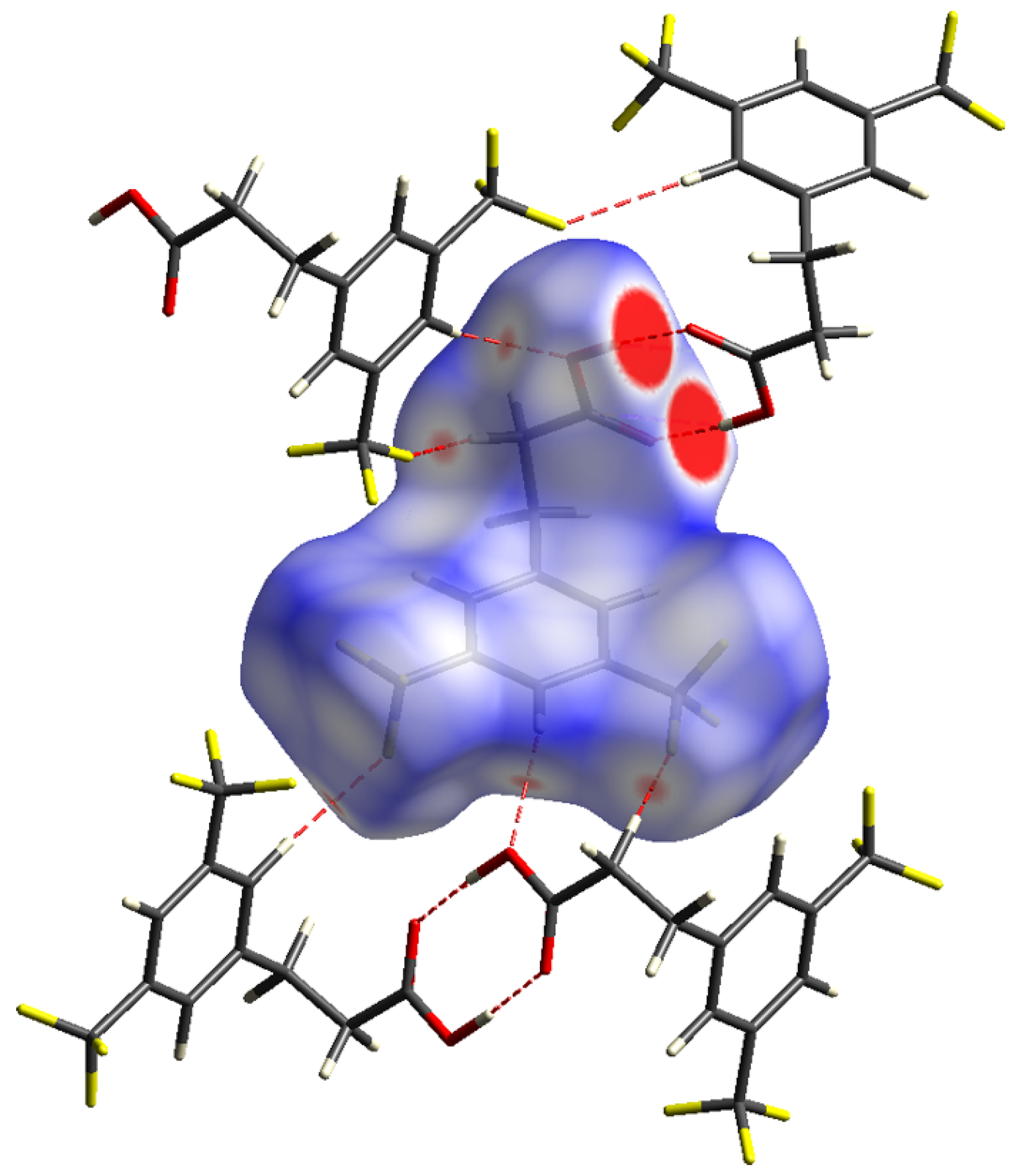
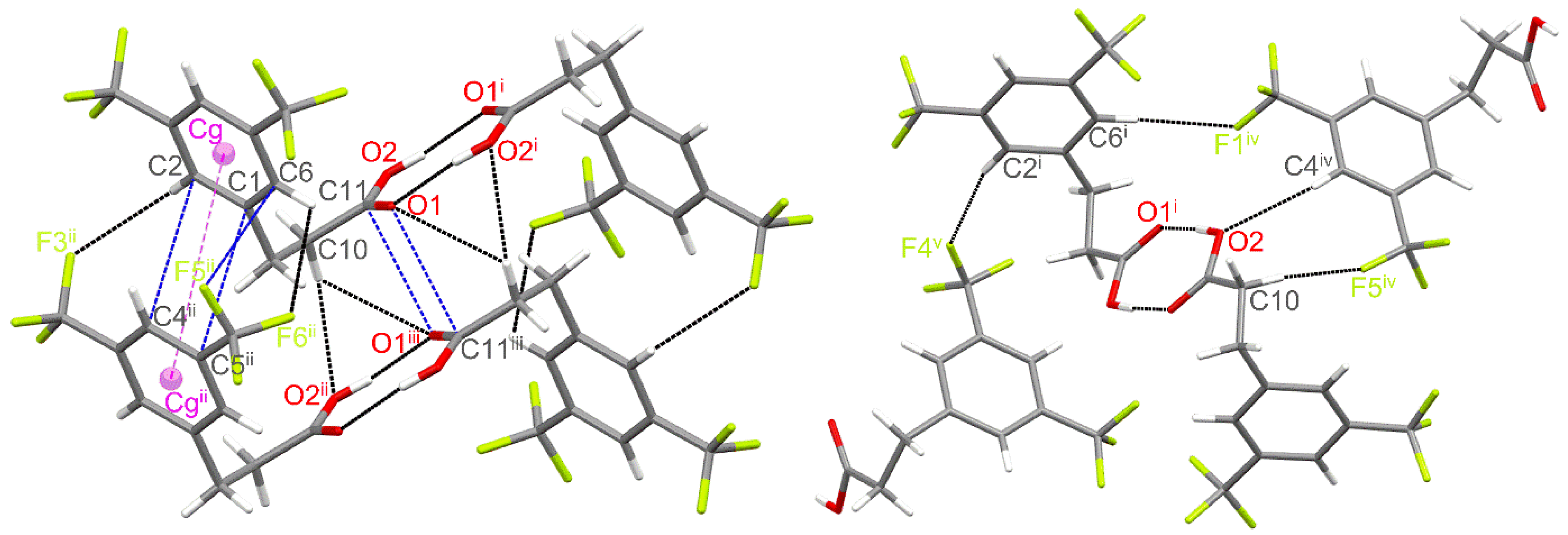
3.1. Crystal Structure and Hirshfeld Surface Analysis
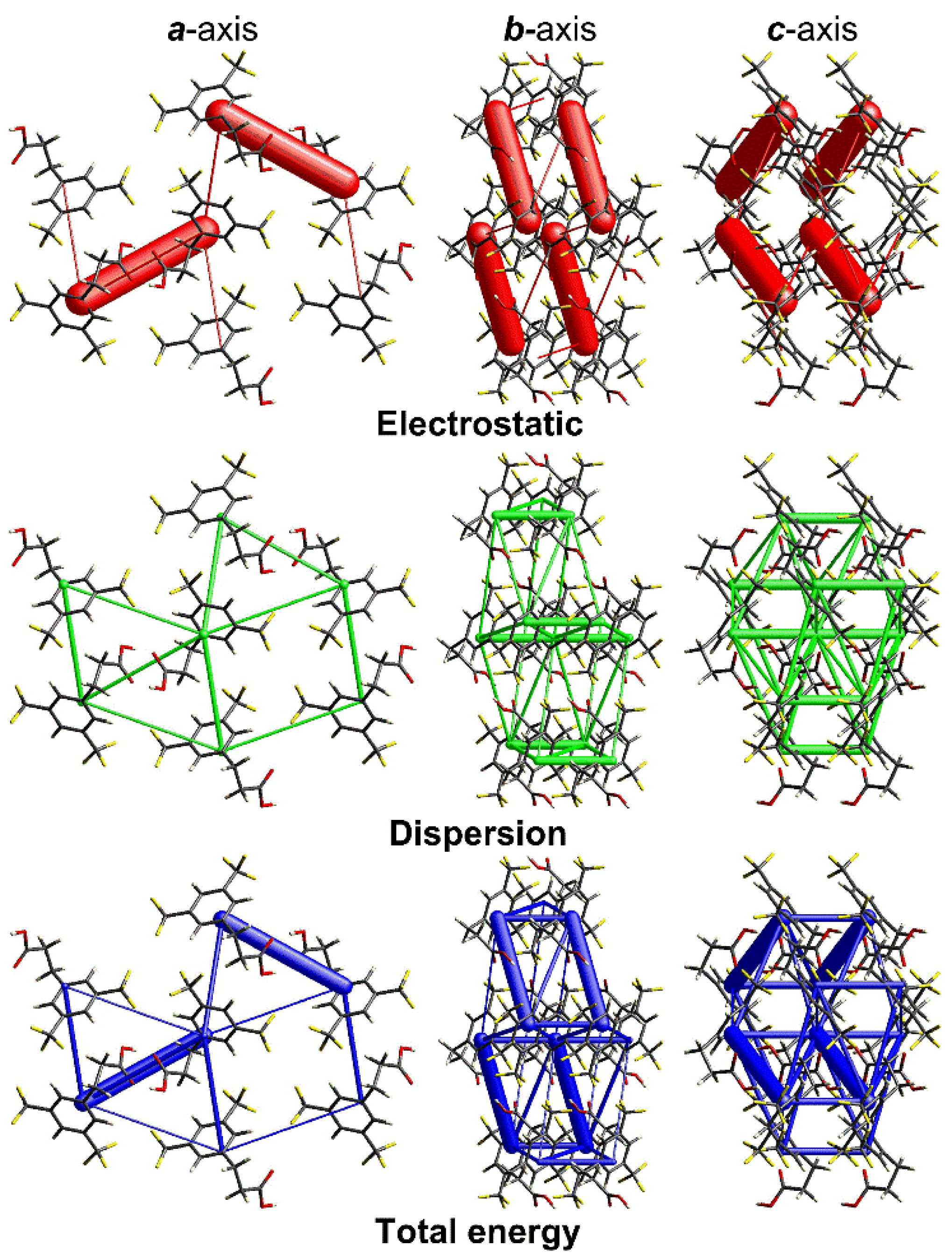
3.2. Intermolecular Interaction Energies and Energy Frameworks
3.3. Comparison with Crystal Structures of Other 3-Phenylpropanoic Acids (3-PPAs)
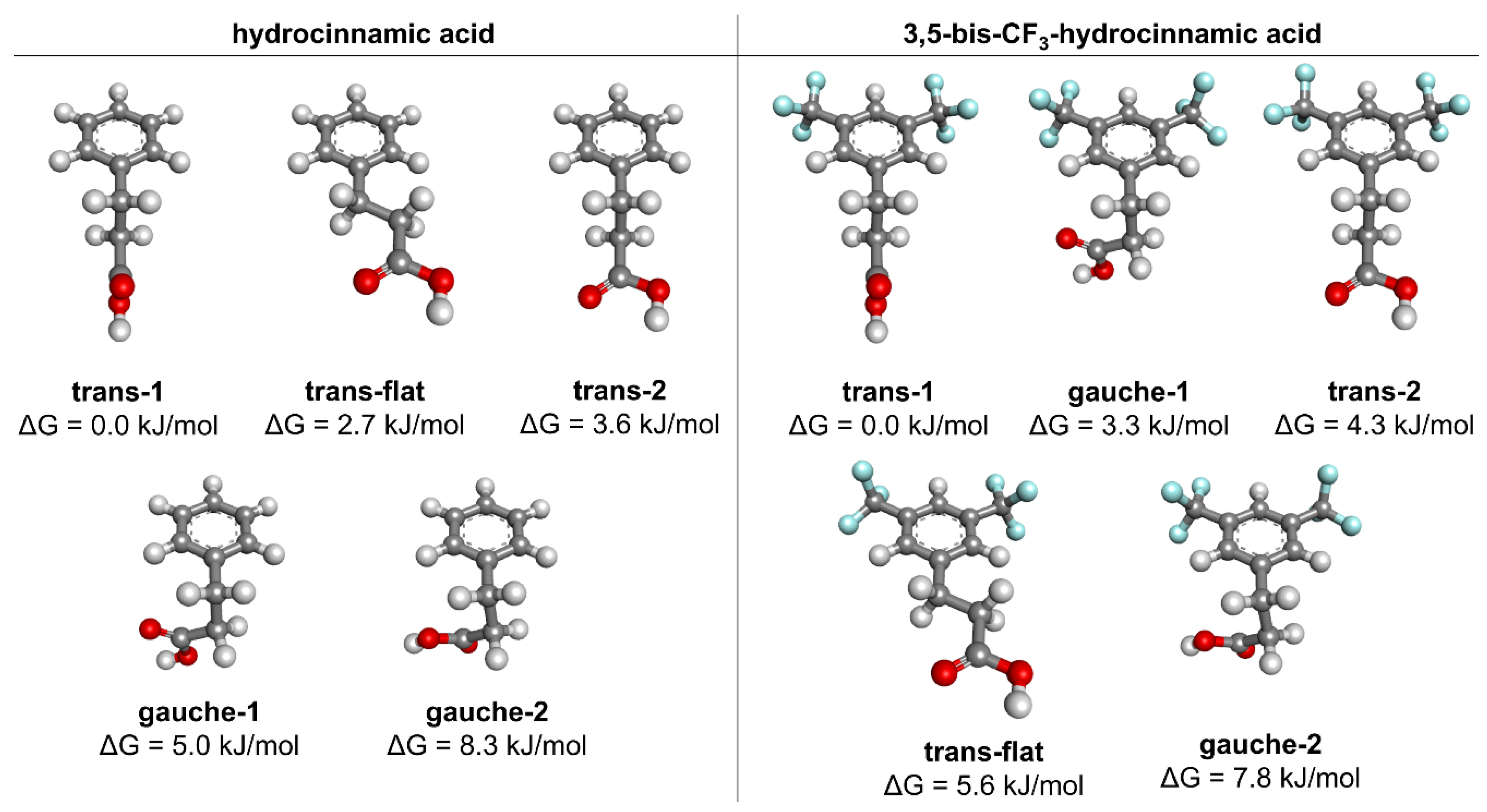
3.4. Quantum Chemical Conformational Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korneev, S. Hydrocinnamic Acids: Application and Strategy of Synthesis. Synthesis 2013, 45, 1000–1015. [Google Scholar] [CrossRef]
- Teall, M.; Harrison, T.; Moseley, J.D.; Owens, A.P.; Sadowski, S.; Cascieri, M.A. Linear amides as substance P antagonists. Bioorg. Med. Chem. Lett. 1996, 6, 1585–1588. [Google Scholar] [CrossRef]
- Ryckmans, T.; Balançon, L.; Berton, O.; Genicot, C.; Lamberty, Y.; Lallemand, B.; Pasau, P.; Pirlot, N.; Quéré, L.; Talaga, P. First dual NK1 antagonists–serotonin reuptake inhibitors: Synthesis and SAR of a new class of potential antidepressants. Bioorg. Med. Chem. Lett. 2002, 12, 261–264. [Google Scholar] [CrossRef]
- Li, F.-N.; Kim, N.-J.; Paek, S.-M.; Kwon, D.-Y.; Min, K.H.; Jeong, Y.-S.; Kim, S.-Y.; Park, Y.-H.; Kim, H.-D.; Park, H.-G.; et al. Design, synthesis, and biological evaluation of novel diarylalkyl amides as TRPV1 antagonists. Bioorg. Med. Chem. 2009, 17, 3557–3567. [Google Scholar] [CrossRef]
- Lemhadri, M.; Doucet, H.; Santelli, M. Direct synthesis of 3-arylpropionic acids by tetraphosphine/palladium catalysed Heck reactions of aryl halides with acrolein ethylene acetal. Tetrahedron 2004, 60, 11533–11540. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, B.-W.; Lee, H.U.; Choi, D.-K.; Yoon, S.-H. Synthesis of Clovamide Analogues That Inhibit NO Production in Activated BV-2 Microglial Cells. Biol. Pharm. Bull. 2017, 40, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Shaala, L.A.; Youssef, D.T.A.; Alzughaibi, T.A.; Elhady, S.S. Antimicrobial Chlorinated 3-Phenylpropanoic Acid Derivatives from the Red Sea Marine Actinomycete Streptomyces coelicolor LY001. Mar. Drugs 2020, 18, 450. [Google Scholar] [CrossRef] [PubMed]
