
Crystallization of Polymers with a Reduced Density of Entanglements


Abstract
:1. Introduction
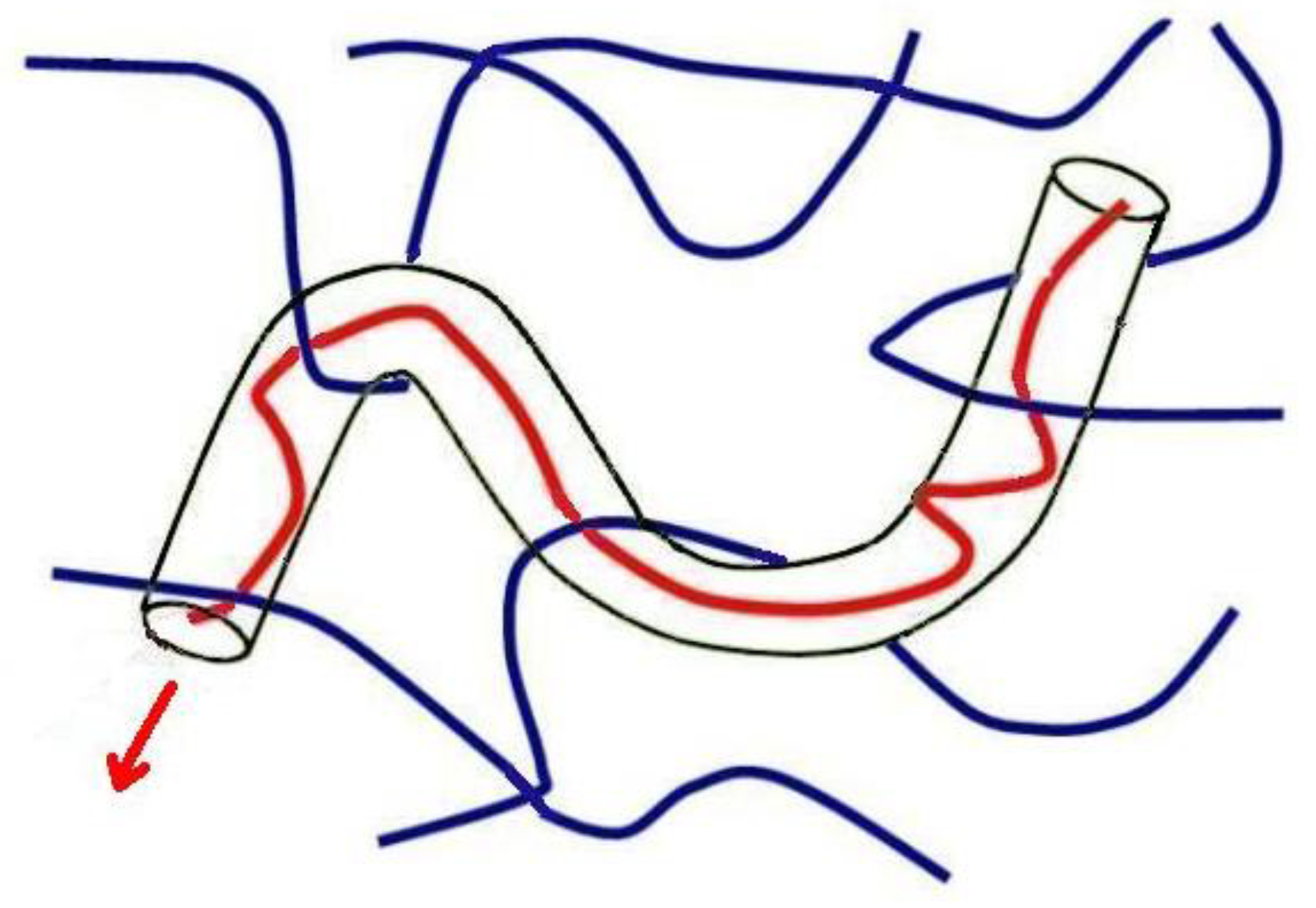
2. Entanglements of Macromolecules
3. Basic Facts about Polymer Crystallization
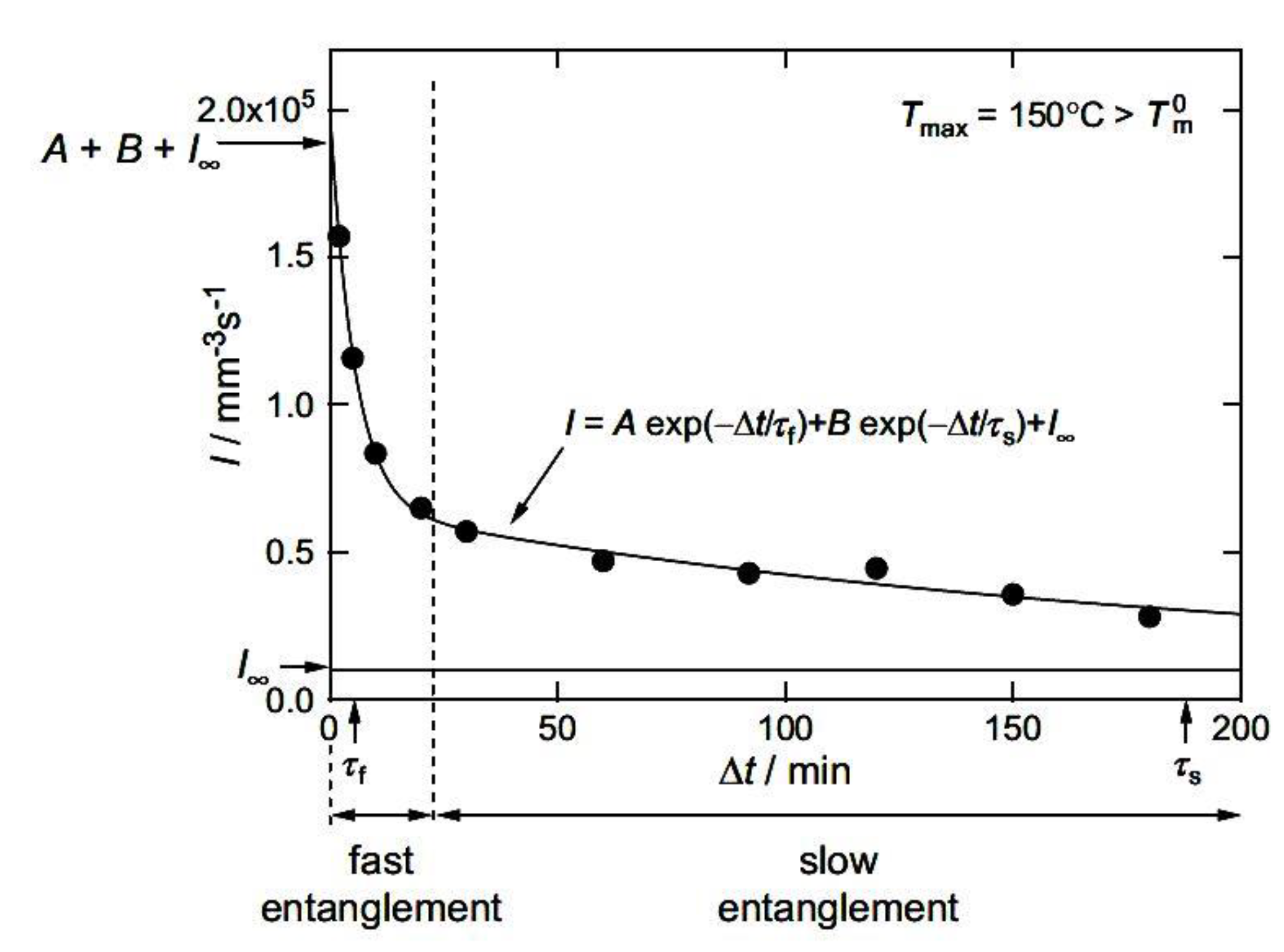
4. Crystallization during Process of Disentangling
5. Non-Isothermal Crystallization of Disentangled and Entangled Polymers
5.1. General Remarks
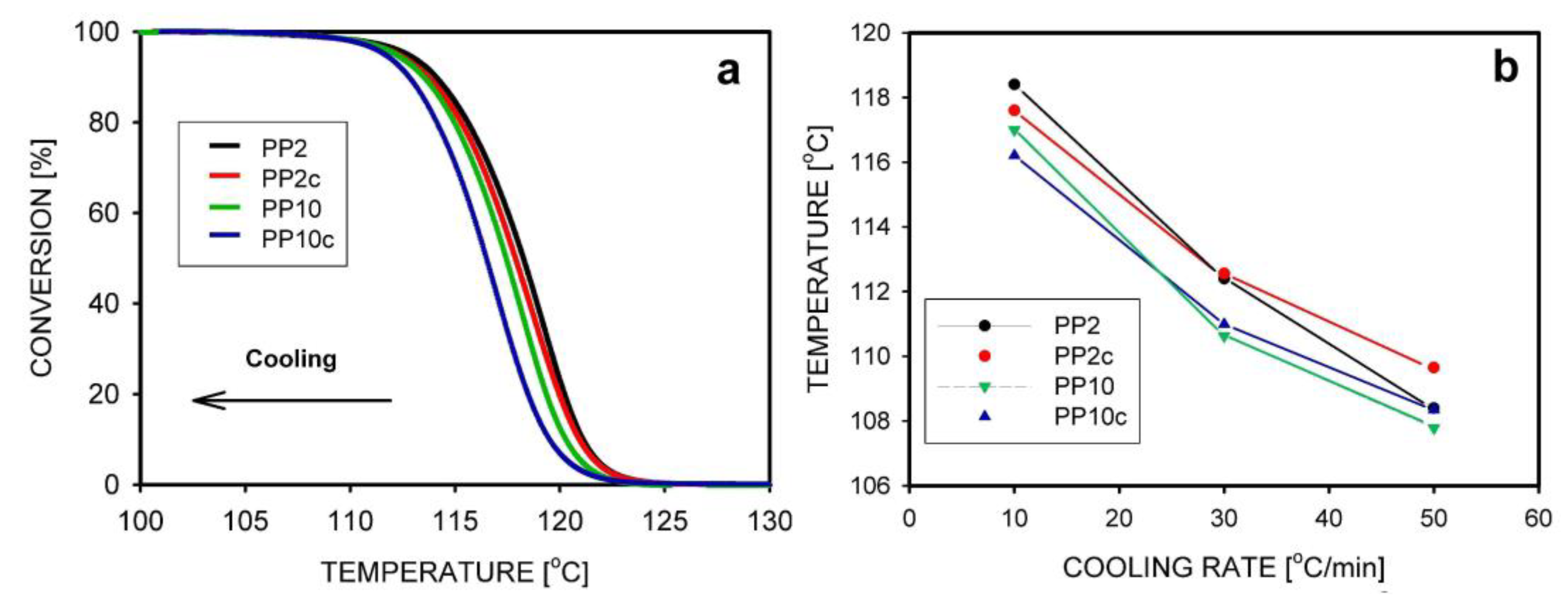
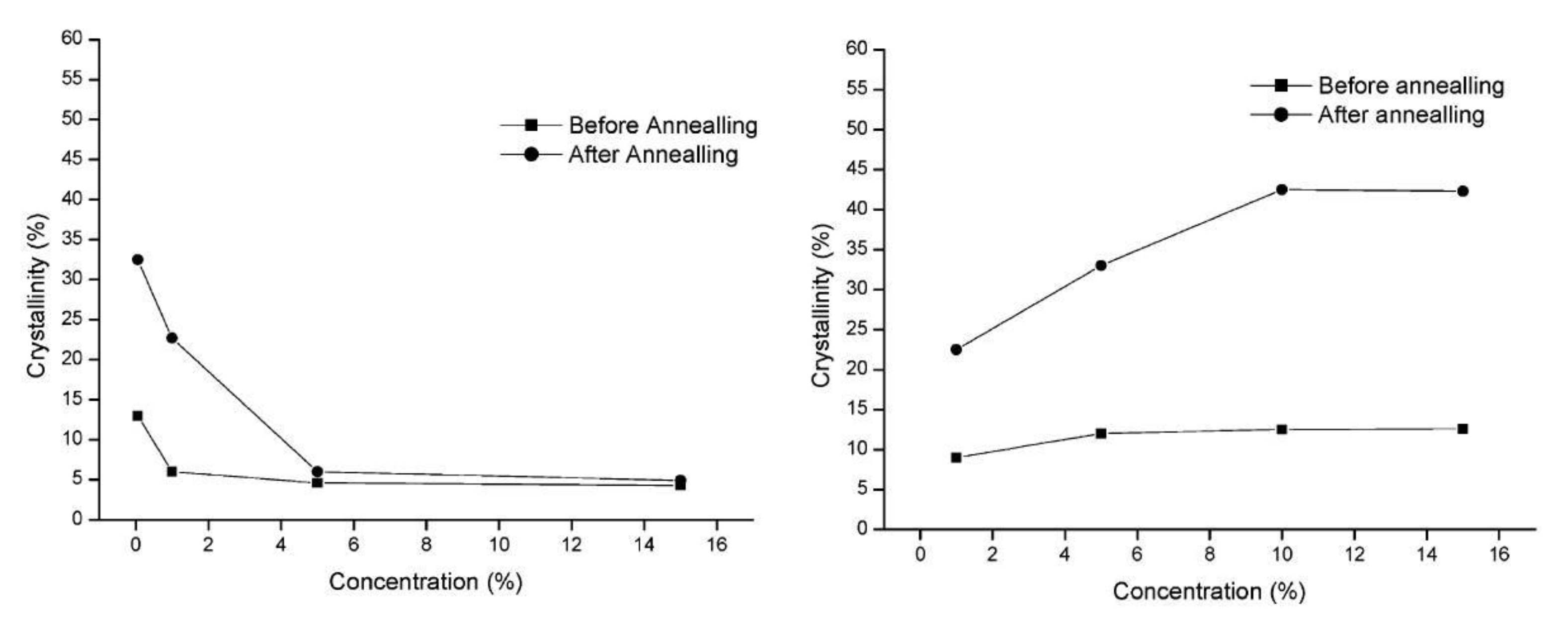
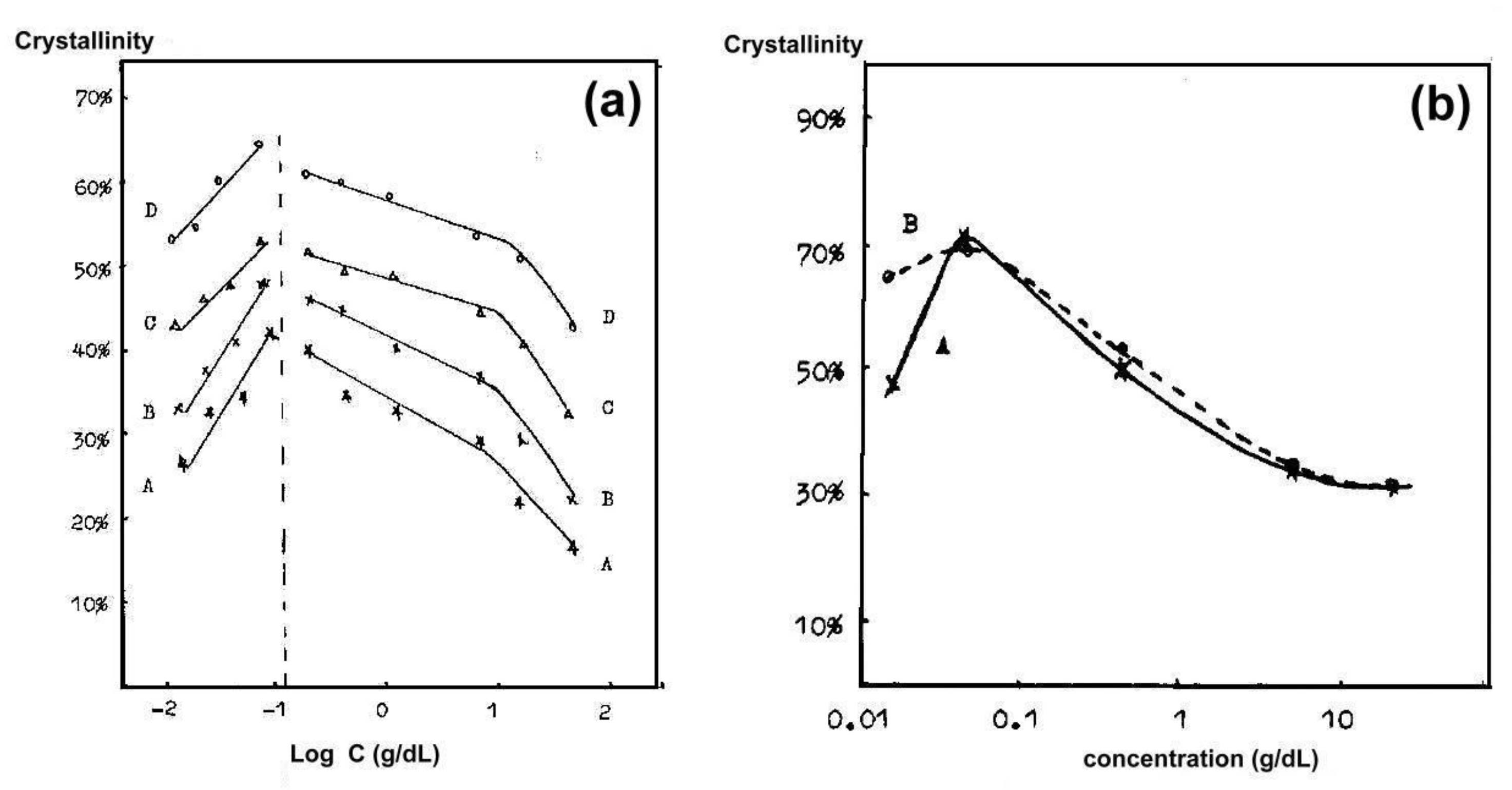
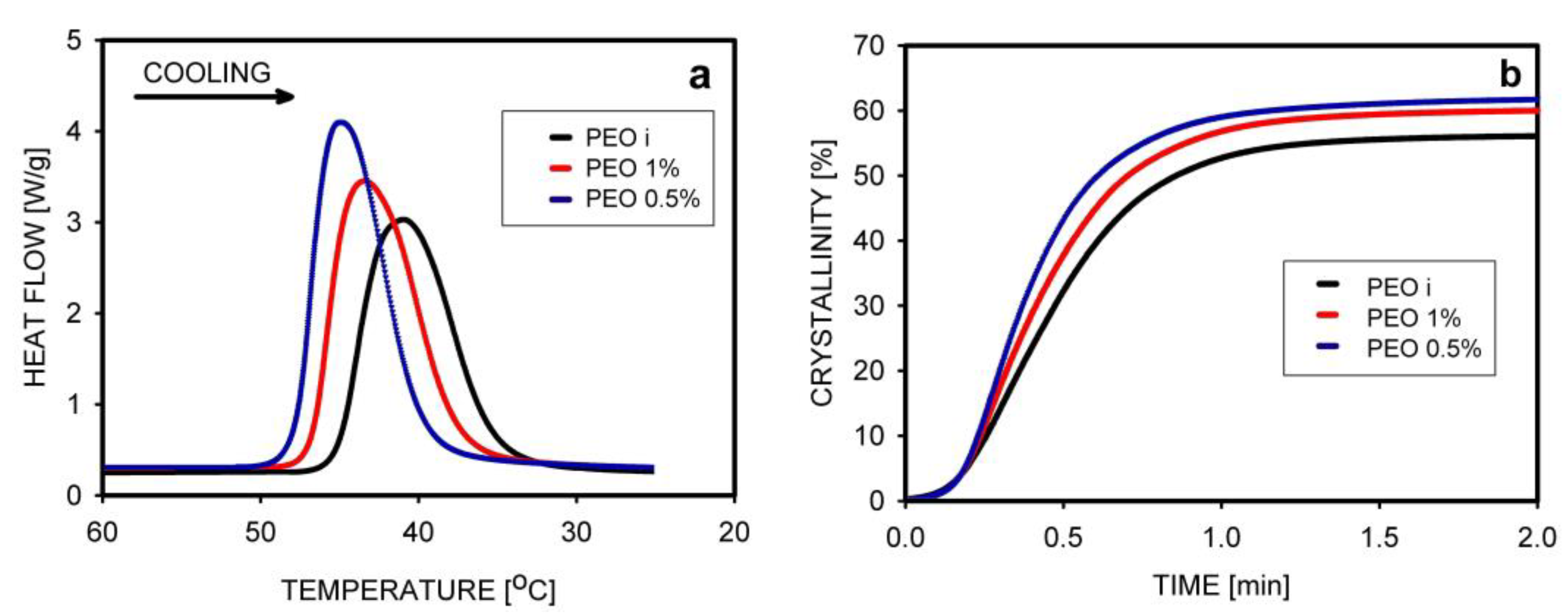
5.2. Crystallization Kinetics, Crystallization Temperature, and the Degree of Crystallinity
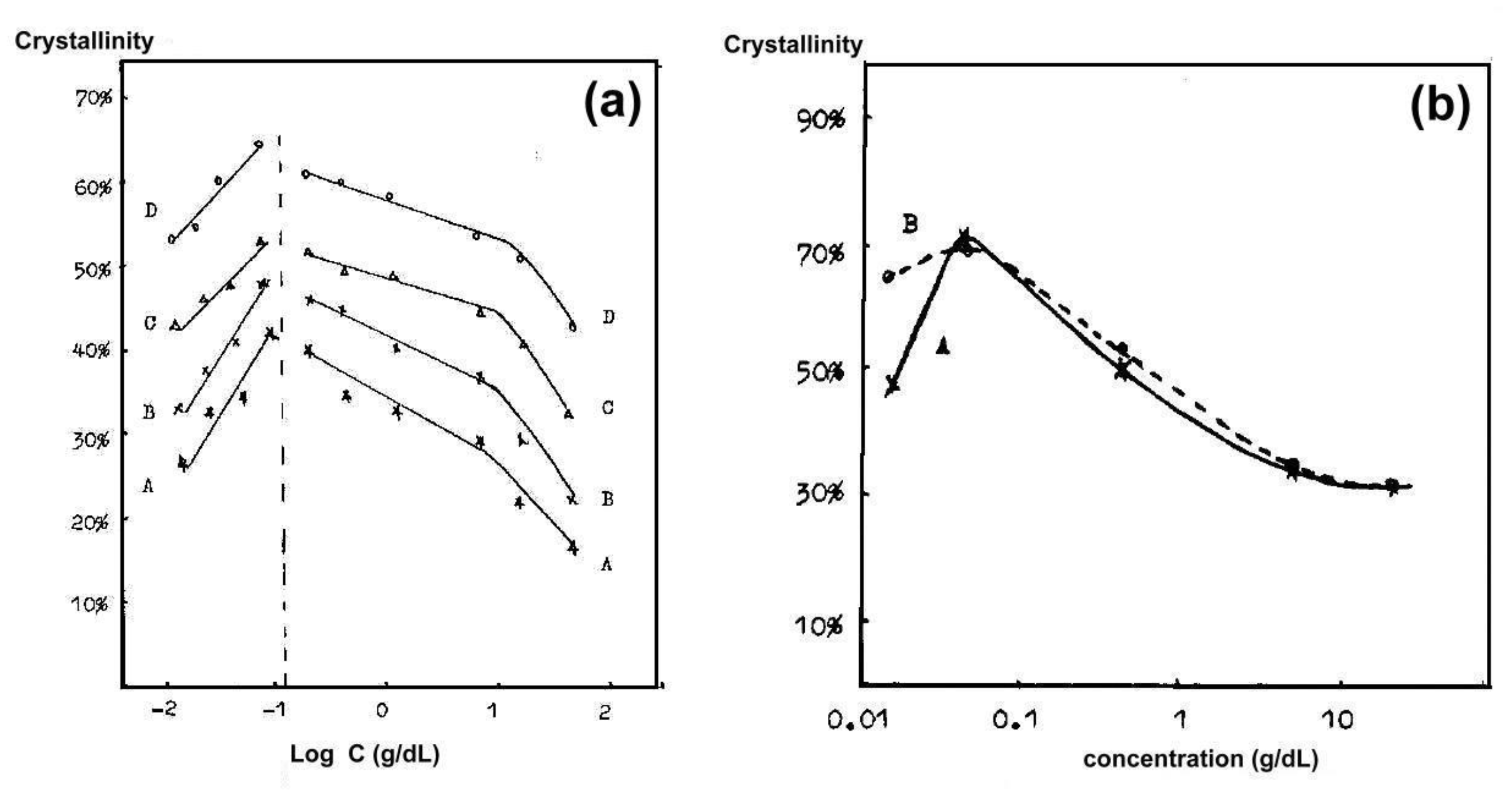
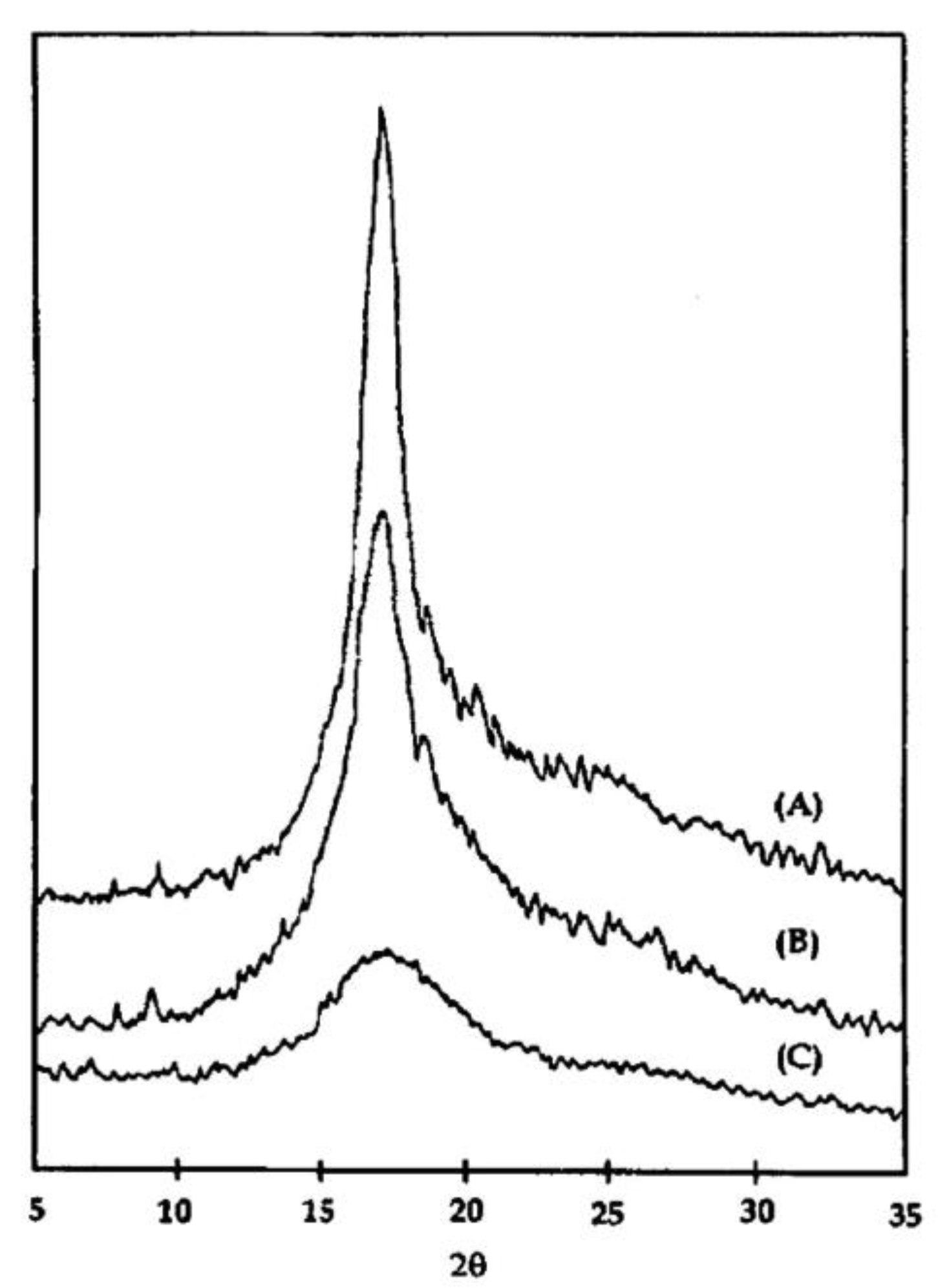
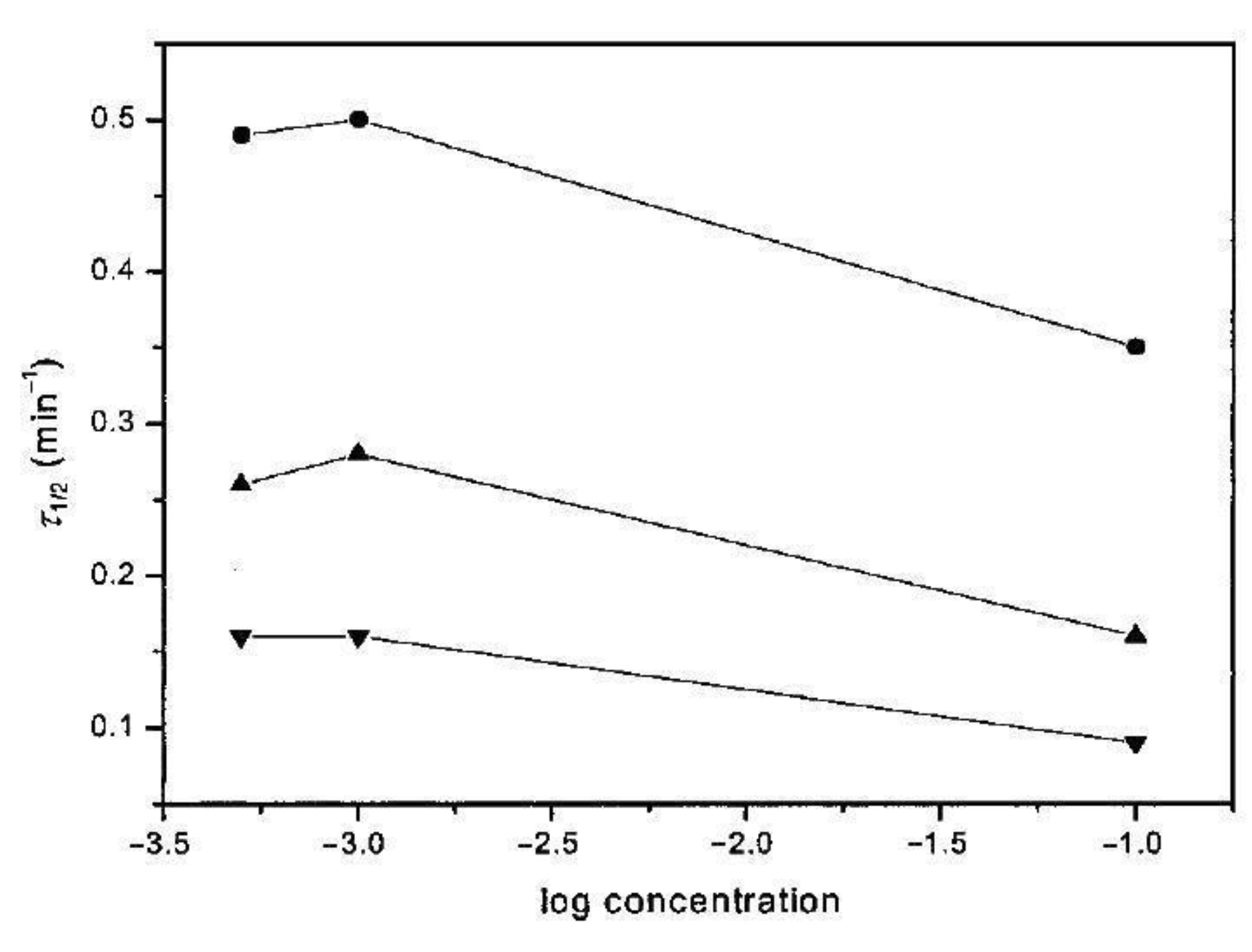
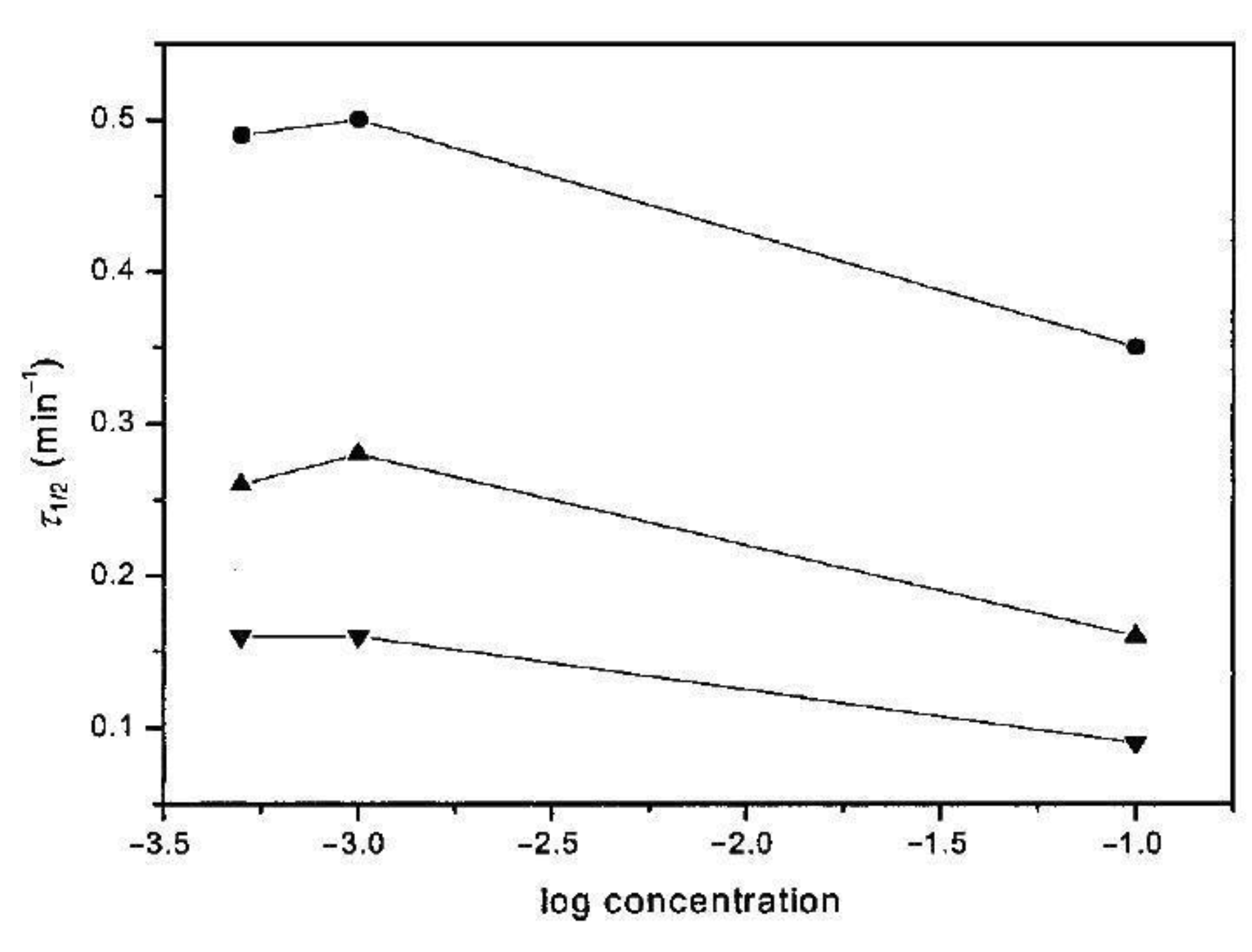
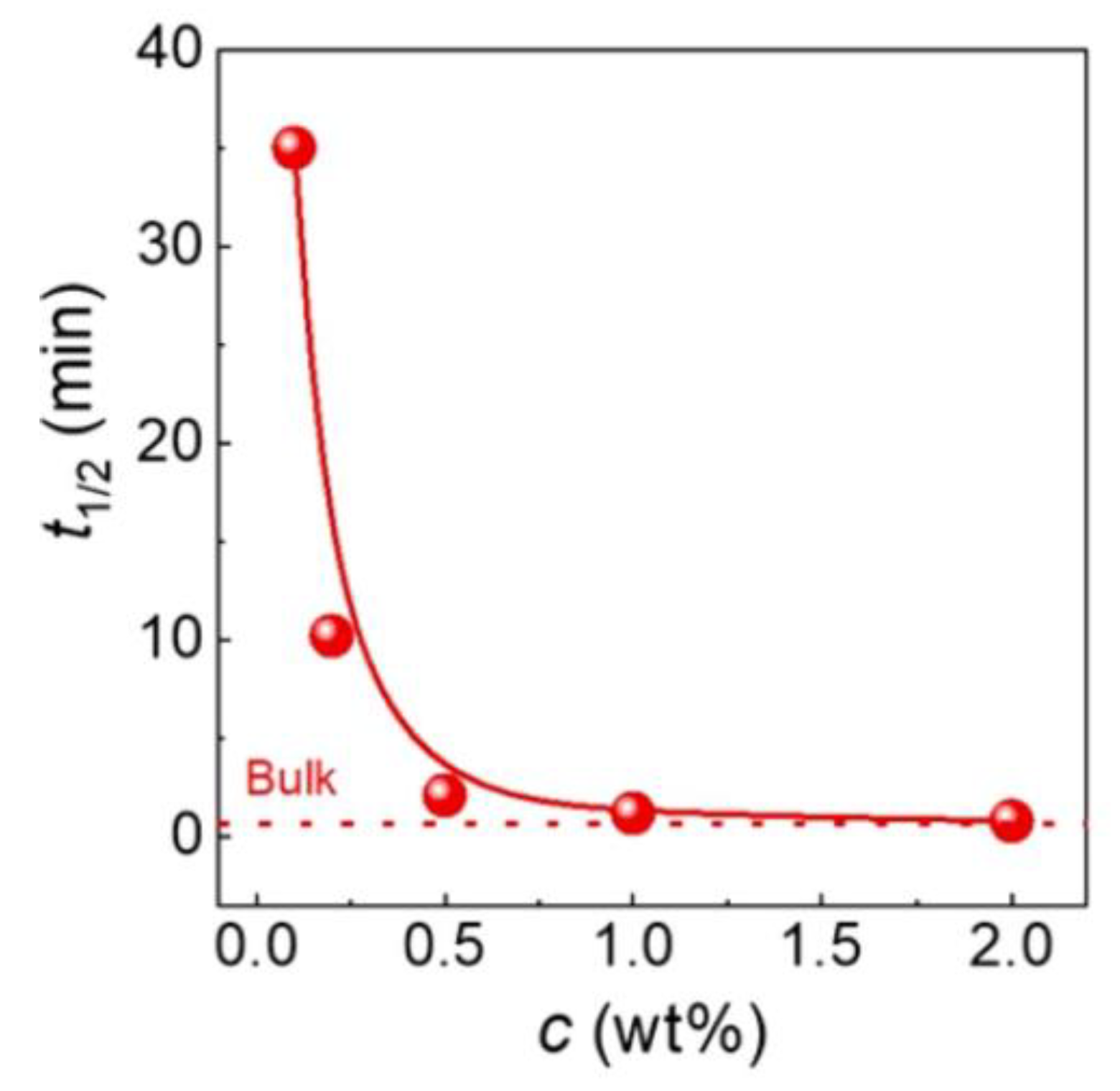
6. Isothermal Crystallization of Partially Disentangled Polymers
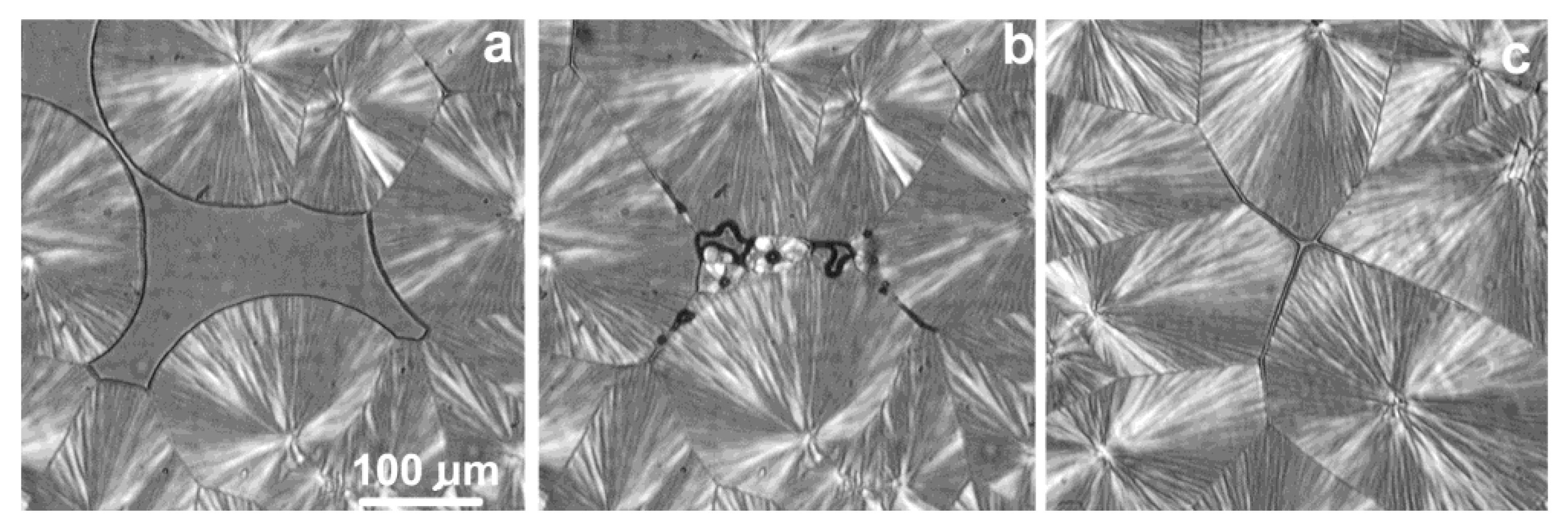
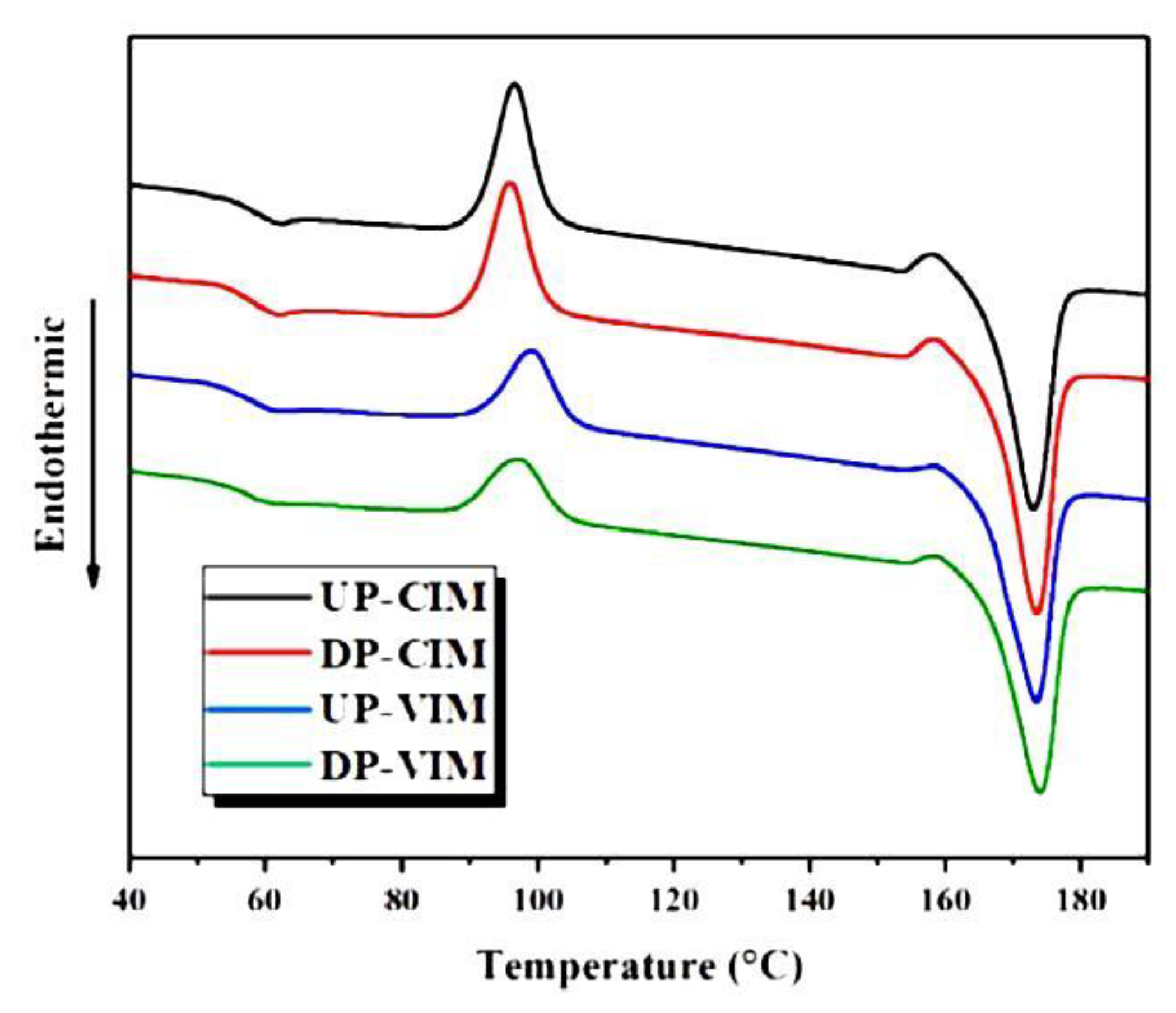
6.1. Formation of Crystal Structure over Time
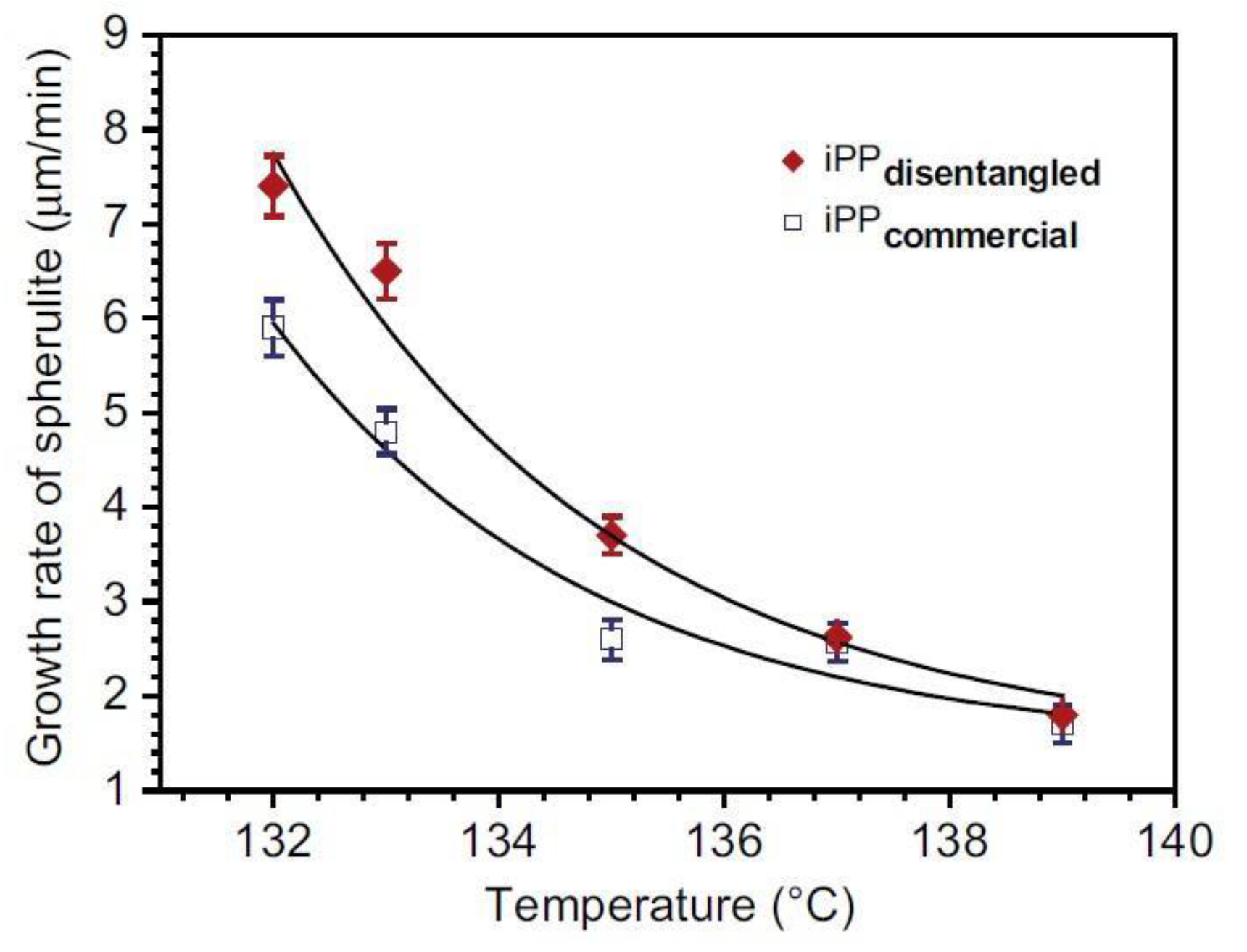
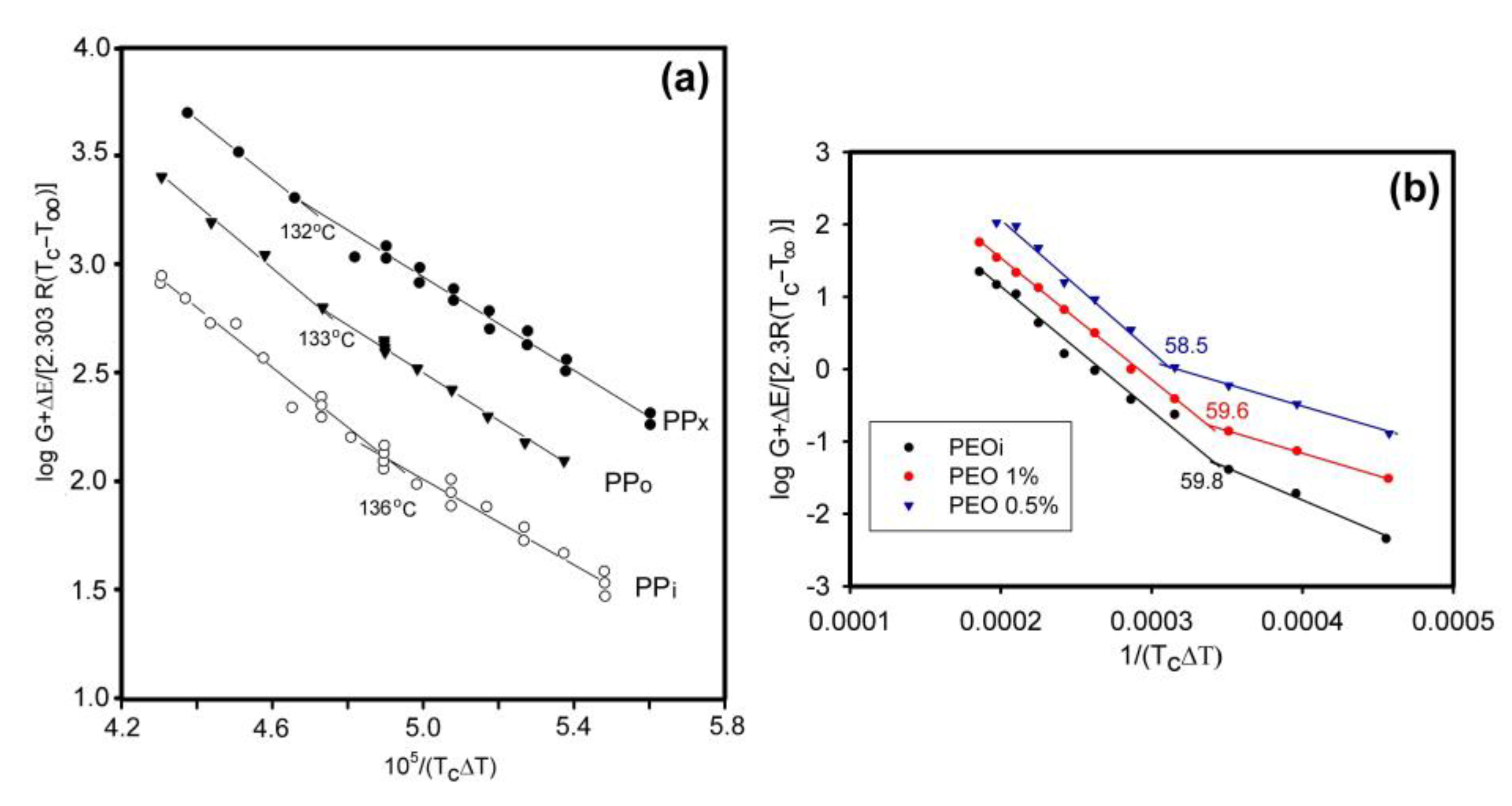
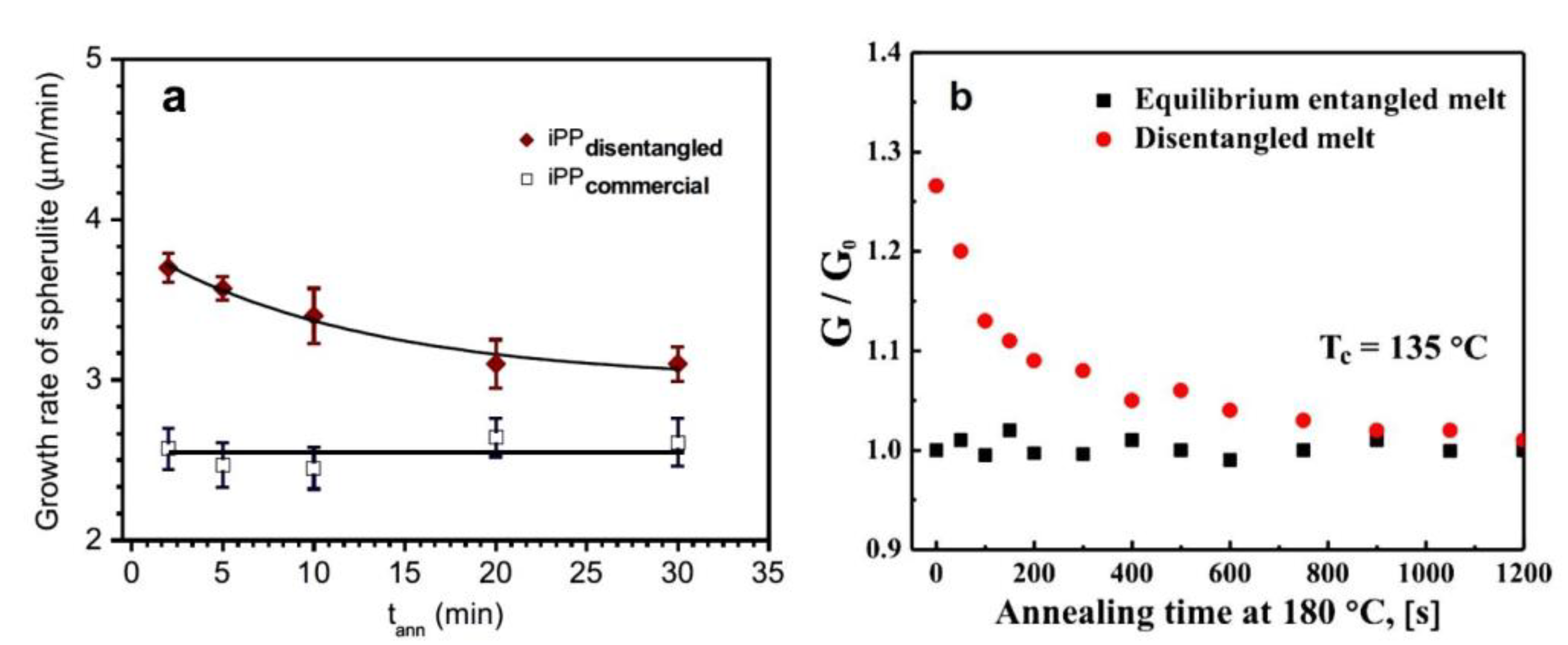
6.2. Spherulite Growth Measurements
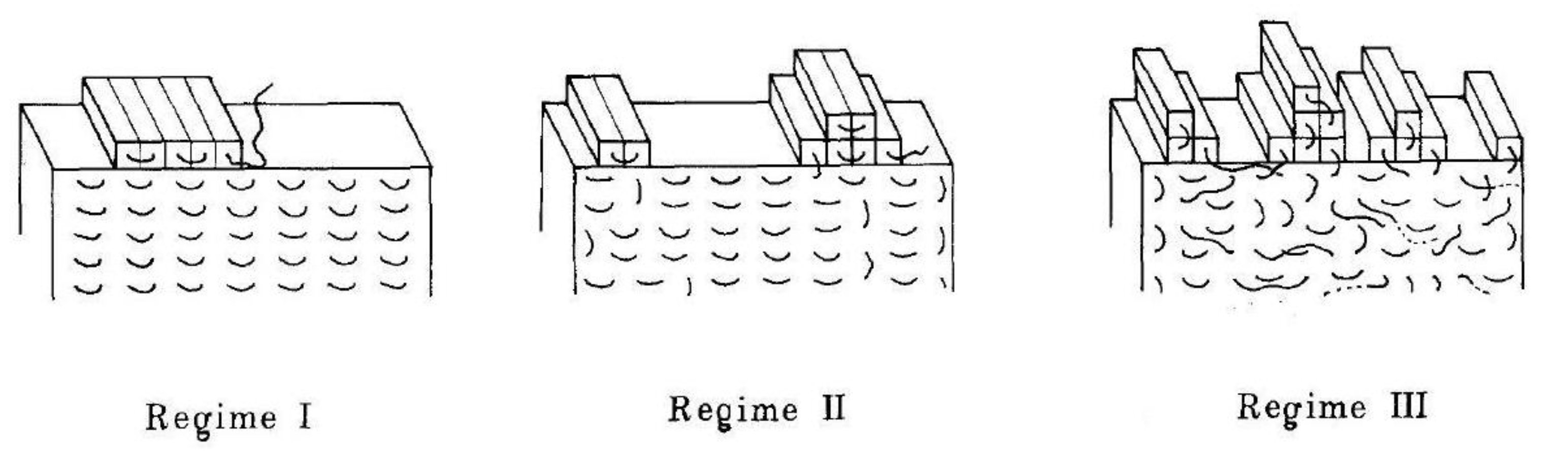
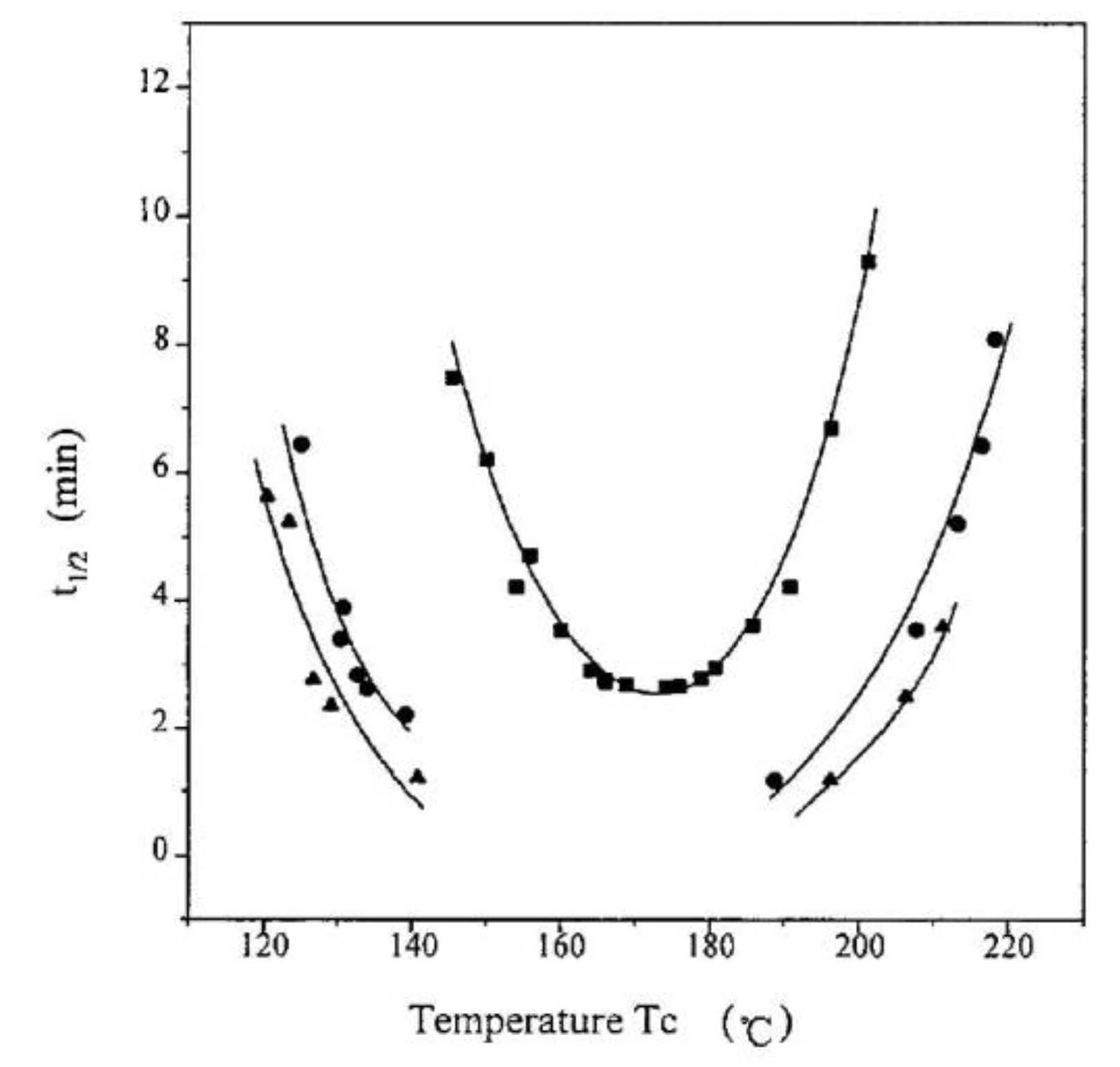
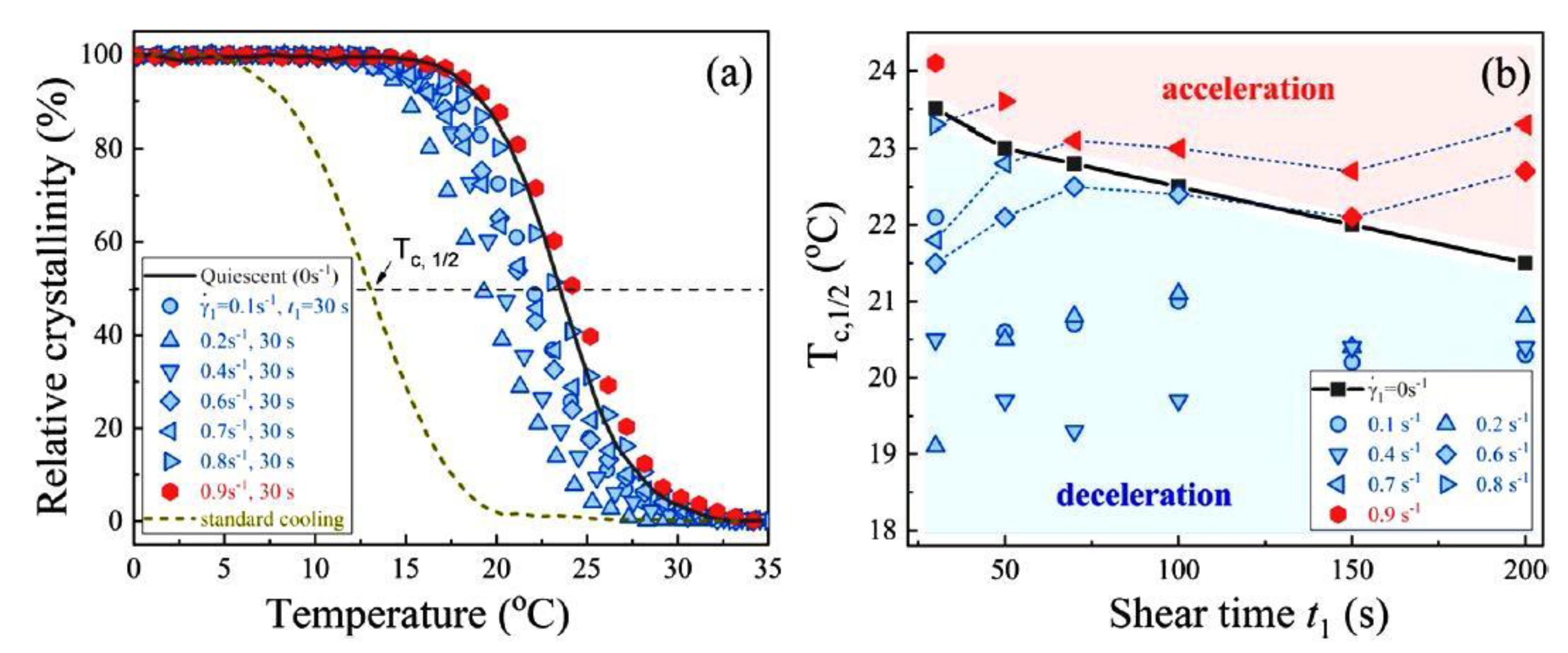
6.3. Regimes of Crystallization
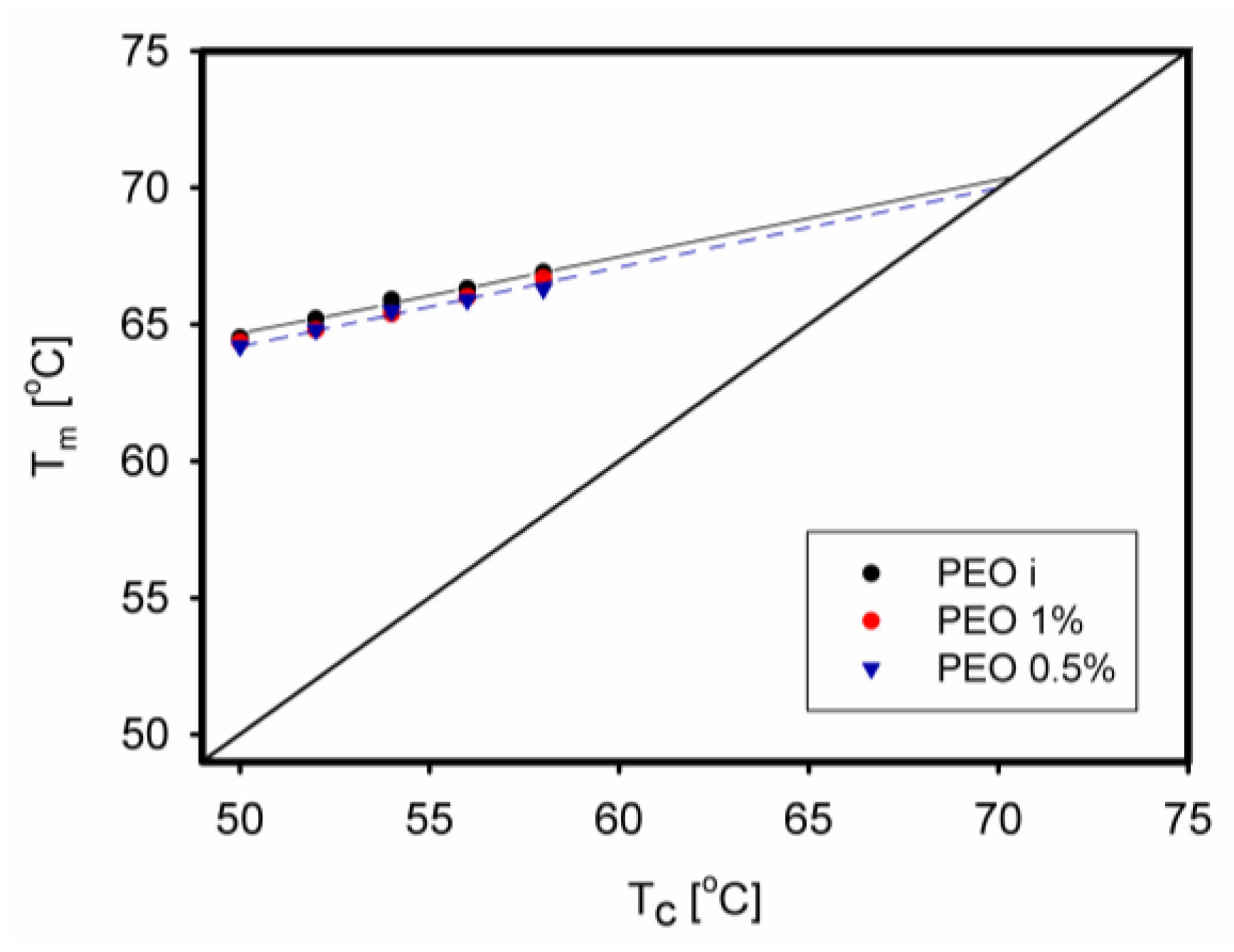
6.4. Other Aspects of Isothermal Crystallization
6.5. Molecular Dynamics Simulations
6.6. Cold Crystallization in Isothermal Conditions
6.7. Conclusions about Isothermal Crystallization
7. Re-Entangling of Polymer Chains
8. Concluding Remarks
Funding
Data Availability Statement
Conflicts of Interest
References
- Berry, G.C.; Fox, T.G. The viscosity of polymers and their concentrated solutions. Adv. Polym. Sci. 1968, 5, 261–357. [Google Scholar]
- Wool, R.P. Polymer entanglements. Macromolecules 1993, 26, 1564–1569. [Google Scholar] [CrossRef]
- Watanabe, H. Viscoelasticity and dynamics of entangled polymers. Prog. Polym. Sci. 1999, 24, 1253–1403. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Ries, M.E.; Baughman, T.W.; Henke, A.; Matloka, P.P. Chain entanglements in polyethylene melts. Why is it studied again? Macromolecules 2013, 46, 541–547. [Google Scholar] [CrossRef]
- Bueche, F. Viscosity, self-diffusion and allied effects in solid polymers. J. Chem. Phys. 1952, 20, 1959–1964. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity, 3rd ed.; Clarendon Press: Oxford, UK, 1975; pp. 64–65. [Google Scholar]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley &Sons: New York, NY, USA, 1981; p. 552. [Google Scholar]
- Zulli, F.; Giordano, M.; Andreozzi, L. Onset of entanglement and reptation in melts of linear homopolymers: Consistent rheological simulations of experiments from oligomers to high polymers. Rheol. Acta 2015, 54, 185–205. [Google Scholar] [CrossRef]
- Ward, M.; Sweeney, J. Mechanical Properties of Solid Polymers; John Wiley & Sons Ltd.: Chichester, UK, 2013; p. 72. [Google Scholar]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Eckstein, A.; Suhm, J.; Friedrich, C.; Maier, R.; Sassmannshausen, J.; Bochmann, M.; Mulhaupt, R. Determination of plateau moduli and entanglement molecular weights of isotactic, syndiotactic, and atactic polypropylenes synthesized with metallocene catalysts. Macromolecules 1998, 31, 1335–1340. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Colby, R.H. Chain dimensions and entanglement spacing. In Physical Properties of Polymers Handbook, 2nd ed.; Mark, J.E., Ed.; Springer Science: New York, NY, USA, 2007; pp. 447–454. [Google Scholar]
- Pawlak, A. The Entanglements of macromolecules and Their Influence on the Properties of Polymers. Macromol. Chem. Phys. 2019, 220, 1900043. [Google Scholar] [CrossRef]
- Raju, V.R.; Menezes, E.V.; Marin, G.; Graessley, W.W.; Fetter, L.J. Concentration and molecular weight dependence of viscoelastic properties in linear and star polymers. Macromolecules 1981, 14, 1668–1676. [Google Scholar] [CrossRef]
- Wu, S. Chain structure and entanglement. J. Polym. Sci. Part B Polym. Phys. 1989, 27, 723–741. [Google Scholar] [CrossRef]
- Nobile, M.R.; Cocchini, F. Evaluation of molecular weight distribution from dynamic moduli. Rheol. Acta 2011, 40, 111–119. [Google Scholar] [CrossRef]
