
Crystal Structures, Molecular Docking and In Vitro Investigations of Two 4-Substituted 2-(5,5-dimethyl-3-styrylcyclohex-2-enylidene)malononitrile Derivatives as Potential Topoisomerase II Inhibitors
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Synthesis
2.3. Single-Crystal X-ray Diffraction (SCXRD)
2.4. Molecular Modeling
2.4.1. Protein Preparation and Ligand-Protein Docking
2.4.2. HYDE Scoring and Visual Assessment
2.5. Evaluation of Cell Viability
3. Results and Discussion
3.1. Synthesis
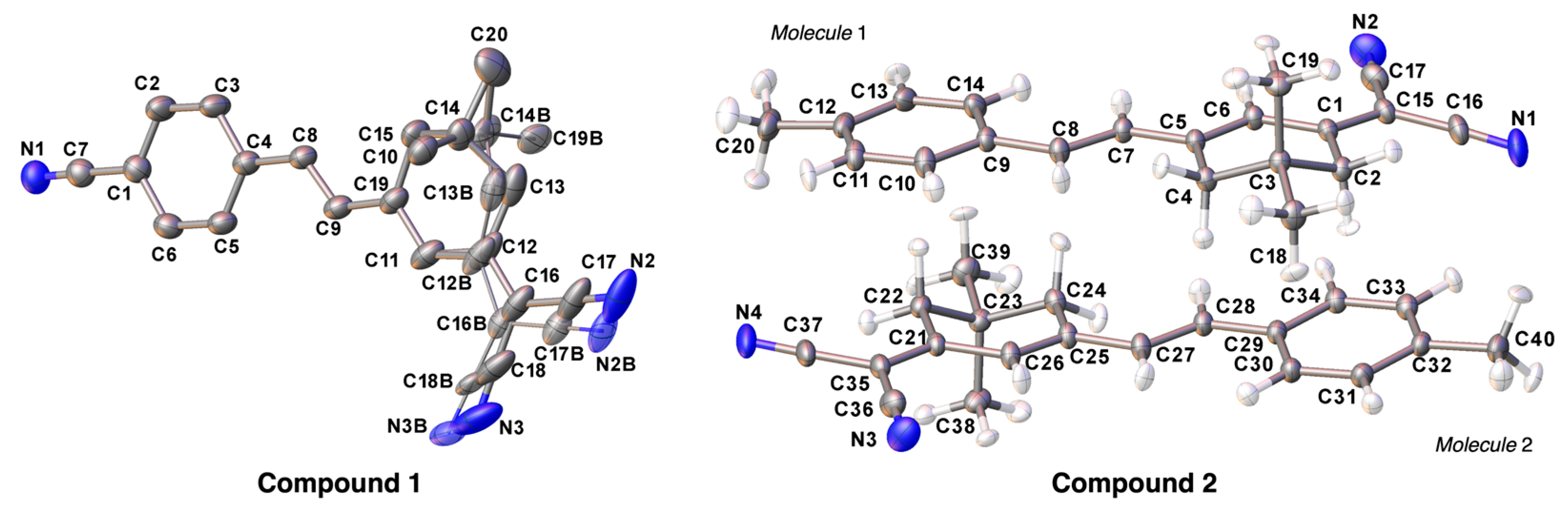
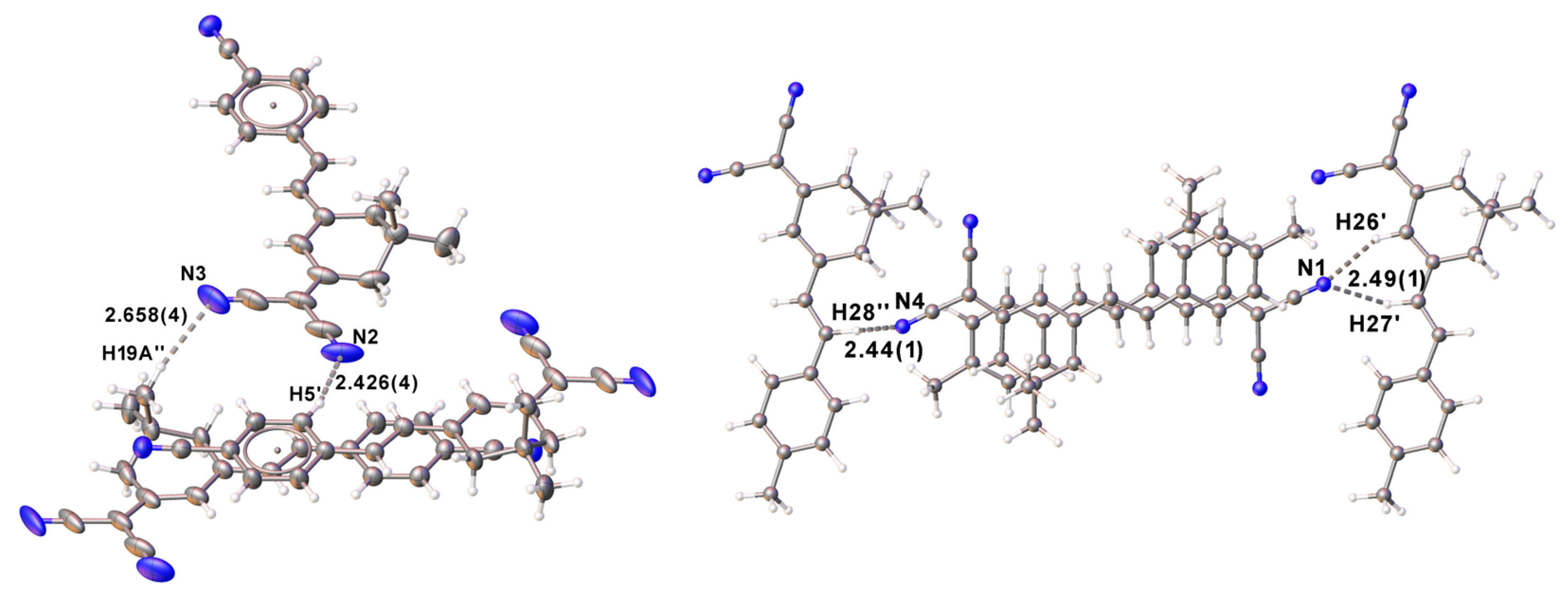
3.2. Crystal Structures of Compounds 1 and 2
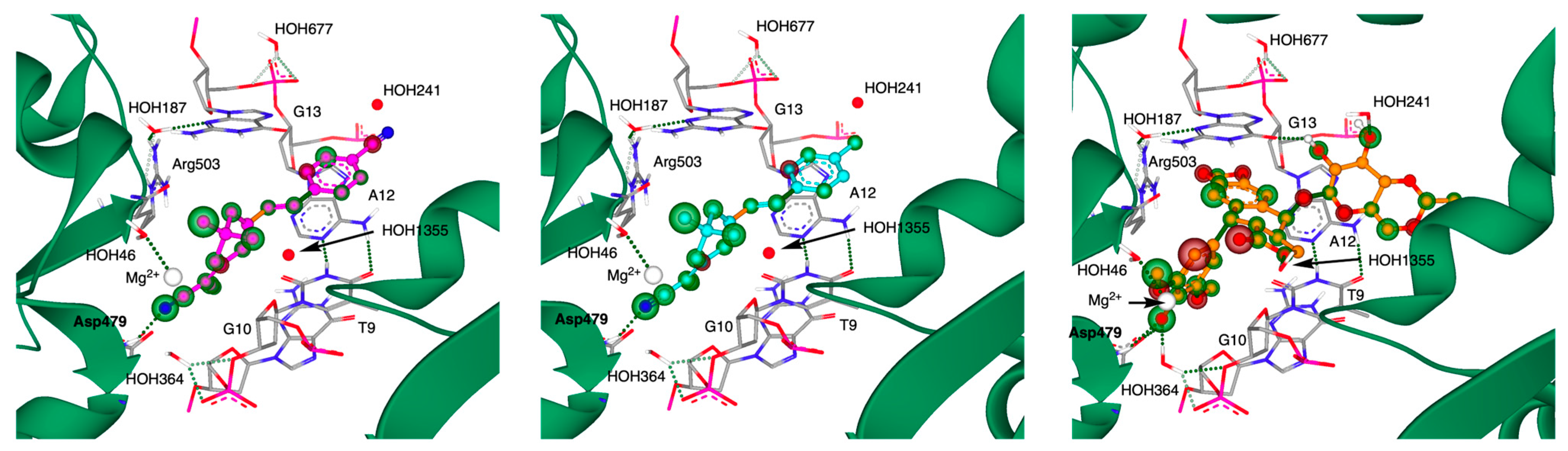
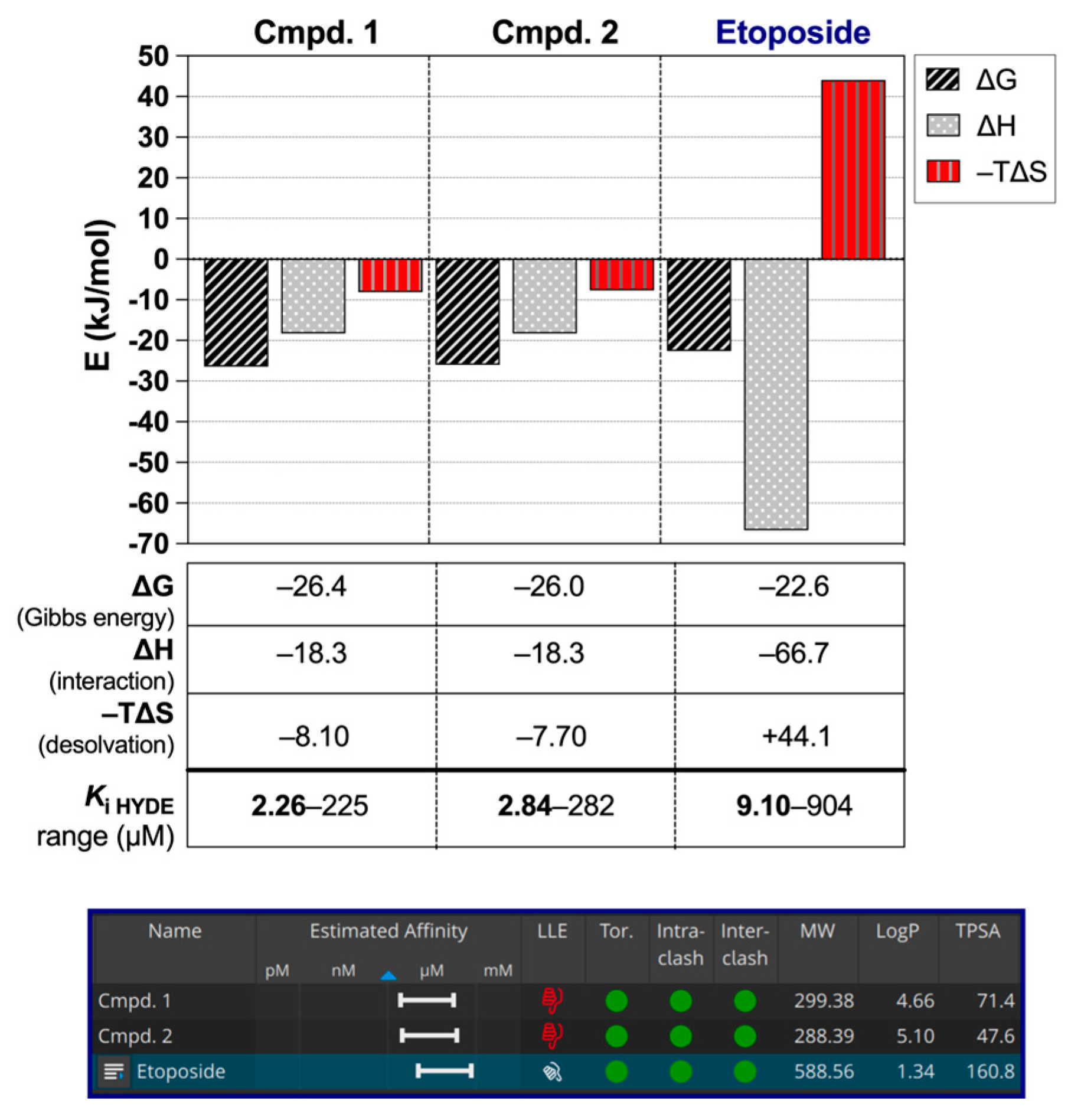
3.3. Molecular Modeling Experiments
3.4. Evaluation of Cytotoxicity and Antiproliferation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Hassanin, H.M.; Serya, R.A.T.; Elmonean, W.R.A.; Mostafa, M.A. Synthesis and molecular docking studies of some novel Schiff bases incorporating 6-butylquinolinedione moiety as potential topoisomerase IIβ inhibitors. R. Soc. Open Sci. 2018, 5, 172407. [Google Scholar] [CrossRef] [PubMed]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA topoisomerases as molecular targets for anticancer drugs. J. Enz. Inh. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef]
- Cattrini, C.; Capaia, M.; Boccardo, F.; Barboro, P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: Data from a translational study. Cancer Treat. Res. Commun. 2020, 25, 100221. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.L.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112, 3611–3640. [Google Scholar] [CrossRef] [PubMed]