- Narayana, K.J.P.; Prabhakar, P.; Vijayalakshmi, M.; Venkateswarlu, Y.; Krishna, P.S.J. Biological activity of phenylpropionic acid isolated from a terrestrial Streptomycetes. Pol. J. Microbiol. 2007, 56, 191–197. [Google Scholar] [PubMed]
- Guzman, J. Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef]
- Okamoto, A.; Ishii, S.; Hirotsu, K.; Kagamiyama, H. The Active Site of Paracoccus denitrificans Aromatic Amino Acid Aminotransferase Has Contrary Properties: Flexibility and Rigidity. Biochemistry 1999, 38, 1176–1184. [Google Scholar] [CrossRef]
- Bosshart, P.D.; Kalbermatter, D.; Bonetti, S.; Fotiadis, D. The making of a potent L-lactate transport inhibitor. Commun. Chem. 2021, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.F.J.; Matalińska, J. NK1 receptor binding of a few low molecular weight 3,5-bistrifluoromethylbenzene derivatives. Acta Pol. Pharm. Drug Res. 2023, 80, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Galano, J.J.; Alías, M.; Pérez, R.; Velázquez-Campoy, A.; Hoffman, P.S.; Sancho, J. Improved Flavodoxin Inhibitors with Potential Therapeutic Effects against Helicobacter pylori Infection. J. Med. Chem. 2013, 56, 6248–6258. [Google Scholar] [CrossRef] [PubMed]
- Lassalas, P.; Gay, B.; Lasfargeas, C.; James, M.J.; Tran, V.; Vijayendran, K.G.; Brunden, K.R.; Kozlowski, M.C.; Thomas, C.J.; Smith, A.B.; et al. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem. 2016, 59, 3183–3203. [Google Scholar] [CrossRef] [PubMed]
- Widler, L.; Altmann, E.; Beerli, R.; Breitenstein, W.; Bouhelal, R.; Buhl, T.; Gamse, R.; Gerspacher, M.; Halleux, C.; John, M.R.; et al. 1-Alkyl-4-phenyl-6-alkoxy-1 H -quinazolin-2-ones: A Novel Series of Potent Calcium-Sensing Receptor Antagonists. J. Med. Chem. 2010, 53, 2250–2263. [Google Scholar] [CrossRef] [PubMed]
- Kuranov, S.O.; Luzina, O.A.; Onopchenko, O.; Pishel, I.; Zozulya, S.; Gureev, M.; Salakhutdinov, N.F.; Krasavin, M. Exploring bulky natural and natural-like periphery in the design of p-(benzyloxy)phenylpropionic acid agonists of free fatty acid receptor 1 (GPR40). Bioorg. Chem. 2020, 99, 103830. [Google Scholar] [CrossRef] [PubMed]