- Liu, C.; He, J.; van Ruymbeke, E.; Keunings, R.; Bailly, C. Evaluation of different methods for the determination of the plateau modulus and the entanglement molecular weight. Polymer 2006, 47, 4461–4479. [Google Scholar] [CrossRef]
- Rouse, P.E. A theory of the linear viscoelastic properties of dilute solutions of coiling polymers. J. Chem. Phys. 1953, 21, 1272–1280. [Google Scholar] [CrossRef]
- Doi, M. Viscoelastic and rheological properties. In Materials Science and Technology. A Comprehensive Treatment. Volume 12 Structure and Properties of Polymers; Cahn, R.W., Haasen, P., Kramer, E.J., Thomas, E.L., Thomas, E.L., Eds.; VCH Verlag: Weinheim, Germany, 1993; pp. 389–425. [Google Scholar]
- DeGennes, P.G. Reptation of polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. 4. Rheological properties. J. Chem. Soc. Faraday Trans. II 1979, 75, 38–54. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon: Oxford, UK, 1986. [Google Scholar]
- Pawlak, A.; Krajenta, J. Progress in Studies of Disentangled Polymers and Composites. J. Compos. Sci. 2023, 7, 521. [Google Scholar] [CrossRef]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Cao, J.; Gu, F.M.; Liu, Y.J.; Bi, S.G.; Bu, H.S. Single-chain single crystals. Macromol. Symp. 1997, 124, 89–109. [Google Scholar] [CrossRef]
- Sun, B.; Lu, Y.; Ni, H.; Wang, C. Highly crystallizable poly(ethylene terephthalate) prepared by freeze-extracting solutions. Polymer 1998, 39, 159–163. [Google Scholar] [CrossRef]
- Huang, Q.; Mednova, O.; Rasmussen, H.K.; Alvarez, N.J.; Skov, A.L.; Almdal, K.; Hassager, O. Concentrated polymer solutions are different from melts: Role of entanglement molecular weight. Macromolecules 2013, 46, 5026–5035. [Google Scholar] [CrossRef]
- Xie, Z.P.; Liu, D.; Zhu, P.P.; Yang, H.Y. Crystallization behavior of chain-disentangled poly(ethylene terephthalate). Acta Polym. Sin. 2010, 5, 522–529. [Google Scholar] [CrossRef]
- Zhou, D.; Li, L.; Che, B.; Cao, Q.; Lu, Y.; Xue, G. Metastable isotactic poly(methyl methacrylate) prepared by freeze-extracting solutions in poly(ethylene glycol). Macromolecules 2004, 37, 4744–4747. [Google Scholar] [CrossRef]
- Wang, X.H.; Liu, R.; Wu, M.; Wang, Z.; Huang, Y. Effect of chain disentanglement on melt crystallization behaviour of isotactic polypropylene. Polymer 2009, 50, 5824–5827. [Google Scholar] [CrossRef]
- Liu, X.T.; Bao, R.Y.; Li, Y.M.; Yang, W.; Xie, B.H.; Yang, M.B. Effect of chain entanglement on the melt-crystallization behavior of poly(L-lactide) acid. J. Polym. Res. 2016, 23, 164. [Google Scholar] [CrossRef]
- Bu, H.S.; Gu, F.M.; Bao, L.; Chen, M. Influence of entanglements on crystallization of macromolecules. Macromolecules 1998, 31, 7108–7110. [Google Scholar] [CrossRef]
- Kotliar, A.M.; Kumar, R.; Back, R.A. Rheology of Polymers that are initially in the disentangled state. J. Polym. Sci. Part B Polym. Phys. 1990, 28, 1033–1045. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, D.; Wang, X.; Xue, G. Crystallization and relaxation behavior of partially disentangled poly(vinyl chloride) prepared from large molecule solvent dioctyl phthalate. Macromolecules 2002, 35, 7089–7092. [Google Scholar] [CrossRef]
- Graessley, W.W. The entanglement concept in polymer rheology. Adv. Polym. Sci. 1974, 16, 1–179. [Google Scholar]
- Heo, Y.; Larson, R.G. Universal scaling of linear and nonlinear rheological properties of semidilute and concentrated polymer solutions. Macromolecules 2008, 41, 8903–8915. [Google Scholar] [CrossRef]
- Luo, C.F.; Kroger, M.; Sommer, J.U. Entanglements and Crystallization of Concentrated Polymer Solutions: Molecular Dynamics Simulations. Macromolecules 2016, 49, 9017–9025. [Google Scholar] [CrossRef]
- Plazek, D.J.; Raghupathi, N.; O’Rourke, V.M. New evidence for molecular entanglements. J. Polym. Sci. Part B Polym. Phys. 1980, 18, 1837–1846. [Google Scholar] [CrossRef]
- Tian, N.; Liu, D.; Wei, H.; Liu, Y.; Kong, J. Crystallization of polycaprolactone with reduced entanglement. Europ. Polym. J. 2018, 102, 38–44. [Google Scholar]
- Qian, R. Intermolecular excimer interactions in polymers. In Macromolecules; Benoit, H., Rempp, R., Eds.; Pergamon Press: Oxford, UK, 1982; pp. 139–154. [Google Scholar]
