
Novel Salts of Heterocyclic Polyamines and 5-Sulfosalicylic Acid: Synthesis, Crystal Structure, and Hierarchical Supramolecular Interactions


Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis of Compounds 1–6
2.2. Single-Crystal X-ray Diffraction
2.3. Computational Details
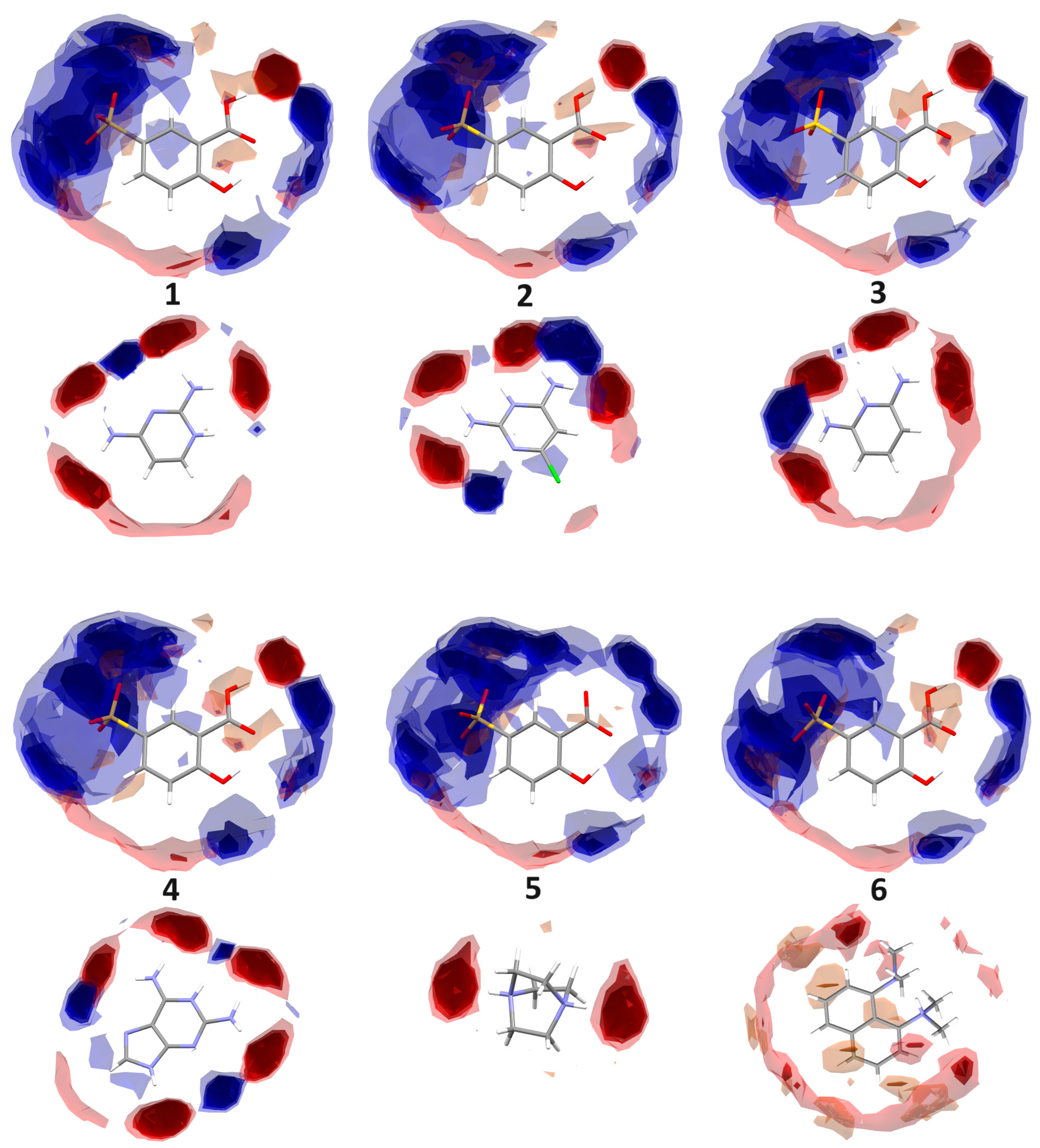