- Laghezza, A.; Pochetti, G.; Lavecchia, A.; Fracchiolla, G.; Faliti, S.; Piemontese, L.; Di Giovanni, C.; Iacobazzi, V.; Infantino, V.; Montanari, R.; et al. New 2-(Aryloxy)-3-phenylpropanoic Acids as Peroxisome Proliferator-Activated Receptor α/γ Dual Agonists Able To Upregulate Mitochondrial Carnitine Shuttle System Gene Expression. J. Med. Chem. 2013, 56, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Chattopadhyay, B.; Mukherjee, M.; Mukherjee, A.K. Structure Analysis of Molecular Compounds with Z ′ = 2 Using Laboratory X-ray Powder Diffraction Data: 3-Phenylpropionic Acid and Its Derivatives. Cryst. Growth Des. 2012, 12, 466–474. [Google Scholar] [CrossRef]
- Begum, N.S.; Jain, M.; Chandrasekhar, S.; Venkatesan, K. Structure of 3-(2-hydroxyphenyl)propionic acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1992, 48, 1076–1078. [Google Scholar] [CrossRef]
- Glusker, J.P.; Zacharias, D.E.; Carrell, H.L. Structure refinement and molecular packing of p-chloro-trans-cinnamic acid and β-(p-chlorophenyl)propionic acid. J. Chem. Soc. Perkin Trans. 2 1975, 4, 68–74. [Google Scholar] [CrossRef]
- Okabe, N.; Suga, T. 3-(p-Hydroxyphenyl)propionic Acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1995, 51, 1324–1325. [Google Scholar] [CrossRef]
- Gerkin, R.E. 3-(2-Methylphenyl)propanoic acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1999, 55, 1857–1859. [Google Scholar] [CrossRef]
- Christiansen, E.; Urban, C.; Merten, N.; Liebscher, K.; Karlsen, K.K.; Hamacher, A.; Spinrath, A.; Bond, A.D.; Drewke, C.; Ullrich, S.; et al. Discovery of Potent and Selective Agonists for the Free Fatty Acid Receptor 1 (FFA 1/GPR40), a Potential Target for the Treatment of Type II Diabetes. J. Med. Chem. 2008, 51, 7061–7064. [Google Scholar] [CrossRef]
- Guan, J.-N.; Kong, X.-J.; Xu, B.; Liang, J.-H.; Song, N. 3-[4-(Trifluoromethyl)phenyl]propanoic acid. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o844. [Google Scholar] [CrossRef]
- Das, U.; Chattopadhyay, B.; Mukherjee, M.; Mukherjee, A.K. Crystal structure and electronic properties of three phenylpropionic acid derivatives: A combined X-ray powder diffraction and quantum mechanical study. Chem. Phys. Lett. 2011, 501, 351–357. [Google Scholar] [CrossRef]