- Lippits, D.R.; Rastogi, S.; Talebi, S.; Bailly, C. Formation of entanglements in initially disentangled polymer melts. Macromolecules 2006, 39, 8882–8885. [Google Scholar] [CrossRef]
- Chen, P.; Yang, H.; Chen, T.; Li, W. Weakly entangled ultrahigh molecular weight polyethylene prepared via ethylene extrusion polymerization. Ind. Eng. Chem. Res. 2015, 54, 11024–11032. [Google Scholar] [CrossRef]
- Romano, D.; Tops, N.; Bos, J.; Rastogi, S. Correlation between thermal and mechanical response of nascent semicrystalline UHMWPEs. Macromolecules 2017, 50, 2033–2042. [Google Scholar] [CrossRef]
- Psarski, M.; Piorkowska, E.; Galeski, A. Crystallization of Polyethylene from Melt with Lowered Chain Entanglements. Macromolecules 2000, 33, 916–932. [Google Scholar] [CrossRef]
- Monasse, B. Nucleation and anisotropic crystalline growth of polyethylene under shear. J. Mater. Sci. 1995, 30, 5002–5012. [Google Scholar] [CrossRef]
- Rozanski, A.; Monasse, B.; Szkudlarek, E.; Pawlak, A.; Piorkowska, E.; Galeski, A.; Haudin, J.M. Shear-induced crystallization of isotactic polypropylene based nanocomposites with montmorillonite. Eur. Polym. J. 2009, 45, 88–101. [Google Scholar] [CrossRef]
- Chen, Q.; Fan, Y.; Zheng, Q. Rheological scaling and modeling of shear-enhanced crystallization rate of polypropylene. Rheol. Acta 2006, 46, 305–316. [Google Scholar] [CrossRef]
- Jiang, S.; An, L.; Jiang, B. Crystallization Kinetics in Shearing-Induced Oriented and Stretched Poly(ethylene oxide). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 656–665. [Google Scholar] [CrossRef]
- Nazari, B.; Rhoades, A.M.; Schaake, R.P.; Colby, R.H. Flow-Induced Crystallization of PEEK: Isothermal Crystallization Kinetics and Lifetime of Flow-Induced Precursors during Isothermal Annealing. ACS Macro Lett. 2016, 5, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Parisi, D.; Gohn, A.M.; Han, A.; Song, L.; Liu, Y.; Schaake, R.P.; Rhoades, A.M.; Colby, R.H. Flow-Induced Crystallization of Poly(ether ether ketone): Universal Aspects of Specific Work Revealed by Corroborative Rheology and X-ray Scattering Studies. Macromolecules 2020, 53, 10040–10050. [Google Scholar] [CrossRef]
- Paukszta, D.; Bednarek, W.H. In situ optical microscope studies at isotactic polypropylene crystallization induced by shear forces—A review. Polym. Test. 2018, 72, 238–243. [Google Scholar] [CrossRef]
- Nie, C.; Peng, F.; Cao, R.; Cui, K.; Sheng, J.; Chen, W.; Li, L. Recent progress in flow-induced polymer crystallization. J. Polym. Sci. 2022, 60, 3149–3175. [Google Scholar] [CrossRef]
- Sheng, J.; Chen, W.; Cui, K.; Li, L. Polymer crystallization under external flow. Rep. Prog. Phys. 2022, 85, 036601. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G. Flow induced crystallization. Chem. Soc. Rev. 2014, 43, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Efficient disentanglement of polycarbonate melts under complex shear field. Polymer 2020, 201, 122610. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Chen, J. Delay of re-entanglement kinetics by shear-induced nucleation precursors in isotactic poly-propylene melt. Polymer 2020, 210, 123000. [Google Scholar] [CrossRef]
- Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Investigating the disentanglement of long chain branched polypropylene under different shear fields. J. Appl. Polym. Sci. 2022, 139, 51642. [Google Scholar] [CrossRef]
- Kamkar, M.; Salehiyan, R.; Goudoulas, T.B.; Abbasi, M.; Saengow, C.; Erfanian, E.; Sadeghi, S.; Natale, G.; Rogers, S.A.; Giacomini, A.J.; et al. Large amplitude oscillatory shear flow: Microstructural assessment of polymeric systems. Prog. Polym. Sci. 2022, 132, 101580. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Zhou, N.-Q.; Liu, B.; Jin, G. Improved Mechanical Properties and Structure of Polypropylene Pipe Prepared under Vibration Force Field. J. Appl. Polym. Sci. 2009, 114, 3612–3620. [Google Scholar] [CrossRef]
- Ibar, J.P. Processing Polymer Melts under Rheo-Fluidification Flow Conditions, Part 1: Boosting Shear-Thinning by Add-ing Low Frequency Nonlinear Vibration to Induce Strain Softening. J. Macromol. Sci. B Phys. 2013, 52, 407–441. [Google Scholar] [CrossRef]