2.3.1. Full Interaction Maps
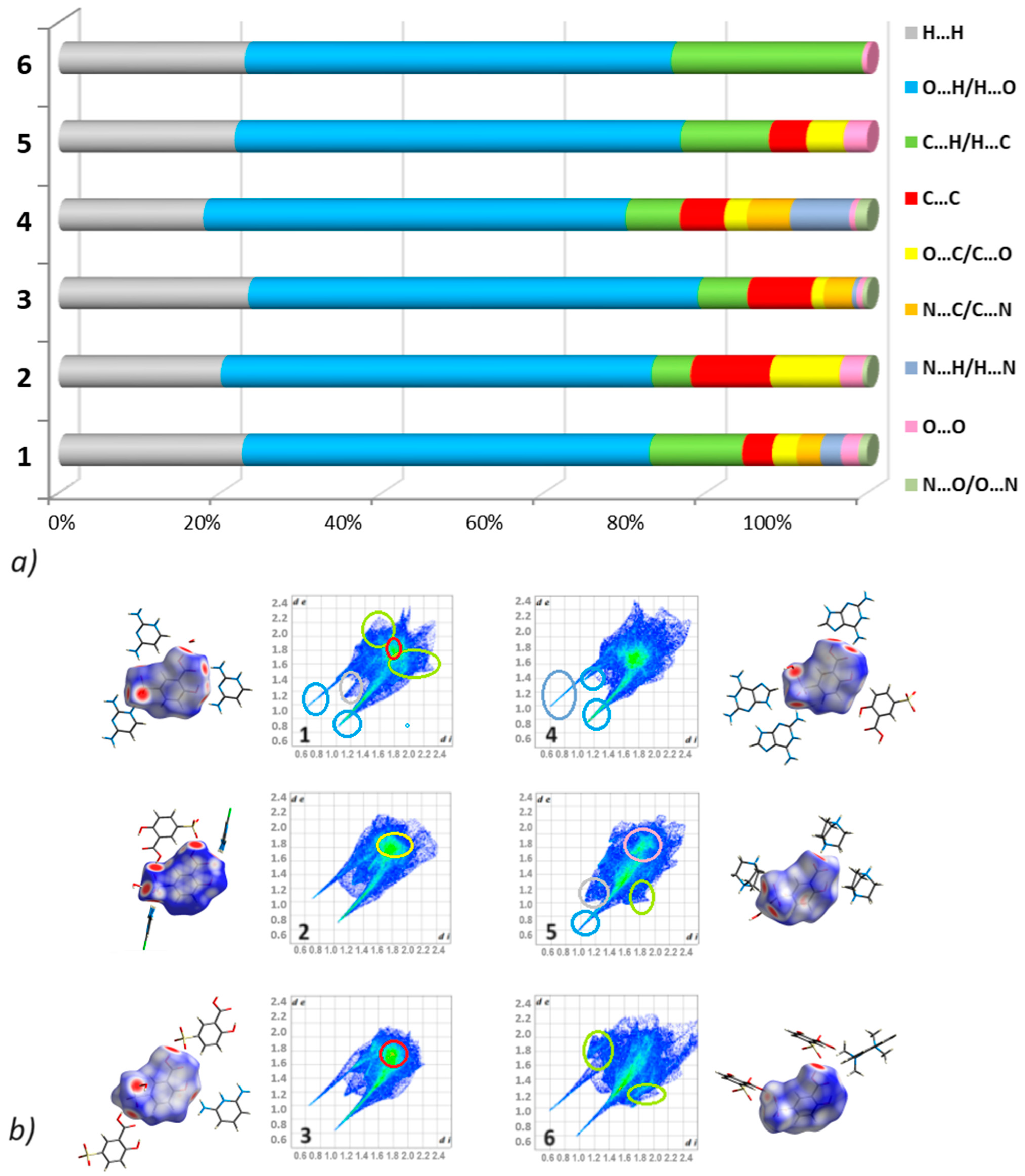
2.3.2. Hirshfeld Surface Analysis
2.3.3. Molecular Electrostatic Potential
2.3.4. The Enrichment Ratio
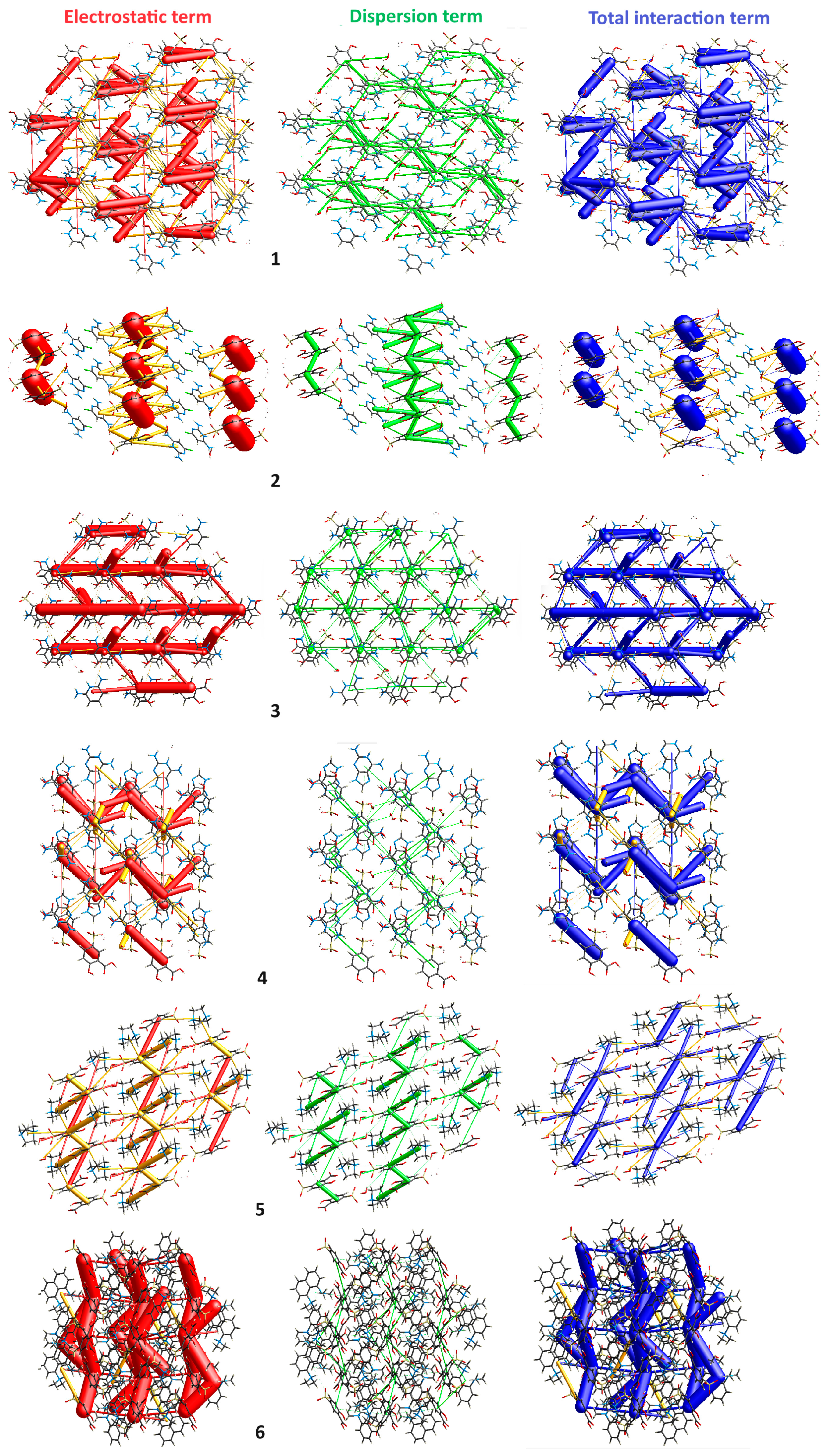
2.3.5. Energy Frameworks
3. Results and Discussion
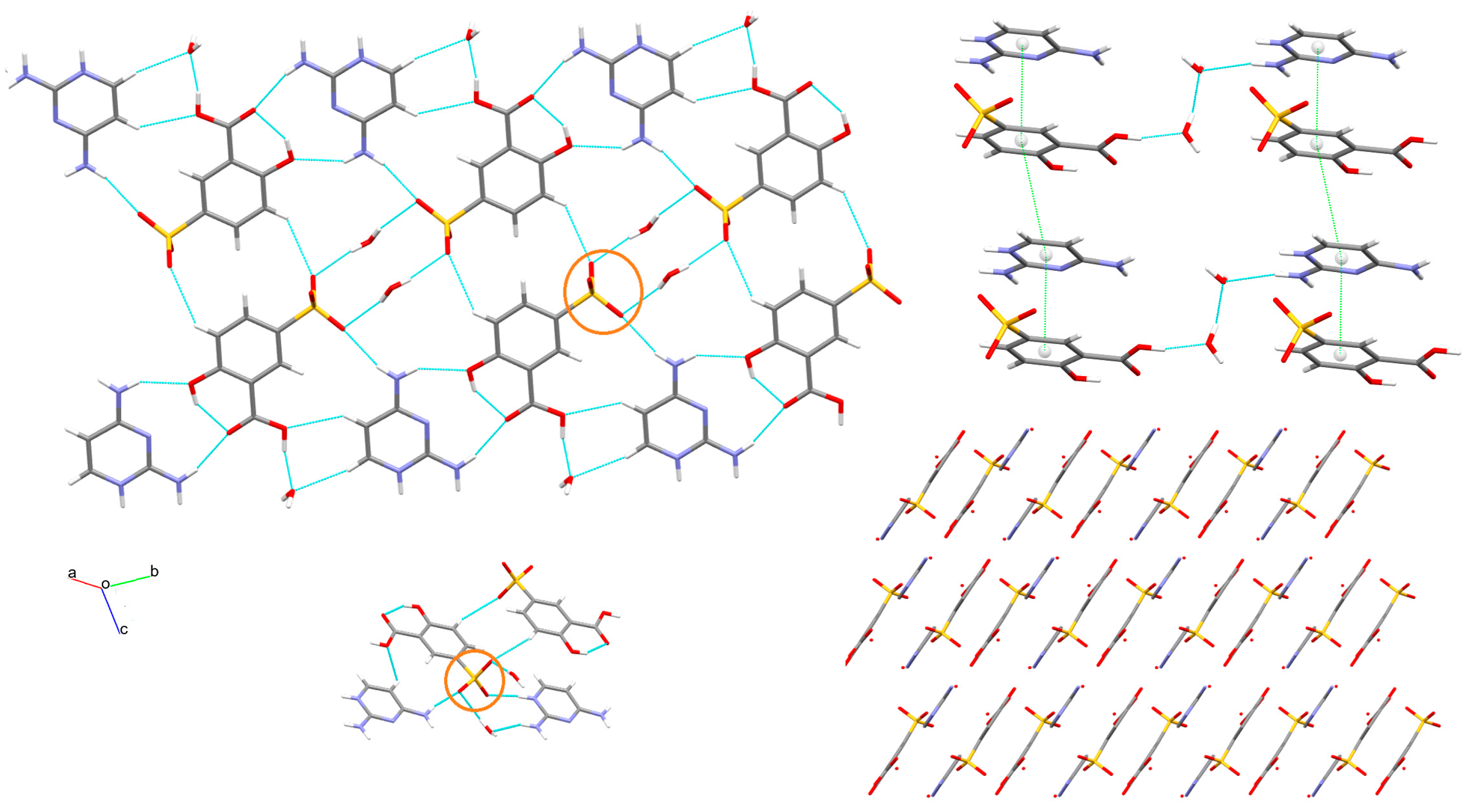
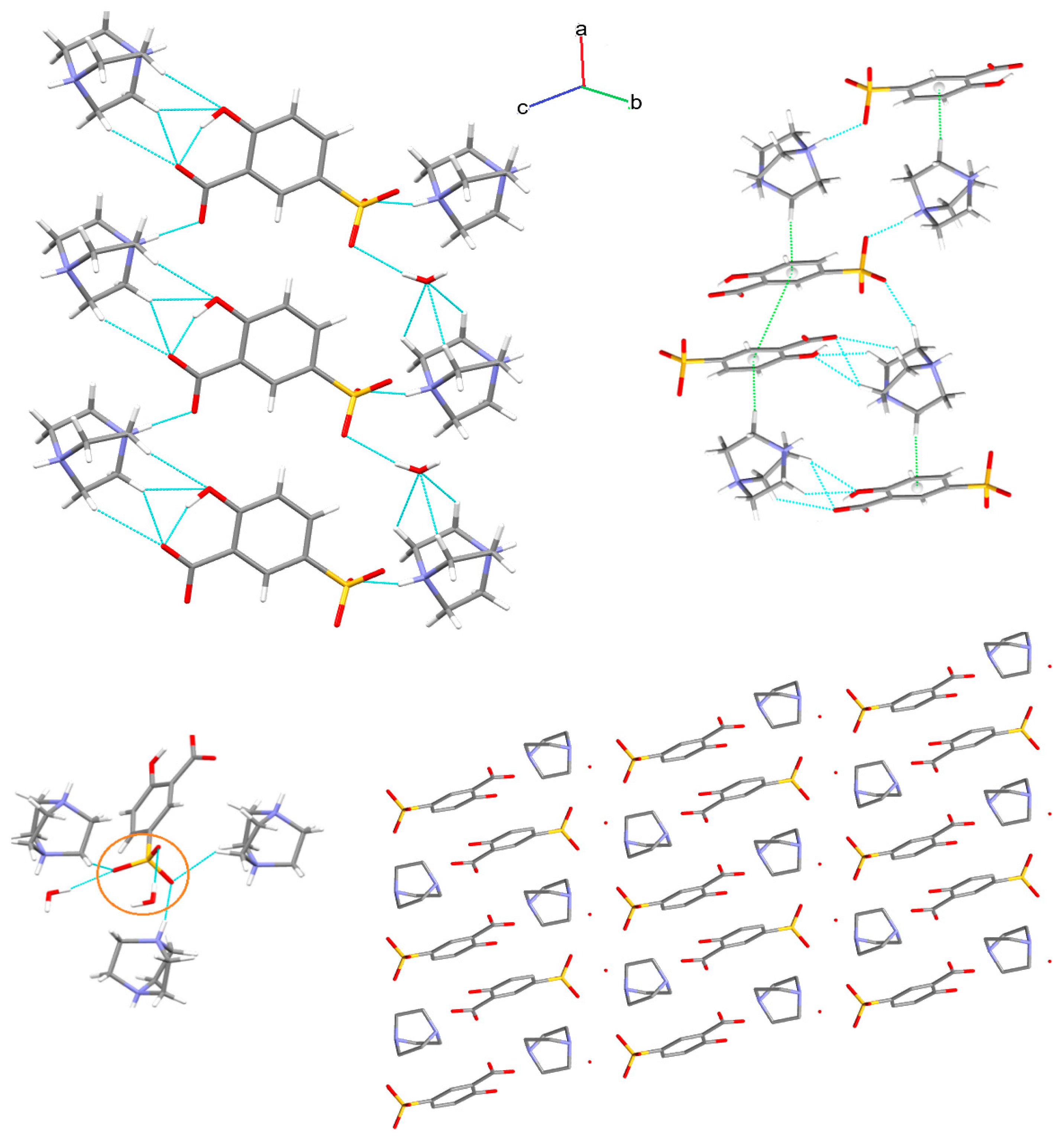