- Tan, S.-S.; Wen, H.-L.; Liu, C.-B.; Peng, X.-P.; Li, X.-S. Crystal structure of 3-(4-(carboxymethoxy)phenyl)propanoic acid, C11H12O5. Z. Für Krist. New Cryst. Struct. 2007, 222, 137–138. [Google Scholar] [CrossRef]
- Lu, L.; Wu, W.P.; Ma, A.Q.; Xie, B.; Wu, Y. Synthesis, structural, and luminescence of two coordination polymers. Russ. J. Coord. Chem. 2015, 41, 573–578. [Google Scholar] [CrossRef]
- Bugenhagen, B.; Al Jasem, Y.; AlAzani, M.; Thiemann, T. Crystal structure of 3-(2,5-dimethoxyphenyl)propionic acid. Acta Crystallogr. Sect. E Crystallogr. Commun. 2015, 71, o337–o338. [Google Scholar] [CrossRef]
- Liu, W.; Ren, W.; Li, J.; Shi, Y.; Chang, W.; Shi, Y. A Ligand-Directed Catalytic Regioselective Hydrocarboxylation of Aryl Olefins with Pd and Formic Acid. Org. Lett. 2017, 19, 1748–1751. [Google Scholar] [CrossRef]
- Pan, R.; Shi, C.; Zhang, D.; Tian, Y.; Guo, S.; Yao, H.; Lin, A. Nickel-Catalyzed Reductive 1,2-Dialkynylation of Alkenes Bearing an 8-Aminoquinoline Directing Group. Org. Lett. 2019, 21, 8915–8920. [Google Scholar] [CrossRef]
- Drebushchak, T.N.; Boldyreva, E.V.; Fucke, K. Hydrogen bonding of catechol groups in the crystal structure of dihydrocaffeic acid. J. Struct. Chem. 2013, 54, 368–372. [Google Scholar] [CrossRef]
- Antonenko, T.A.; Shpakovsky, D.B.; Berseneva, D.A.; Gracheva, Y.A.; Dubova, L.G.; Shevtsov, P.N.; Redkozubova, O.M.; Shevtsova, E.F.; Tafeenko, V.A.; Aslanov, L.A.; et al. Cytotoxic activity of organotin carboxylates based on synthetic phenolic antioxidants and polycyclic bile acids. J. Organomet. Chem. 2020, 909, 121089. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Cordes, D.B.; Slawin, A.M.Z.; O’Hagan, D. Total Synthesis of a Reported Fluorometabolite from Streptomyces sp. TC1 Indicates an Incorrect Assignment. The Isolated Compound Did Not Contain Fluorine. J. Nat. Prod. 2014, 77, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Jaivel, N.; Uvarani, C.; Rajesh, R.; Velmurugan, D.; Marimuthu, P. Correction to Natural Occurrence of Organofluorine and Other Constituents from Streptomyces sp. TC1. J. Nat. Prod. 2015, 78, 343. [Google Scholar] [CrossRef]