- An, F.Z.; Gao, X.Q.; Lei, J.; Deng, C.; Li, Z.M.; Shen, K.Z. Vibration assisted extrusion of polypropylene. Chin. J. Polym. Sci. 2015, 33, 688–696. [Google Scholar] [CrossRef]
- Nusser, K.; Schneider, G.J.; Pyckhout-Hintzen, W.; Richter, D. Viscosity decrease and reinforcement in polymer-silsesquioxane composites. Macromolecules 2011, 44, 7820–7830. [Google Scholar] [CrossRef]
- Chai, S.-C.; Xu, T.-Y.; Cao, X.; Wang, G.; Chen, Q.; Li, H.-L. Ultrasmall Nanoparticles Diluted Chain Entanglement in Polymer Nanocomposites. Chin. J. Polym. Sci. 2019, 37, 797–805. [Google Scholar] [CrossRef]
- Senses, E.; Ansar, S.M.; Kitchens, C.L.; Mao, Y.; Narayanan, S.; Natarajan, B.; Faraone, A. Small Particle Driven Chain Dis-entanglements in Polymer Nanocomposites. Phys. Rev. Lett. 2017, 118, 147801. [Google Scholar] [CrossRef]
- Romo-Uribe, A. Dispersion at Single Unit TiO2 Nanoparticles Reduced Tg, Induced Chain Disentanglement and Reduced Tensile Modulus in Waterborne Acrylic Coatings. Macromol. Mater. Eng. 2021, 306, 200059. [Google Scholar] [CrossRef]
- Sui, Y.; Yui, Y.; Wei, P.; Cong, C.; Meng, X.; Ye, H.-M.; Zhou, Q. Nanoscale effects of TiO2 nanoparticles on the rheological behaviors of ultra-high molecular weight polyethylene (UHMWPE). Soft Matter 2023, 19, 5459–5467. [Google Scholar] [CrossRef] [PubMed]
- Krajenta, J.; Polinska, M.; Lapienis, G.; Pawlak, A. The crystallization of poly(ethylene oxide) with limited density of macromolecular entanglements. Polymer 2020, 197, 122500. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physcial Polymer Science; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Chapter 6. [Google Scholar]
- Wunderlich, B. Macromolecular Physics; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Mandelkern, L. Crystallization of Polymers. Vol 2. Kinetics and Mechanisms; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Phillips, P.J. Polymer crystals. Rep. Prog. Phys. 1990, 53, 549–604. [Google Scholar] [CrossRef]
- Michell, R.M.; Mugica, A.; Zubitur, M.; Muller, A.J. Self-Nucleation of Crystalline Phases within Homopolymers, Polymer Blends, Copolymers, and Nanocomposites. Adv. Polym. Sci. 2017, 276, 215–256. [Google Scholar]
- Sangroniz, L.; Cavallo, D.; Müller, A.J. Self-nucleation effects on polymer crystallization. Macromolecules 2020, 53, 4581–4604. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Davis, G.T.; Lauritzen, J.I., Jr. The Rate of crystallization of linear Polymers with Chain Folding. In Treatise on Solid State Chemistry. Volume 3. Crystalline and Noncrystalline Solids; Hannay, N.B., Ed.; Plenum Press: New York, NY, USA, 1976; pp. 497–614. [Google Scholar]
- Hoffman, J.D. Role of reptation in the rate of crystallization of polyethylene fractions from the melt. Polymer 1982, 23, 656–670. [Google Scholar] [CrossRef]
- Hoffman, J.D. Regime III crystallization in melt-crystallized polymers: The variable cluster model of chain folding. Polymer 1983, 24, 3–26. [Google Scholar] [CrossRef]
- Jin, F.; Huang, Z.; Zheng, Y.; Sun, C.; Kafle, N.; Ma, J.; Pan, P.; Miyoshi, T. Impact of Entanglement on Folding of Semicrystalline Polymer during Crystallization. ACS Macro Lett. 2023, 12, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.J.; Hoffman, J.D. Regime III Crystallization in Polypropylene. Macromolecules 1984, 17, 878–885. [Google Scholar] [CrossRef]
- Monasse, B.; Haudin, J.M. Growth transition and morphology change in polypropylene. Coll. Polym. Sci. 1985, 263, 822–831. [Google Scholar] [CrossRef]
- Pelzbauer, Z.; Galeski, A. Growth Rate and Morphology of Polyoxymethylene Supermolecular Structures. J. Polym. Sci. Part C 1972, 38, 23–32. [Google Scholar] [CrossRef]
- Crist, B. Yet Another Visit to the Melting of Polyethylene Crystals. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 3231–3236. [Google Scholar] [CrossRef]
- Strobl, G. The Physics of Polymers; Springer: Berlin/Heidelberg, Germany, 2007; Chapter 5. [Google Scholar]
- Crist, B.; Schultz, J.M. Polymer spherulites: A critical review. Prog. Polym. Sci. 2016, 56, 1–63. [Google Scholar] [CrossRef]
- Galeski, A.; Piorkowska, E. Localized volume deficiencies as an effect of spherulite growth. I. The two-dimensional case. J. Polym. Sci. Polym. Phys. Ed. 1983, 21, 1313–1322. [Google Scholar] [CrossRef]