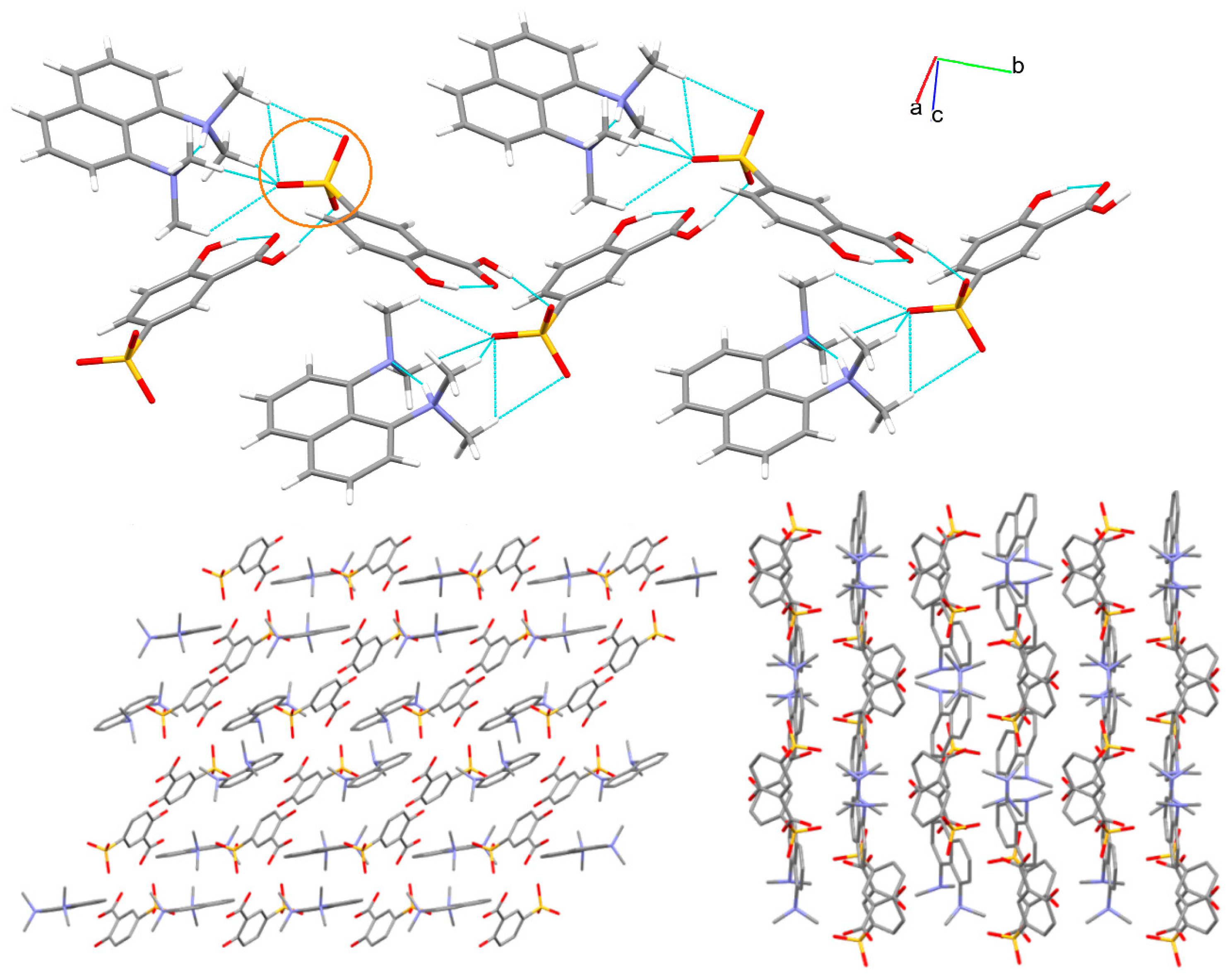
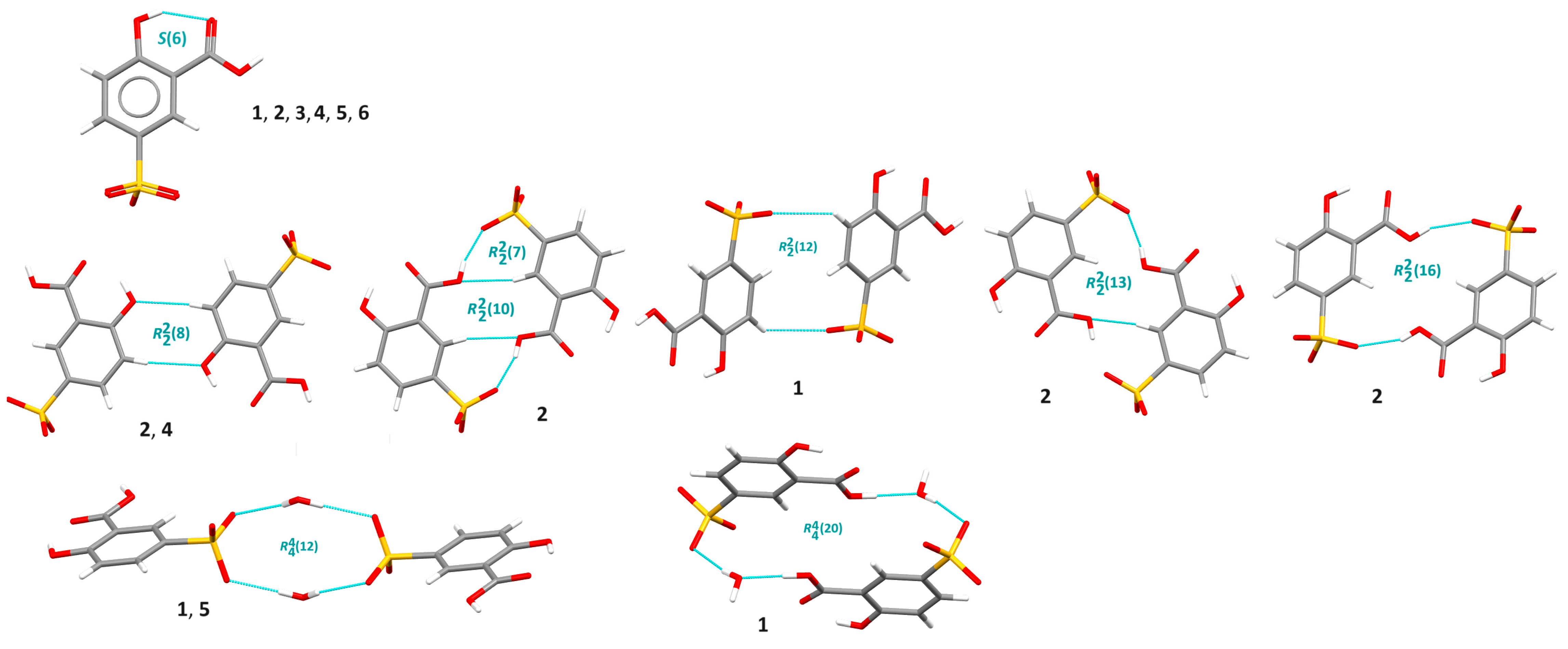
3.1. X-ray Structure and Supramolecular Features
3.2. Hirshfeld Surface Analysis
3.2.1. Molecular Electrostatic Potential
3.2.2. Fingerprints
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ganie, A.A.; Vishnoi, P.; Dar, A.A. Utility