- Fucke, K.; Myz, S.A.; Shakhtshneider, T.P.; Boldyreva, E.V.; Griesser, U.J. How good are the crystallisation methods for co-crystals? A comparative study of piroxicam. New J. Chem. 2012, 36, 1969. [Google Scholar] [CrossRef]
- Dickinson, J.A.; Joireman, P.W.; Randall, R.W.; Robertson, E.G.; Simons, J.P. Conformational Structures of 3-Phenyl-1-propionic Acid, Its p -Hydroxy Derivative, and Its Hydrated Clusters. J. Phys. Chem. A 1997, 101, 513–521. [Google Scholar] [CrossRef]
- Martinez, S.J.; Alfano, J.C.; Levy, D.H. The electronic spectroscopy of tyrosine and phenylalanine analogs in a supersonic jet: Acidic analogs. J. Mol. Spectrosc. 1991, 145, 100–111. [Google Scholar] [CrossRef]
- Joireman, P.W.; Kroemer, R.T.; Pratt, D.W.; Simons, J.P. Conformationally induced rotation of a molecular electronic transition moment. J. Chem. Phys. 1996, 105, 6075–6077. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Proceedings of the International Conference on Computational Science 2003, Petersburg, Russia, 2–4 June 2003; pp. 142–151. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01 2013; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 7, 3814. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Chopra, D.; Row, T.N.G. Role of organic fluorine in crystal engineering. CrystEngComm 2011, 13, 2175. [Google Scholar] [CrossRef]
- Dey, D.; Bhandary, S.; Thomas, S.P.; Spackman, M.A.; Chopra, D. Energy frameworks and a topological analysis of the supramolecular features in in situ cryocrystallized liquids: Tuning the weak interaction landscape via fluorination. Phys. Chem. Chem. Phys. 2016, 18, 31811–31820. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, B.; Spiteller, M. CCDC 900046: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2017. [Google Scholar]
- Cody, V.; Erman, M.; De Jarnette, F.E. Crystal Structure of the Active Thyroxine Analogue, 3,5-Diiodothyropropionic Acid (3-(4-Hydroxy-phenoxy-3,5-di-iodophenyl) Propanoic Acid). J. Chem. Res. 1977, 126, 1444. [Google Scholar]