- Galeski, A.; Piorkowska, E. Localized volume deficiencies as an effect of spherulite growth. II. The three-dimensional case. J. Polym. Sci. Polym. Phys. Ed. 1983, 21, 1323–1339. [Google Scholar] [CrossRef]
- Pawlak, A.; Piorkowska, E. Effect of Negative Pressure on Melting Behavior of Spherulites in Thin Films of Several Crystalline Polymers. J. Appl. Polym. Sci. 1999, 74, 1380–1385. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A. Stability of Spherulite Growth. J. Polym. Sci. Part B Polym. Phys. 1990, 28, 1813–1821. [Google Scholar] [CrossRef]
- Piorkowska, E.; Galeski, A. Crystallization of isotactic polypropylene and high-density polyethylene under negative pressure resulting from uncompensated volume change. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 1285–1291. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II Transformation-Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Menczel, J.D.; Judovits, L.; Prime, R.B.; Bair, H.E.; Reading, M.; Swier, S. Differential scanning calorimetry. In Thermal Analysis of Polymers; Menczel, J.D., Prime, R.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer-Verlag: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Chalmers, J.M.; Edwards, H.G.M.; Lees, J.S.; Long, D.A.; Mackenzie, M.W.; Willis, H.A. Raman spectra of polymorphs of isotactic polypropylene. J. Raman Spectr. 1991, 22, 613–618. [Google Scholar] [CrossRef]
- Beckett, D.R.; Chalmers, J.M.; Mackenzie, M.W.; Willis, H.A.; Edwards, H.G.M.; Lees, J.S.; Long, D.A. The far-infra-red spectra of crystalline isotactic polypropylene polymorphs. Eur. Polym. J. 1985, 21, 849–852. [Google Scholar] [CrossRef]
- Xue, G.; Wang, Y.; Liu, S.; Liao, Y.T. FT-IR Study of Concentration Dependence for Crystallization of Isotactic Polystyrene Arising from Freeze-Drying Dilute Solutions. Macromolecules 1995, 28, 4344–4346. [Google Scholar] [CrossRef]
- Ji, G.; Ni, H.; Wang, C.; Xue, G.; Liao, Y.-T. Concentration Dependence of Crystalline Poly(ethylene terephthalate) Prepared by Freeze-Extracting Solutions. Macromolecules 1996, 29, 2691–2693. [Google Scholar] [CrossRef]
- Xue, G.; Wang, Y.X.; Liu, S.; Liao, Y.T. Concentration Dependence of Crystalline Poly(vinylidene fluoride) Prepared by Freeze-Extracting Solutions. Macromolecules 1995, 28, 3476–3478. [Google Scholar] [CrossRef]
- Xue, G.; Wang, Y.; Gu, X.; Lu, Y. Rapid Crystallization of Isotactic Polystyrene by Shock-Cooling and Subsequent Freeze-Drying of Its Very Dilute Solution. Macromolecules 1994, 27, 4016–4017. [Google Scholar] [CrossRef]
- Wissler, G.E.; Crist, B., Jr. Glass transition in semicrystalline polycarbonate. J. Polym. Sci. Polym. Phys. 1980, 18, 1257–1270. [Google Scholar] [CrossRef]
- Ji, G.; Li, F.; Zhu, W.; Dai, Q.; Xue, G.; Gu, X. Rapid Crystallization of Polycarbonate by Shock-Cooling and Freeze-Drying Method. J. Macromol. Sci. Part A Pure and App. Chem. 1997, 34, 369–376. [Google Scholar] [CrossRef]
- Ji, G.; Xue, G.; Ma, J.; Dong, C.; Gu, X. Concentration dependence of crystallinity of polycarbonate by shock-cooling and subsequent freeze-drying of its various solutions. Polymer 1996, 37, 3255–3258. [Google Scholar] [CrossRef]
- Hu, S.; Yang, F.; Cao, Y.; Yan, Y.; Wu, T.; Fu, Q. Influence of chain entanglements and melt memory effect on the crystallization behavior of polypropylene sulfide. Polymer 2023, 285, 126390. [Google Scholar] [CrossRef]
- Krajenta, J.; Pawlak, A.; Galeski, A. Deformation of Disentangled Polypropylene Crystalline Grains into Nanofibers. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1983–1994. [Google Scholar] [CrossRef]
- Alexander, L.E. X-Ray Diffraction Methods in Polymer Sciences; Wiley-Interscience: New York, NY, USA, 1970; p. 335. [Google Scholar]
- Sasaki, T.; Yamauchi, N.; Irie, S.; Sakurai, K. Differential Scanning Calorimetry Study on Thermal Behaviors of Freeze-Dried Poly(L-lactide) from Dilute Solutions. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 115–124. [Google Scholar] [CrossRef]