- Thakur, D. Topoisomerase II inhibitors in cancer treatment. Int. J. Pharm. Sci. Nanotechnol. 2011, 3, 1173–1181. [Google Scholar] [CrossRef]
- Najar, I.A.; Johri, R.K. Pharmaceutical and pharmacological approaches for bioavailability enhancement of etoposide. J. Biosci. 2014, 39, 139–144. [Google Scholar] [CrossRef]
- Arthur, D.E. Molecular docking studies of some topoisomerase II inhibitors: Implications in designing of novel anticancer drugs. Radiol. Infect. Dis. 2019, 6, 68–79. [Google Scholar] [CrossRef]
- Herrlich, P.; Morrison, H.; Sleeman, J.; Orian-Rousseau, V.; König, H.; Weg-Remers, S.; Ponta, H. CD44 acts as a growth- and invasiveness-promoting molecule and as a tumor-suppressing cofactor. Ann. N. Y. Acad. Sci. 2000, 910, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.I.; Slevin, M.L. The clinical pharmacology of etoposide and teniposide. Clin. Pharmacokinet. 1987, 12, 223–252. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, W.; Zhang, P.; Li, N. Topoisomerase II trapping agent teniposide induces apoptosis and G2/M or S phase arrest of oral cell carcinoma. World J. Surg. Oncol. 2006, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11 (Suppl. S5), 87. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.T.; Peeva, M.I.; Georgieva, M.G.; Deneva, V.; Balacheva, A.A.; Bogdanov, I.P.; Ponticelli, M.; Milella, L.; Kirilov, K.; Matin, M.; et al. Favipiravir vs. deferiprone: Tautomeric, photophysical, in vitro biological studies, and binding interactions with SARS-CoV-2-Mpro/ACE2. Curr. Res. Biotechnol. 2024, 7, 100176. [Google Scholar] [CrossRef]