- Ramesh, P.; Sreenivasulu, C.; Kishore, D.R.; Srinivas, D.; Gorantla, K.R.; Mallik, B.S.; Satyanarayana, G. Recyclable Aliphatic Nitrile-Template Enabled Remote meta -C–H Functionalization at Room Temperature. J. Org. Chem. 2022, 87, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Deepa, P.; Vijay Solomon, R.; Angeline Vedha, S.; Kolandaivel, P.; Venuvanalingam, P. The nature of hydrogen bonding in R 2 2 (8) crystal motifs—A computational exploration. Mol. Phys. 2014, 112, 3195–3205. [Google Scholar] [CrossRef]
- Allen, F.H.; Samuel Motherwell, W.D.; Raithby, P.R.; Shields, G.P.; Taylor, R. Systematic analysis of the probabilities of formation of bimolecular hydrogen-bonded ring motifs in organic crystal structures. New J. Chem. 1999, 23, 25–34. [Google Scholar] [CrossRef]
- Spassov, S.L.; Simova, S.D. Conformational analysis of 3-arylpropanoic acids and their methyl esters by 1H nuclear magnetic resonance spectroscopy. J. Chem. Soc. Perkin Trans. 2 1978, 7, 1113. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical formula | C11H8F6O2 |
| Formula weight | 286.17 |
| Crystal system | Monoclinic |
| space group | P21/c |
| a, b, c (Å) | 5.00408(19), 13.7194(5), 16.6021(5) |
| α, β, γ (°) | 90, 97.603(3), 90 |
| V (Å) | 1129.76(7) |
| Z | 4 |
| Dcalc (g∙cm−3) | 1.682 |
| μ (mm−1) | 1.625 |
| F (000) | 576 |
| Crystal size (mm) | 0.12 × 0.06 × 0.04 |
| Reflections collected | 4063 |
| Unique reflections | 2090 |
| Reflections I > 2σ(I) | 1793 |
| Rint | 0.0197 |
| Restraints/parameters | 0/177 |
| Goodness-of-fit | 1.051 |
| R1, wR2 (I > 2σ(I)) | 0.0340, 0.0820 |
| R1, wR2 (all data) | 0.0407, 0.0869 |
| Max. peak/hole (e∙A−3) | 0.304, −0.291 |
| D−H∙∙∙A | D−H | H∙∙∙A | D∙∙∙A | <(D−H∙∙∙A) |
|---|---|---|---|---|
| O2−H2∙∙∙O1 i | 0.87(2) | 1.794(1) | 2.655(1) | 173.4(1) |
| C2−H2A∙∙∙F3 ii | 0.95 | 2.764 | 3.213(2) | 109.8 |
| C10−H10A∙∙∙O2 ii | 0.99 | 2.672 | 3.577(2) | 152.1 |
| C6−H6∙∙∙F6 ii | 0.95 | 2.755 | 3.281(2) | 115.8 |
| C10−H10A∙∙∙O1 iii | 0.99 | 2.840 | 3.417(2) | 117.9 |
| C10−H10B∙∙∙F5 iv | 0.99 | 2.517 | 3.439(2) | 154.8 |
| C4 iv−H4 iv∙∙∙O2 | 0.95 | 2.606 | 3.489(2) | 154.9 |
| C6 i−H6 i∙∙∙F1 iv | 0.95 | 2.556 | 3.485(2) | 165.9 |
| C2 i−H2A i∙∙∙F4v | 0.95 | 2.581 | 3.505(2) | 164.3 |
| Molecule Paired to Molecule 0 | N | Sym op | R | Eele | Epol | Edis | Erep | Etot |