- Sasaki, T.; Morino, D.; Tabata, N. Origin of enhanced cold crystallization rate for freeze-dried poly(L-lactide) from solutions. Polym. Eng. Sci. 2011, 51, 1858–1865. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, Y.; Xu, S.; Ni, L.; Li, X.; Shan, G.; Bao, Y.; Pan, P. Role of Chain Entanglements in the Stereocomplex Crystallization between Poly(lactic acid) Enantiomers. ACS Macro. Lett. 2021, 10, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Barangizi, H.; Pawlak, A. Crystallization of partially disentangled polypropylene in nanocomposites with aluminum oxide. Polymer 2022, 254, 125049. [Google Scholar] [CrossRef]
- Pandey, A.; Toda, A.; Rastogi, S. Influence of Amorphous Component on Melting of Semicrystalline Polymers. Macromolecules 2011, 44, 8042–8055. [Google Scholar] [CrossRef]
- Xue, J.; Lu, Y.; Wang, B.; Chen, J.; Shen, C.; Zhang, B. The Isothermal Melting Kinetics of Ultra-High Molecular Weight Polyethylene Crystals. Macromol. Rapid Commun. 2024, 225, 2300704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, J.; Fu, Q.; Zhang, J. Novel Strategy to Improve the Performance of Poly(L-lactide): The Synergistic Effect of Disentanglement and Strong Shear Field. ACS Sustain. Chem. Eng. 2023, 11, 9630–9642. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hikosaka, M.; Gu, F.; Ghosh, S.K.; Arakaki, M.; Toda, A. Effect of entanglement on nucleation rate of poly-ethylene. Polymer J. 2001, 33, 906–908. [Google Scholar] [CrossRef]
- Robelin-Souffache, E.; Rault, J. Origin of the long period and crystallinity in quenched semicrystalline polymers. Macromolecules 1989, 22, 3581–3594. [Google Scholar] [CrossRef]
- Bartczak, Z.; Kozanecki, M. Influence of molecular parameters on high-strain deformation of polyethylene in the plane-strain compression. Part I. Stress—Strain behavior. Polymer 2005, 46, 8210–8221. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Zhong, L.W.; Yang, S.; Liang, D.H.; Chen, E.Q. Memory effect on solution crystallization of high molecular weight poly(ethylene oxide). Polymer 2012, 53, 3621–3628. [Google Scholar] [CrossRef]
- Flory, P.J.; Yoon, D.Y. Molecular morphology in semicrystalline polymers. Nature 1978, 272, 226–229. [Google Scholar] [CrossRef]
- Jani, F.; Sepahi, A.; Afzali, S.K.; Moyad, S.H. Experimental study on the effect of molecular weight and chemical composition distribution on the mechanical response of high-density polyethylene. Polym. Eng. Sci. 2023, 63, 176–188. [Google Scholar] [CrossRef]
- Peters, G.W.M.; Balzano, L.; Steenbakkers, R.J.A. Flow-induced Crystallization. In Handbook of Polymer Crystallization; Piorkowska, E., Rutledge, G.C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Chapter 17; pp. 399–431. [Google Scholar]
- Martins, J.A.; Zhang, W.; Brito, A.M. Origin of the melt memory effect in polymer crystallization. Polymer 2010, 51, 4185–4194. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. The Crystallization of Polypropylene with Reduced Density of Entanglements. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 748–756. [Google Scholar] [CrossRef]
- Krajenta, J.; Safandowska, M.; Pawlak, A. The re-entangling of macromolecules in polypropylene. Polymer 2019, 175, 215–226. [Google Scholar] [CrossRef]
- Hong, H.; Rongjie, L.; Yaling, Z. Concentration Dependence of Crystalline Poly(L-lactide) Prepared by Freeze-drying Solutions. Polym. Polym. Compos. 2009, 17, 31–35. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, J.; Liu, M.; Fu, Q.; Zhang, J. Understanding the effect of chain entanglement state on melt crystallization of the polymer freeze-extracted from solution: The role of critical overlap concentration. Polymer 2019, 178, 121588. [Google Scholar] [CrossRef]
- Barangizi, H.; Krajenta, J.; Pawlak, A. The influence of entanglements of macromolecules on the mechanical and thermal properties of polylactide composites with carbon nanotubes. Exp. Polym. Lett. 2023, 17, 738–758. [Google Scholar] [CrossRef]
- Ni, L.; Xu, S.; Sun, C.; Qin, Y.; Zheng, Y.; Zhou, J.; Yu, C.; Pan, P. Retarded Crystallization and Promoted Phase Transition of Freeze-Dried Polybutene-1: Direct Evidence for the Critical Role of Chain Entanglement. ACS Macro Lett. 2022, 11, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Chen, J.; Yang, T.; Wang, P.; Min, J.; Fu, Q.; Zhang, J. Regulation of Entanglement Networks under Different Shear Fields and Its Effect on the Properties of Poly(L-lactide). Ind. Eng. Chem. Res. 2023, 62, 7434–7446. [Google Scholar] [CrossRef]