- Kosilkin, I.V.; Hillenbrand, E.A.; Tongwa, P.; Fonari, A.; Zazueta, J.; Fonari, M.S.; Antipin, M.; Dalton, L.R.; Timofeeva, T. Synthesis, structure, thermal and nonlinear optical properties of a series of novel D-π-A chromophores with varying alkoxy substituents. J. Mol. Struct. 2011, 1006, 356–365. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Cryst. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Lidgley, M.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef] [PubMed]
- SeeSAR Package Version 13.1 from BioSolveIT GmbH, St. Augustin 2024, Germany. Available online: http://www.biosolveit.de/SeeSAR (accessed on 17 May 2023).
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration energies in protein-ligand complexes: Methods behind the HYDE scoring function. J. Comput. Aided Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Schärfer, C.; Schulz-Gasch, T.; Hert, J.; Heinzerling, L.; Schulz, B.; Inhester, T.; Stahl, M.; Rarey, M. CONFECT: Conformations from an expert collection of torsion patterns. ChemMedChem 2013, 8, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef] [PubMed]
- Peeva, M.I.; Georgieva, M.G.; Balacheva, A.A.; Pavlov, A.; Tzvetkov, N.T. In vitro investigation of the cytotoxic and antiproliferative effects of Haberlea rhodopensis total extract: A comparative study. Cosmetics 2024, 11, 46. [Google Scholar] [CrossRef]
- Tzvetkov, N.T.; Peeva, M.I.; Tsakovska, I.; Milella, L.; Pajeva, I.; Stammler, H.-G. The crystal structure of (4SR)-7-(3,4-dichlorobenzyl)-4,8,8,-trimethyl-7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6(2H,4H)-dione, C15H16Cl2N4O2. Z. Für Krist.-New Cryst. Struct. 2022, 237, 319–321. [Google Scholar] [CrossRef]