|---|---|---|---|---|---|---|---|---|
| a | 1 | −x + 1, −y + 1, −z + 1 | 9.26 | −115.6 | −25.3 | −15.0 | 139.3 | −67.9 |
| b | 2 | x − 1, y, z; x + 1, y, z | 5.00 | −0.8 | −2.0 | −44.3 | 20.7 | −28.1 |
| c | 2 | −x + 1, y − , −z + ; −x + 1, y + , −z + | 7.23 | −8.0 | −0.8 | −19.9 | 11.5 | −19.3 |
| d | 1 | −x, −y + 1, −z + 1 | 9.96 | −8.7 | −2.1 | −15.9 | 9.2 | −19.0 |
| e | 2 | −x, y − , −z + ; −x, y + , −z + | 7.41 | −4.2 | −1.0 | −21.9 | 11.2 | −17.4 |
| f | 2 | ; x, −, z − | 8.52 | −0.2 | −0.5 | −12.6 | 5.2 | −8.4 |
| g | 1 | −x, −y, −z | 10.13 | −1.4 | −0.1 | −3.4 | 0.3 | −4.3 |
| h | 2 | ; | 9.31 | 0.2 | −0.2 | −3.4 | 0.1 | −2.8 |
| CSD Code | Substituent Pattern | Space Group | Molecule | Side-Chain Dihedral Angles 1 | αrsc 2 | Reference | ||
|---|---|---|---|---|---|---|---|---|
| θ1 | θ2 | θ3 | ||||||
| YASFUV | 3-OMe | P21/a | A | 0 | 180 | 0 | 0 | [19] |
| YASFUV | 3-OMe | P21/a | B | 0 | 180 | 0 | 0 | [19] |
| CUQBEW | 4-C≡C−o-Tol | P21/n | A | −178 | 177 | −2 | 1 | [24] |
| BOPSOO | 2-Me | P21/c | A | 180 | −178 | 0 | 1 | [23] |
| CPPROP | 4-Cl | P21/a | A | 180 | 180 | 3 | 2 | [21] |
| VOQLUJ | 4-CF3 | A | 0 | 177 | 5 | 3 | [25] | |
| WIKRUE | 4-OCH2CO2H | P1 | A | 180 | −177 | 2 | 4 | [27] |
| YASFIJ01 | unsubstituted | P21/n | A | 171 | 180 | 4 | 5 | [15] |
| YARQIU | 2,4-bis-Me | P21/c | A | −178 | 177 | −3 | 5 | [30] |
| YASFIJ02 | unsubstituted | P21/n | A | −170 | 179 | 1 | 10 | [55] |
| YABJUI | 2-OMe | P21/c | A | 178 | −171 | 167 | 10 | [26] |
| YASFIJ | unsubstituted | P21/a | A | 177 | −170 | −177 | 11 | [19] |
| MOWZEG | 3,4-bis-Ph | A | −18 | −179 | −5 | 20 | [31] | |
| DITHPA10 | 3,5-bis-I, 4-OPh(4OH) | P21/c | A | −81 | −177 | 65 | 23 | [56] |
| VOXHOF | 2-OH | P21/c | A | 75 | −174 | 118 | 34 | [20] |
| YABJOC | 2-OH, 4-Me | P21/c | A | 142 | −166 | 71 | 40 | [26] |
| YASFOP | 3-Me | P21/c | A | −96 | 173 | −55 | 41 | [19] |
| YASFOP | 3-Me | P21/c | B | 144 | −151 | 166 | 42 | [19] |
| AFUFIS | 3,4-bis-OH | P21/c | A | −70 | 179 | −175 | 67 | [32] |
| YUYGEE | 4-OH | P21/c | A | 113 | 178 | −2 | 68 | [22] |
| YUYGEE01 | 4-OH | P21/c | A | −113 | −178 | 1 | 68 | [28] |