- Liu, X.; Yu, W. Weak Shear-Induced Slowdown in Crystallization of Less-Entangled Poly(ε-caprolactone). Macromolecules 2021, 54, 3347–3357. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Bu, H. Influence of Intermolecular Entanglements on Crystallization Behavior of Ultra-High Molar Mass Polyethylene. Polym. Eng. Sci. 2003, 43, 607–614. [Google Scholar] [CrossRef]
- Sun, Q.; Fu, Q.; Xue, G.; Chen, W. Crystallization Behavior of Syndiotactic Poly(propylene) Freeze-Dried from Toluene at Very Dilute Concentration. Macromol. Rapid Comm. 2001, 22, 1182–1185. [Google Scholar] [CrossRef]
- Xiao, Z.; Sun, Q.; Xue, G.; Yuan, Z.; Dai, Q.; Hu, Y. Thermal behavior of isotactic polypropylene freeze-extracted from solutions with varying concentrations. Europ. Polym. J. 2003, 39, 927–931. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Zhang, X.; Zhang, B.; Chen, J. Evolution of chain entanglements under large amplitude oscillatory shear flow and its effect on crystallization of isotactic polypropylene. Polymer 2020, 186, 121899. [Google Scholar] [CrossRef]
- Krajenta, J.; Safandowska, M.; Pawlak, A.; Galeski, A. All-polymer composites—A new approach with the use of disentangled semi-crystalline polymers. Part 1. Disentangling and properties of disentangled polylactide. Polimery 2020, 65, 167–173. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hikosaka, M.; Toda, A.; Wataoka, I.; Gu, F. Role of entanglement in nucleation and ‘melt relaxation’ of polyethylene. Polymer 2002, 43, 6585–6593. [Google Scholar] [CrossRef]
- Gaonkar, A.A.; Murudkar, V.V.; Deshpande, V.D. Comparison of crystallization kinetics of polyethylene terephthalate (PET) and reorganized PET. Thermoch. Acta 2020, 683, 178472. [Google Scholar] [CrossRef]
- Wang, X.H.; Wang, Z.; Luo, K.; Huang, Y. Translation and Rotation of Spherulites during the Crystallization of Isotactic Polypropylene with Reduced Chain Entanglements. Macromolecules 2011, 44, 2844–2851. [Google Scholar] [CrossRef]
- Nowacki, R.; Kolasinska, J.; Piorkowska, E. Cavitation during isothermal crystallization of isotactic polypropylene. J. Appl. Polym. Sci. 2001, 79, 2439–2448. [Google Scholar] [CrossRef]
- Gu, F.; Bu, H.; Zhang, Z. Unique morphologies found in freeze-dried poly(ethy1ene oxide) prepared from dilute solutions. Macromol. Chem. Phys. 1998, 199, 2597–2600. [Google Scholar] [CrossRef]
- Gu, F.M.; Bu, H.S.; Zhang, Z. A unique morphology of freeze-dried poly(ethylene oxide) and its transformation. Polymer 2000, 41, 7605–7609. [Google Scholar] [CrossRef]
- Luo, C.; Sommer, J.-U. Frozen Topology: Entanglements Control Nucleation and Crystallization in Polymers. Phys. Rev. Let. 2014, 112, 195702. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Zhang, W. Molecular Dynamics Simulations of the Effects of Entanglement on Polymer Crystal Nucleation. Macromolecules 2022, 55, 4899–4906. [Google Scholar] [CrossRef]
- Zhu, C.; Zhao, J. Nucleation and Crystallization of Polymer Melts under Cyclic Stretching: Entanglement Effect. Macromolecules 2023, 56, 5490–5501. [Google Scholar] [CrossRef]
- Zhai, Z.; Fusco, C.; Morthomas, J.; Perez, M.; Lame, O. Disentangling and Lamellar Thickening of Linear Polymers during Crystallization: Simulation of Bimodal and Unimodal Molecular Weight Distribution Systems. ACS Nano 2019, 13, 11310–11319. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Gu, F.; Watanabe, K.; Okada, K.; Toda, A.; Hikosaka, M. Two-step formation of entanglement from disentangled polymer melt detected by using nucleation rate. Polymer 2006, 47, 6422–6428. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Y.; Shen, K.; Fu, Q.; Zhang, J. Insight into Shear-Induced Modification for Improving Processability of Polymers: Effect of Shear Rate on the Evolution of Entanglement State. J. Polym. Sci. B Polym. Phys. 2019, 57, 598–606. [Google Scholar] [CrossRef]

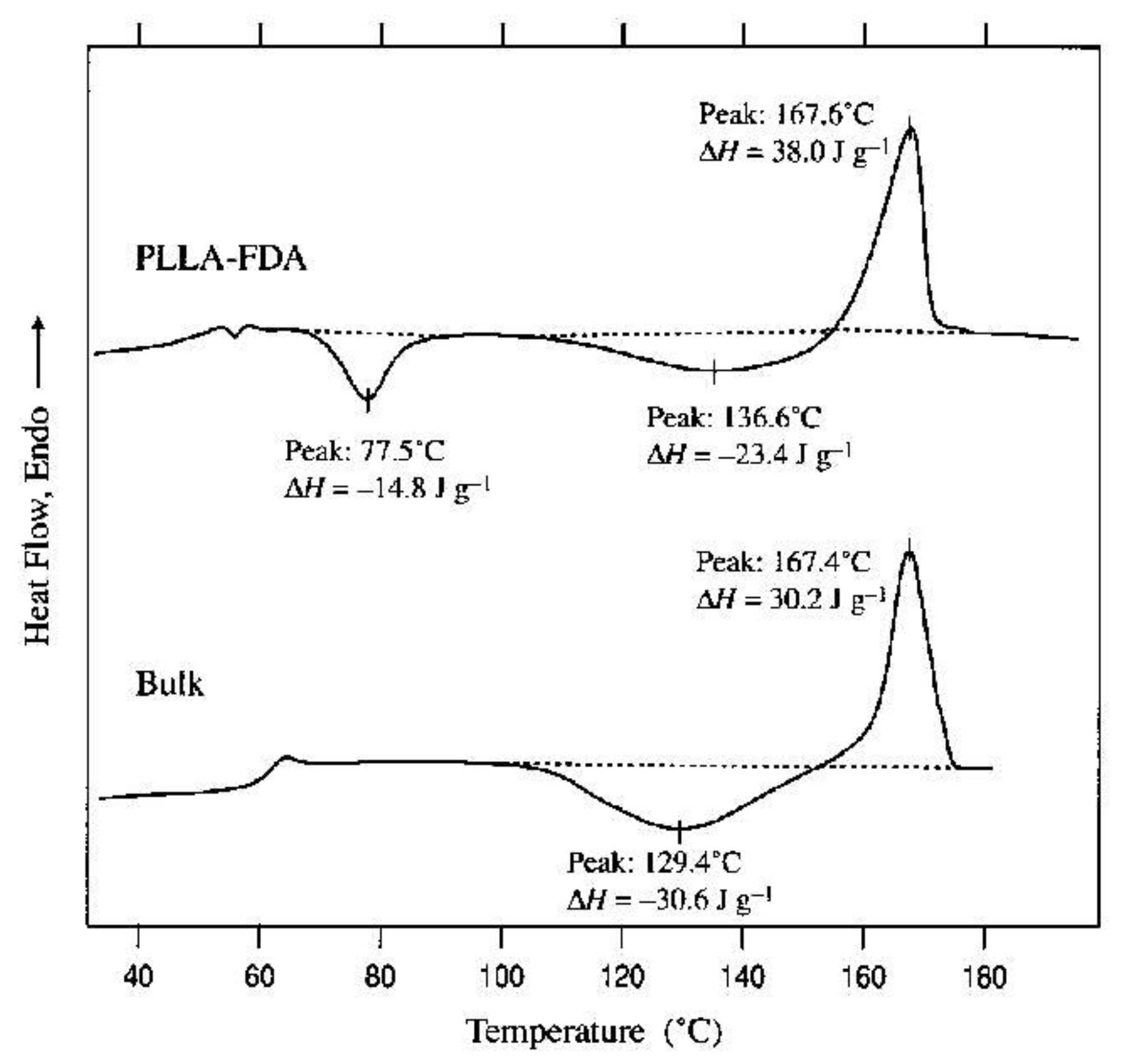
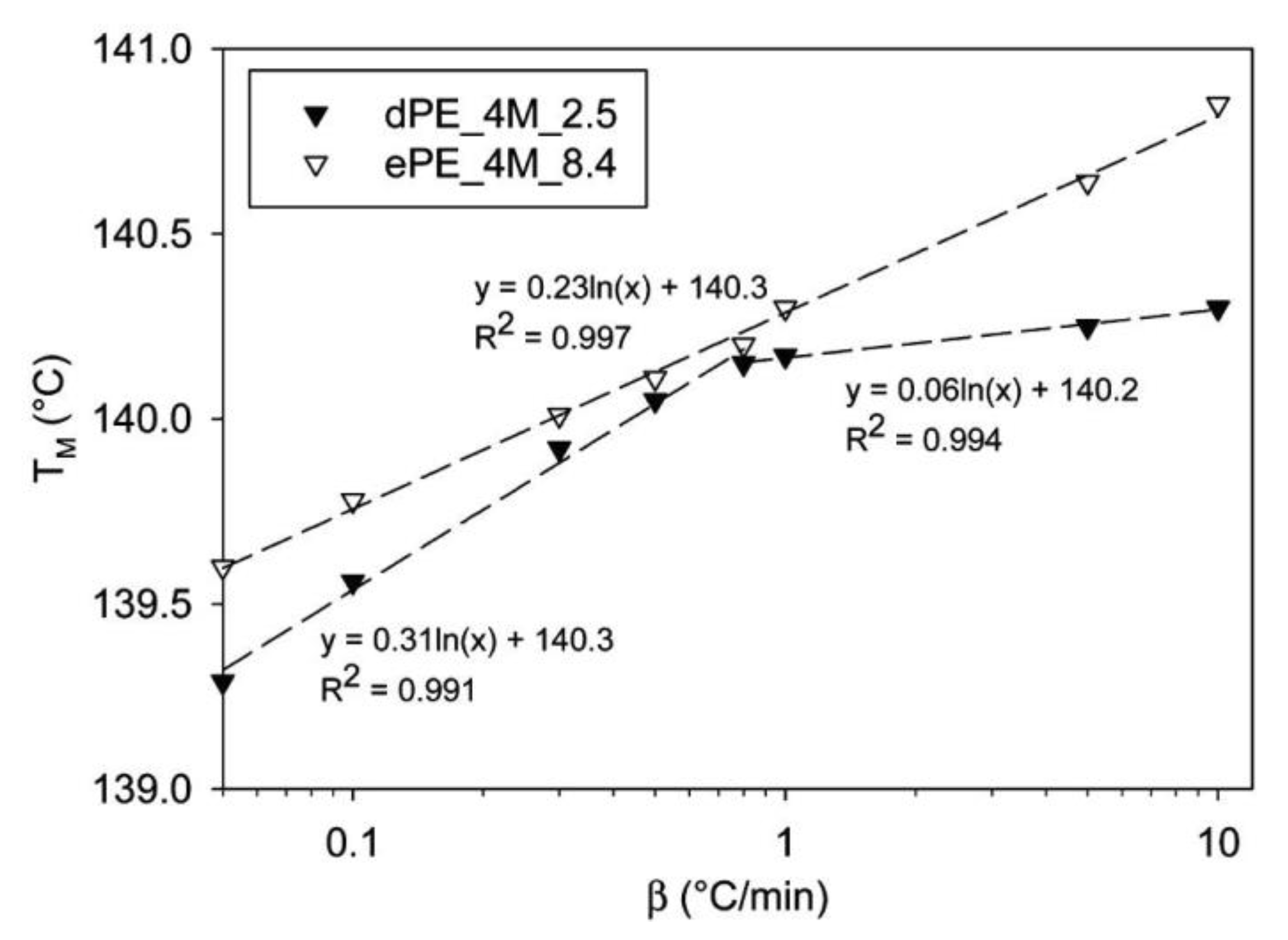
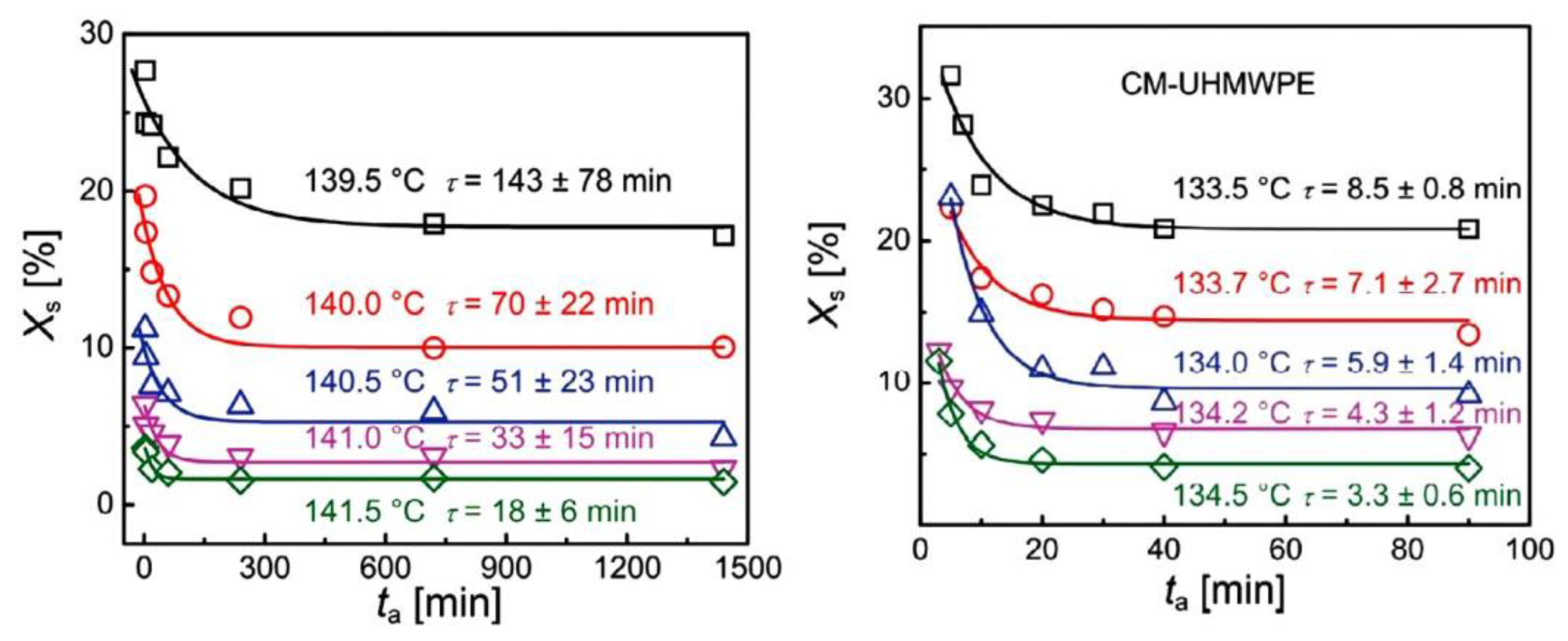
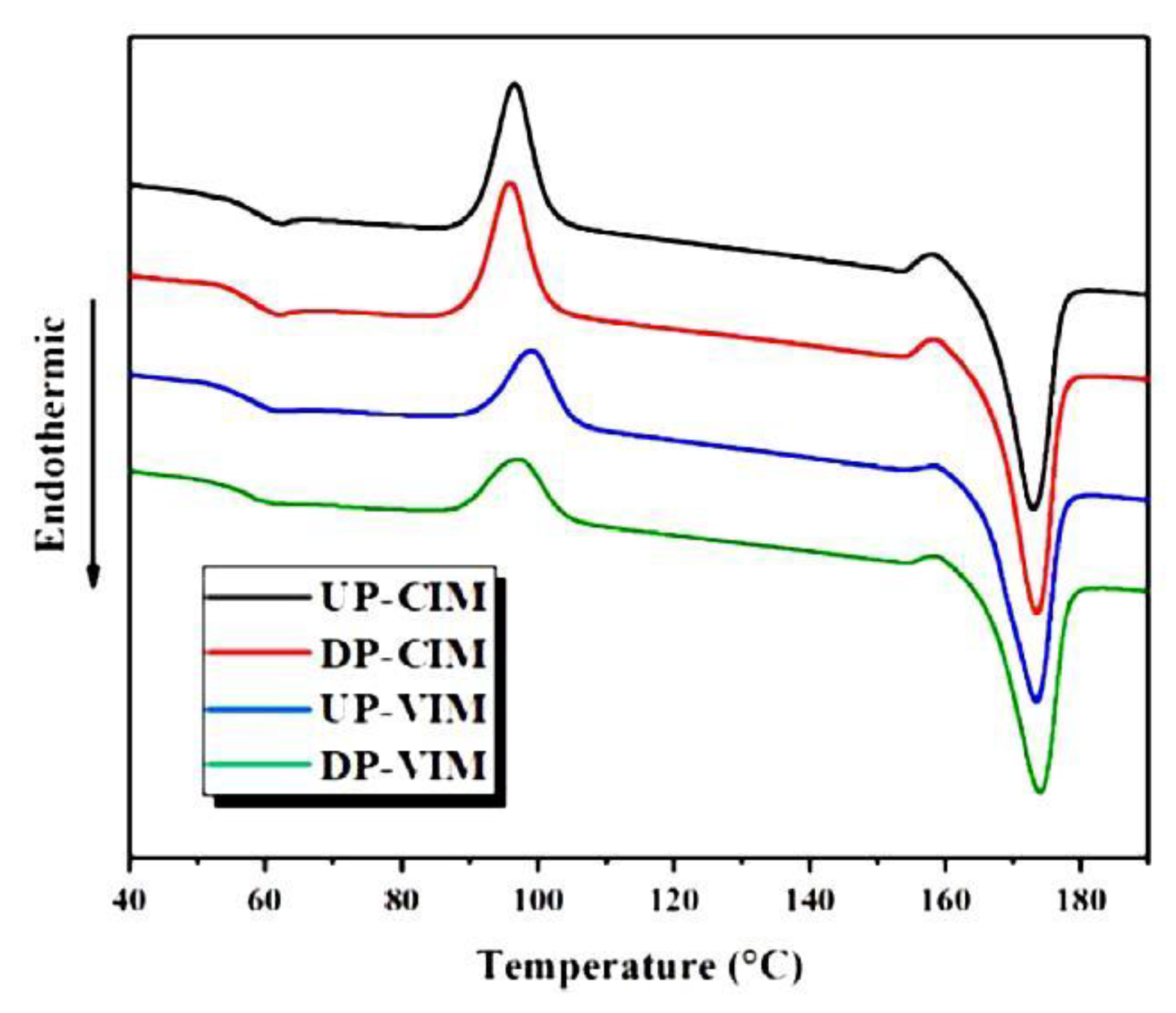






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Me [g/mol] |
|---|---|
| Polyethylene, PE | 830–2600 |
| Isotactic polypropylene, iPP | 5500–8100 |
| Polylactide, PLA | 4000–10,500 |
| Isotactic polystyrene, iPS | 17,500–28,800 |