- Lin, Z.; Will, Y. Evaluation of drugs with specific organ toxicities in organ-specific cell lines. Toxicol. Sci. 2012, 126, 114–127. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Compound 2 | |
|---|---|---|
| CCDC dep. no. | 2350475 | 2350476 |
| Empirical formula | C20H17N3 | C20H20N2 |
| Formula weight (FW, g/mol) | 299.38 | 288.39 |
| Temperature (K) | 100.0 (1) | 95.0 (1) |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | Pna21 (no.33) | P21/n |
| a (Å) | 16.5791 (2) | 13.4133 (2) |
| b (Å) | 15.9039 (2) | 16.0132 (3) |
| c (Å) | 6.38900 (10) | 15.9997 (3) |
| β (°) | 90 | 102.706 (2) |
| Volume (Å3) | 1684.60 (4) | 3352.42 (11) |
| Z | 4 | 8 |
| Dcalc (g/cm3) | 1.180 | 1.143 |
| μ, mm−1 | 0.553 | 0.067 |
| F(000) | 632.0 | 1232.6 |
| Crystal size (mm) | 0.17 × 0.14 × 0.12 | 0.35 × 0.29 × 0.13 |
| Radiation (Å) | Cu Kα (λ = 1.54184) | Mo Kα (λ = 0.71073) |
| Crystal color | Clear orange | Clear yellow |
| 2θ range for data collection, ° | 7.7 to 152.7 | 4.5 to 60.1 |
| Index ranges | −20 ≤ h ≤ 20 | −21 ≤ h ≤ 22 |
| −20 ≤ k ≤ 20 | −26 ≤ k ≤ 26 | |
| −8 ≤ l ≤ 8 | −26 ≤ l ≤ 26 | |
| Reflections collected | 61,297 | 109,135 |
| Independent reflections | 3519 | 9793 |
| R(int) | 0.0300 | 0.0485 |
| No. of parameters | 281 | 757 |
| No. of restraints | 468 | 0 |
| R indexes [I > 2σ(I)] | R1 = 0.0401 | R1 = 0.0334 |
| wR2 = 0.1141 | wR2 = 0.0757 | |
| R indexes | R1 = 0.0410 | R1 = 0.0454 |
| (all data) | wR2 = 0.1155 | wR2 = 0.0810 |
| Goodness-of-fit on F2 | 1.077 | 1.083 |
| Largest diff. peak/hole (eÅ−3) | 0.15/−0.16 | 0.39/−0.21 |