| YASFIJ | unsubstituted | P21/a | B | −100 | 156 | −173 | 77 | [19] |
| YOLPEW | 3,5-bis-tBu, 4-F | P21/c | A | −91 | −178 | 20 | 77 | [35] |
| JUBKUP | 3,5-bis-tBu, 4-OH | P21/c | A | −90 | −178 | 19 | 77 | [33] |
| UHUGUB | 2,5-OMe | C2/c | A | 84 | −172 | 20 | 79 | [29] |
| 3,5-bis-CF3 | P21/c | A | 76 | 67 | 17 | 84 | this work | |
| YASFIJ01 | unsubstituted | P21/n | B | 80 | −178 | 3 | 85 | [15] |
| LIWJEI | 3,5-bis-tBu, 4-F | P21/c | A | 88 | −178 | 6 | 86 | [34] |
| YASFIJ02 | unsubstituted | P21/n | B | 93 | 177 | −2 | 88 | [55] |
| YABKAP | 4-OMe | P21/n | A | 86 | 175 | 16 | 88 | [26] |
| VASDOM | 3-(CH=CH2CO2H), 4-OMe | C2/c | A | 100 | 180 | −14 | 88 | [57] |
| hydrocinnamic Acid | 3,5-Bistrifluoromethylhydrocinnamic Acid | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Side-Chain Dihedral Angles | Side-Chain Dihedral Angles | ||||||||||
| ΔG [kJ/mol] | θ1 | θ2 | θ3 | ΔG [kJ/mol] | θ1 | θ2 | θ3 | ||||
| 1 | trans-1 (c,ap,sp) | 0.0 | 89 | 180 | 0 | 1 | trans-1 (c,ap,sp) | 0.0 | 90 | 180 | 0 |
| 2 | trans-flat (sp,ap,sp) | 2.7 | 0 | 180 | 0 | 2 | gauche-1 (c,sc,sp) | 3.3 | 93 | −74 | −17 |
| 3 | trans-2 (c,ap,c) | 3.6 | 89 | −178 | 93 | 3 | trans-2 (c,ap,c) | 4.3 | 90 | −178 | 92 |
| 4 | gauche-1 (c,sc,sp) | 5.0 | 92 | −74 | −24 | 4 | trans-flat (sp,ap,sp) | 5.6 | 0 | 180 | 0 |
| 5 | gauche-2 (c,sc,ac) | 8.3 | 99 | −65 | 133 | 5 | gauche-2 (c,sc,ac) | 7.8 | 100 | −63 | 137 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipiński, P.F.J.; Łyczko, K. Structural Analysis of 3,5-Bistrifluoromethylhydrocinnamic Acid. Crystals 2024, 14, 342. https://doi.org/10.3390/cryst14040342
Lipiński PFJ, Łyczko K. Structural Analysis of 3,5-Bistrifluoromethylhydrocinnamic Acid. Crystals. 2024; 14(4):342. https://doi.org/10.3390/cryst14040342
Chicago/Turabian StyleLipiński, Piotr F. J., and Krzysztof Łyczko. 2024. "Structural Analysis of 3,5-Bistrifluoromethylhydrocinnamic Acid" Crystals 14, no. 4: 342. https://doi.org/10.3390/cryst14040342
APA StyleLipiński, P. F. J., & Łyczko, K. (2024). Structural Analysis of 3,5-Bistrifluoromethylhydrocinnamic Acid. Crystals, 14(4), 342. https://doi.org/10.3390/cryst14040342