| Polyethylene terephthalate, PET | 1170–1450 |
| Polycarbonate, PC | 1330–1660 |
| 1,4- polybutdiene, PB | 1850 |
| Polymer | Disentangling Procedure | Remarks about Influence of Disentangling on Crystallization and Obtained Crystallinity | Reference |
|---|---|---|---|
| PET | Freeze-drying | Max. crystallinity was obtained from intermediate concentration | [26,28,95] |
| PVDF | Freeze-drying | Max. crystallinity was obtained from intermediate concentration | [96] |
| iPS | Freeze-drying | Only dilute solution enhanced crystallization. The easier cold crystallization from disentangled iPS | [32,94,97] |
| PC | Freeze-drying | Higher crystallinity of disentangled PC, more effective cold crystallization | [98,99] |
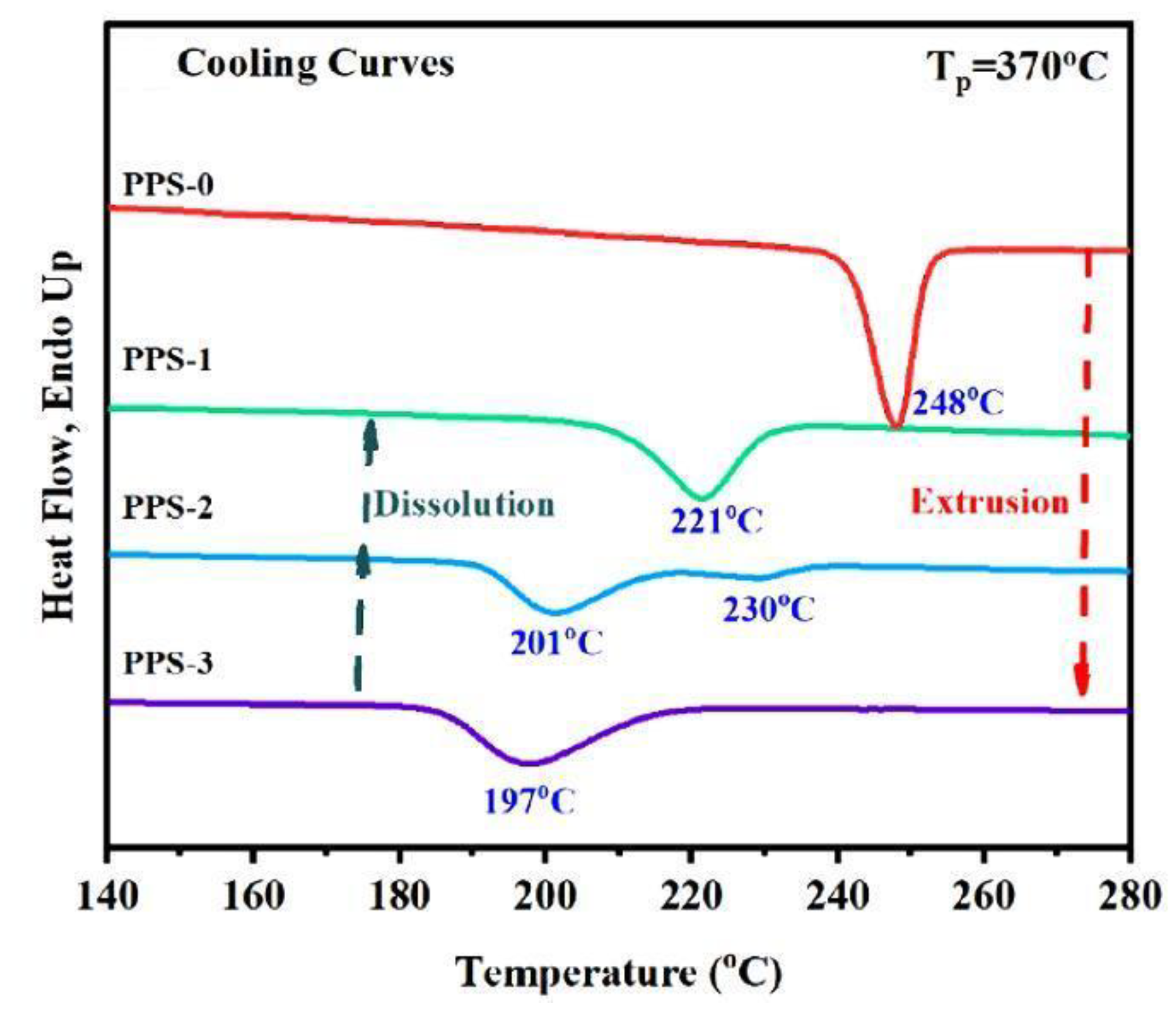
| PPS | Freeze-drying and polymerization | Extended chain crystals grew from polymerized PPS | [101] |
| PP | Freeze-drying | Higher crystallinity, higher Tm | [102] |
| Dissolving with crystallization | No effect on crystallinity, lower Tm | [107] | |
| Shear | No effect on crystallinity, higher Tm, thicker lamellae | [57] | |
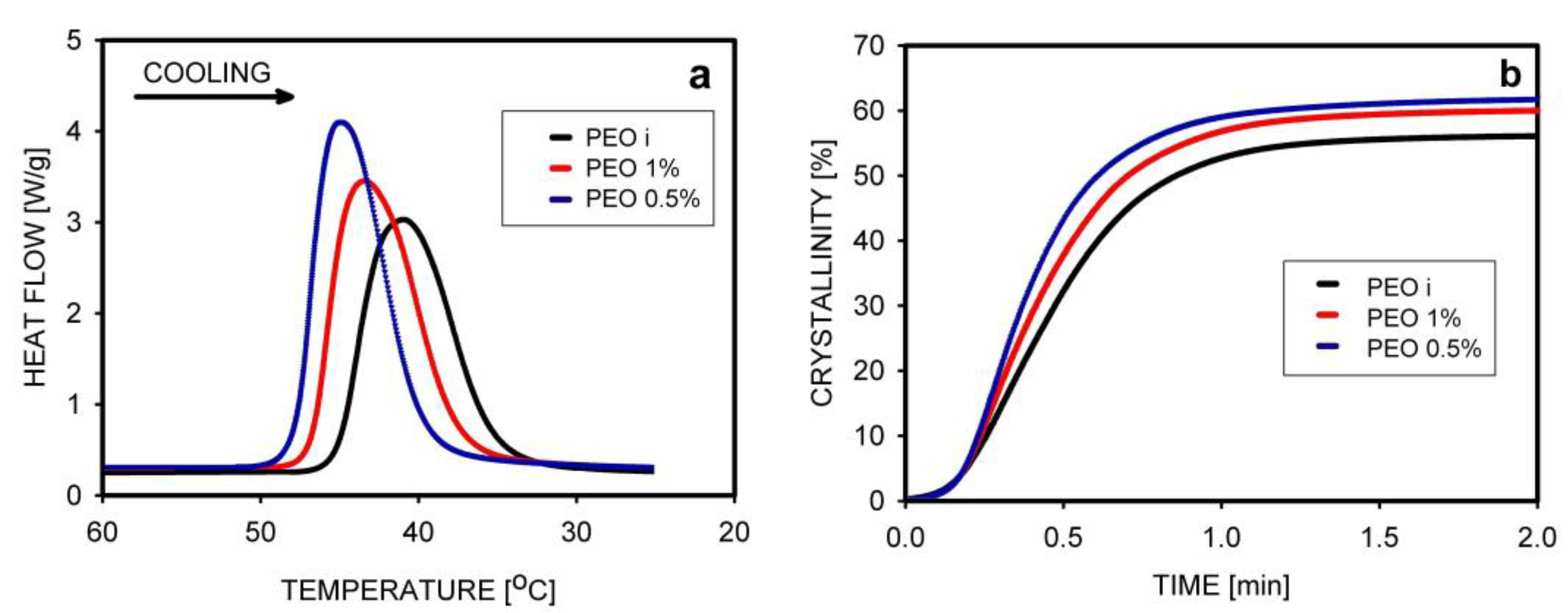
| PEO | Freeze-drying | No effect on crystallinity, lower Tm | [67] |
| PCL | Freeze-drying | Higher crystallinity, higher Tm | [39] |
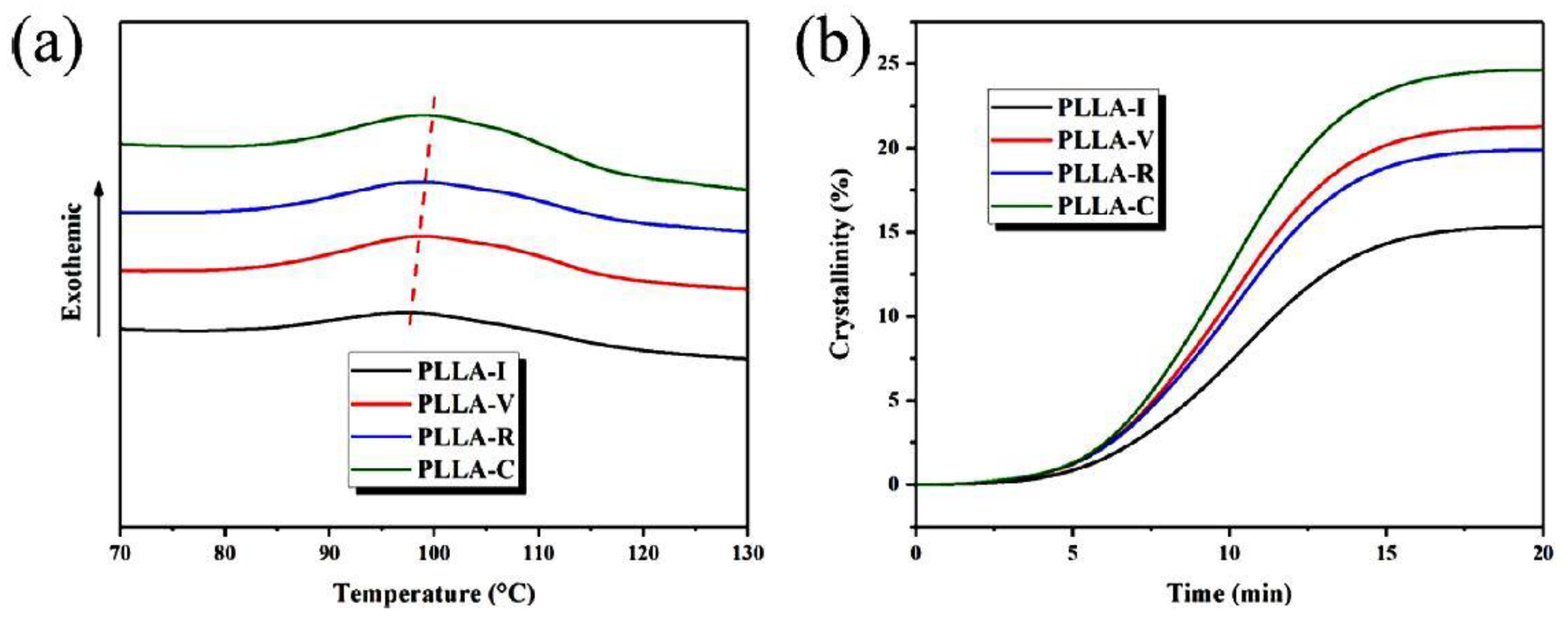
| PLLA | Freeze drying | Cold crystallization initiated at lower temperature | [104,105] |
| Shear during extrusion | Higher crystallinity, higher Tm | [110] | |
| PLLA/PDLA | Freeze-drying | Decreased crystallinity of disentangled complex | [106] |
| HDPE | High pressure crystallization | Higher crystallinity, thicker crystals | [44] |
| UHMWPE | Polymerization | Lower Tm, different way of crystal melting | [108,109] |
| Polymer | Studies of Crystalline Structure after Disentangling | Non-Isothermal Crystallization | Isothermal Crystallization | Re-Entangling Effect |
|---|---|---|---|---|
| PET | [26,28,95] | [133] | ||
| PVDF | [96] | |||
| iPS | [32,94,97] | [32] | [32] | |
| PC | [98,99] | |||
| iPP | [57,102,107] | [57,107,119,120] | [30,57,107,119,129,130,134] | [30,119,120,130] |
| sPP | [128] | |||
| PPS | [101] | [101] | ||
| PEO | [67] | [67] | [67,136,137] | [67] |
| PCL | [39] | [126] | ||
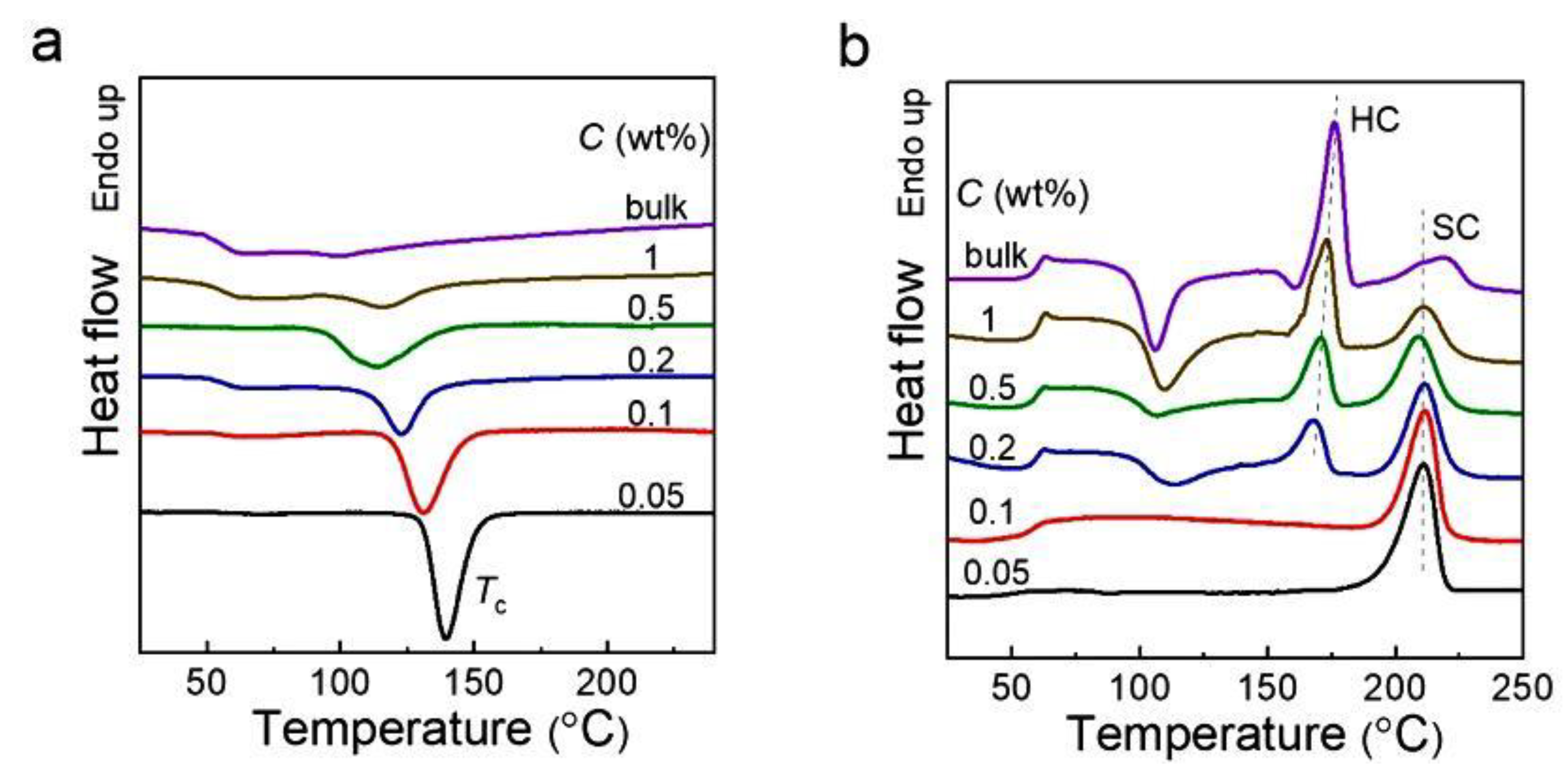
| PLA | [104,105,110] | [120,122,123,125] | [31,105,122,123,125,131] | [31,131] |
| PLLA/PDLA | [106] | [106] | [106] | |
| PE | [44] | [143] | [44,132] | [44,142] |
| UHMWPE | [108,109] | [127] | [127] | |
| PB | [124] | [124] | ||
| PMMA | [29] |
| Property | Non-Isothermal Crystallization | Isothermal Crystallization |
|---|---|---|
| Melt–crystal conversion | Faster [107,119,120,125,126] | Faster [32,44,57,67,106,107,119, 122,125,126,128] Slower [124] |
| Crystallization temperature | Higher [57,67,101,106,107,119,121,122,125,126] Lower [124,127] | -- |
| Melting temperature | Higher [57] Lower [124] | -- |
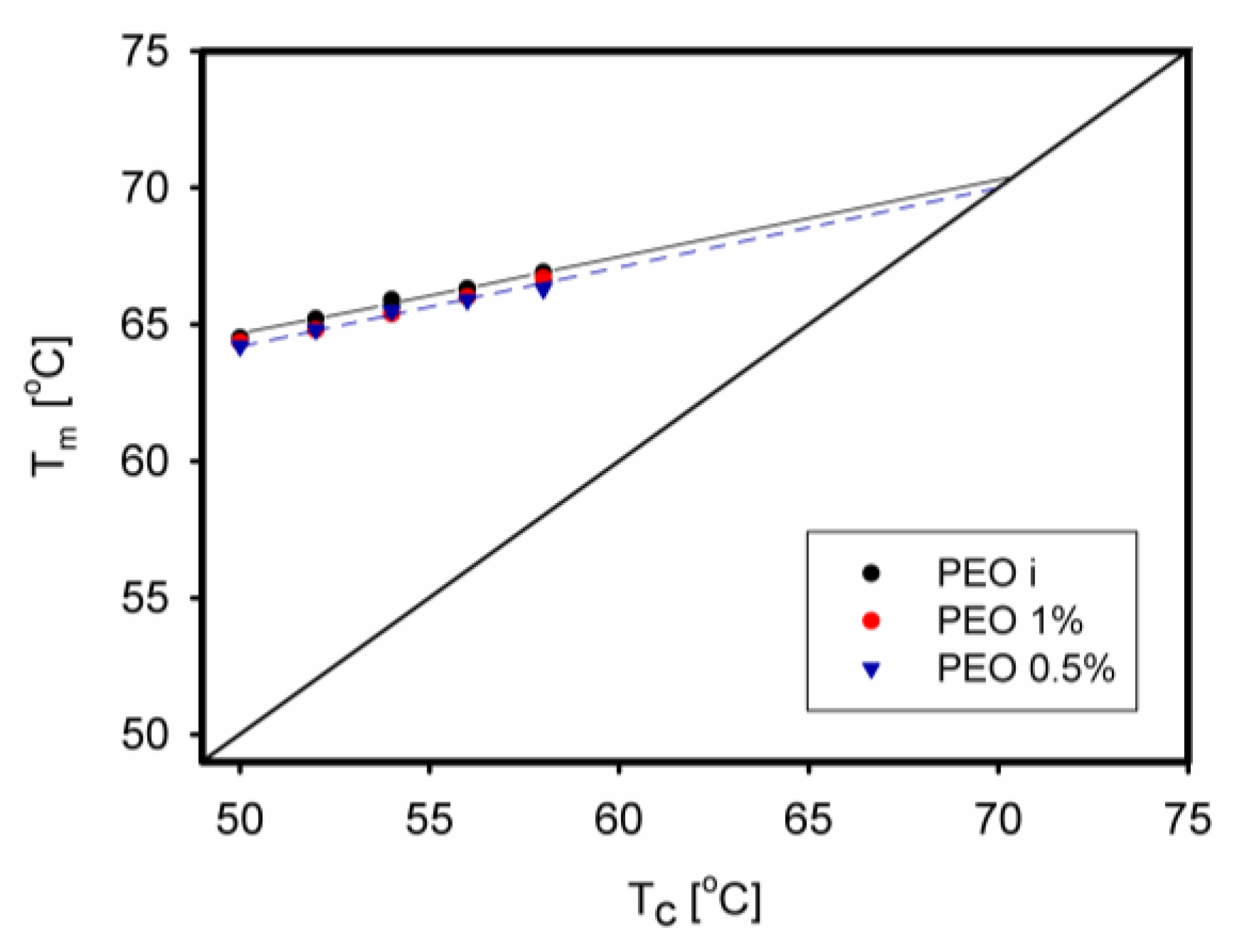
| Equilibrium melting temperature | -- | Higher [133] Lower [67] |
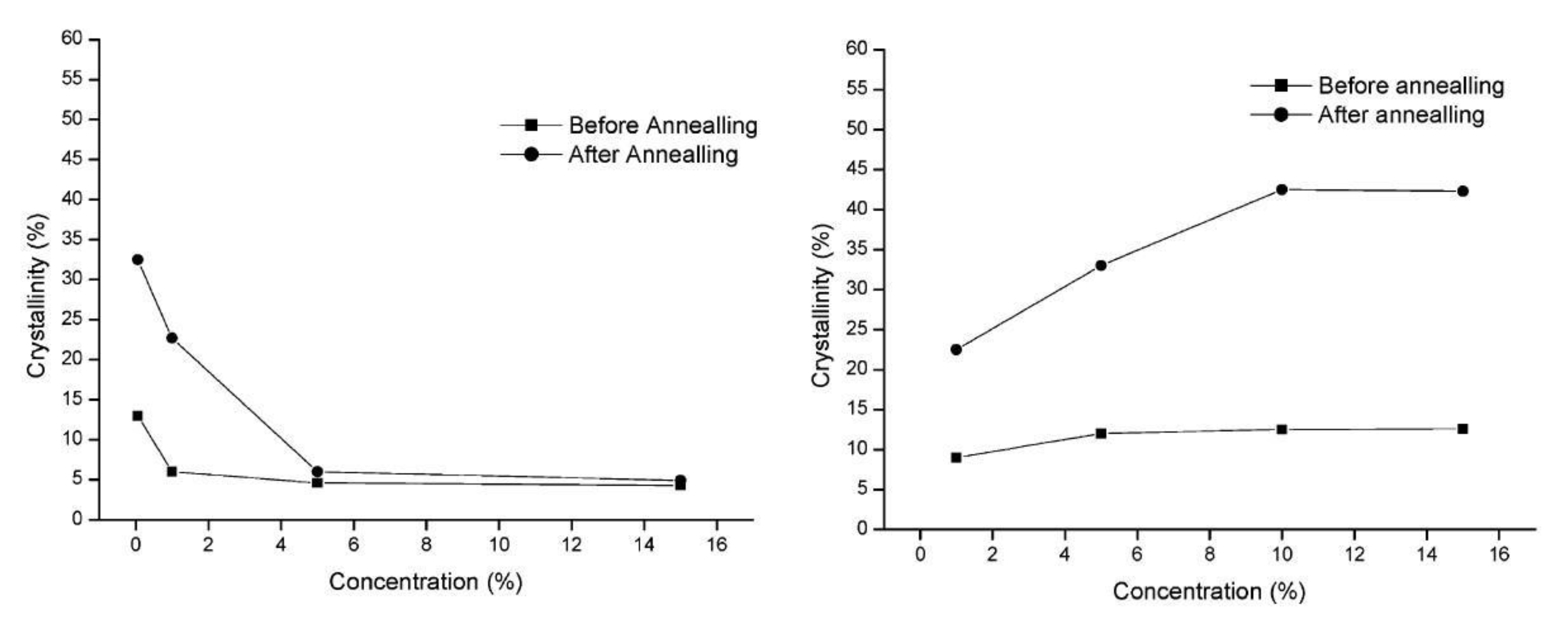
| Crystallinity degree | Higher [57,67,106,119,121,125] Lower [124] | Higher [122] |
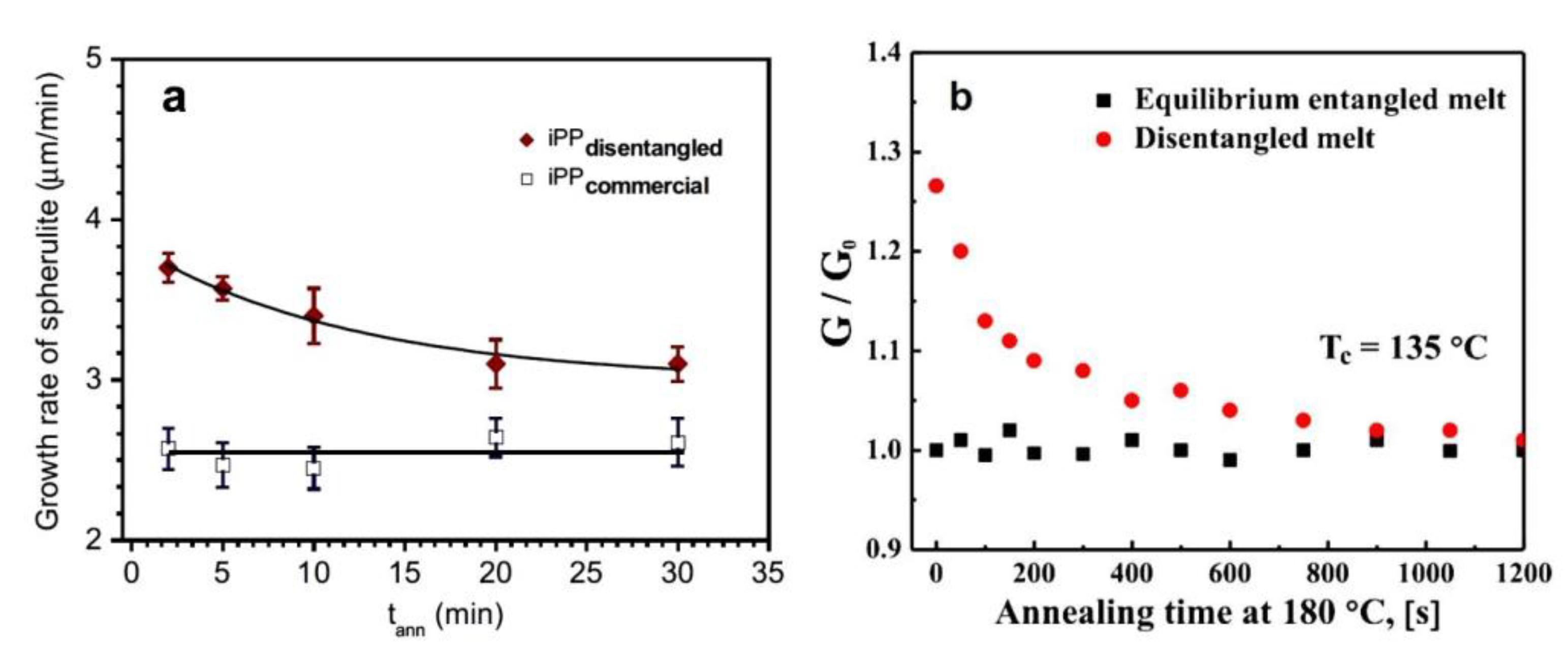
| Growth rate of spherulites | -- | Faster [30,31,44,67,106,107,119,123,130,131] Slower [124] |
| Regimes of crystallization | -- | Shift of temperature [67,119,131,133] |
| Lamellae thickness | Increased [57,143] | Increased [57] |
| Nucleation density | -- | Increased [30,31,67,106,130] Decreased [44,107,124,131] |
| Cold-crystallization rate | -- | Faster [105,106] |
| Avrami’s k coefficient | Increased [122,125] Decreased [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlak, A. Crystallization of Polymers with a Reduced Density of Entanglements. Crystals 2024, 14, 385. https://doi.org/10.3390/cryst14040385
Pawlak A. Crystallization of Polymers with a Reduced Density of Entanglements. Crystals. 2024; 14(4):385. https://doi.org/10.3390/cryst14040385
Chicago/Turabian StylePawlak, Andrzej. 2024. "Crystallization of Polymers with a Reduced Density of Entanglements" Crystals 14, no. 4: 385. https://doi.org/10.3390/cryst14040385
APA StylePawlak, A. (2024). Crystallization of Polymers with a Reduced Density of Entanglements. Crystals, 14(4), 385. https://doi.org/10.3390/cryst14040385