| Flack parameter | 0.5 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peeva, M.I.; Georgieva, M.G.; Balacheva, A.A.; Ponticelli, M.; Bogdanov, I.P.; Kolev, T.; Milella, L.; Stammler, H.-G.; Tzvetkov, N.T. Crystal Structures, Molecular Docking and In Vitro Investigations of Two 4-Substituted 2-(5,5-dimethyl-3-styrylcyclohex-2-enylidene)malononitrile Derivatives as Potential Topoisomerase II Inhibitors. Crystals 2024, 14, 496. https://doi.org/10.3390/cryst14060496
Peeva MI, Georgieva MG, Balacheva AA, Ponticelli M, Bogdanov IP, Kolev T, Milella L, Stammler H-G, Tzvetkov NT. Crystal Structures, Molecular Docking and In Vitro Investigations of Two 4-Substituted 2-(5,5-dimethyl-3-styrylcyclohex-2-enylidene)malononitrile Derivatives as Potential Topoisomerase II Inhibitors. Crystals. 2024; 14(6):496. https://doi.org/10.3390/cryst14060496
Chicago/Turabian StylePeeva, Martina I., Maya G. Georgieva, Aneliya A. Balacheva, Maria Ponticelli, Ivan P. Bogdanov, Tsonko Kolev, Luigi Milella, Hans-Georg Stammler, and Nikolay T. Tzvetkov. 2024. "Crystal Structures, Molecular Docking and In Vitro Investigations of Two 4-Substituted 2-(5,5-dimethyl-3-styrylcyclohex-2-enylidene)malononitrile Derivatives as Potential Topoisomerase II Inhibitors" Crystals 14, no. 6: 496. https://doi.org/10.3390/cryst14060496
APA StylePeeva, M. I., Georgieva, M. G., Balacheva, A. A., Ponticelli, M., Bogdanov, I. P., Kolev, T., Milella, L., Stammler, H.-G., & Tzvetkov, N. T. (2024). Crystal Structures, Molecular Docking and In Vitro Investigations of Two 4-Substituted 2-(5,5-dimethyl-3-styrylcyclohex-2-enylidene)malononitrile Derivatives as Potential Topoisomerase II Inhibitors. Crystals, 14(6), 496. https://doi.org/10.3390/cryst14060496