of Bis-4-pyridines as Supramolecular Linkers for 5-Sulfosalicylic Acid Centers: Structural and Optical Investigations. Cryst. Growth Des. 2019, 19, 2289–2297. [Google Scholar] [CrossRef]
- Wu, Y.; Hao, X.; Li, J.; Guan, A.; Zhou, Z.; Guo, F. New insight into improving the solubility of poorly soluble drugs by preventing the formation of their hydrogen-bonds: A case of dapsone salts with camphorsulfonic and 5-sulfosalicylic acid. CrystEngComm 2021, 23, 6191–6198. [Google Scholar] [CrossRef]
- Du, H.; Xu, L.; Yue, M.; Xu, F.; Wang, Y. Studies on crystal structures, optical, solubility and dyeing properties of two new crystalline dye salts based on berberine with aromatic carboxylic acid. J. Mol. Struct. 2022, 1260, 132856. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Duan, Y.; Liu, L.-X.; Chang, L.; Feng, Y.-R.; Wu, L.-L.; Zhang, L.; Zhang, Y.-J.; Zou, D.-Y.; Liu, Y.-L.; et al. On improving the hygroscopic stability of palmatine chloride with crystalline palmatine sulfosalicylate pharmaceutical salt. J. Struct. Chem. 2022, 63, 52–61. [Google Scholar] [CrossRef]
- Hua, X.-N.; Pan, X.; Zhu, Y.; Cai, Z.; Song, Q.; Li, Y.; Feng, W.; Chen, X.; Zhang, H.; Sun, B. Novel pharmaceutical salts of cephalexin with organic counterions: Structural analysis and properties. RSC Adv. 2022, 12, 34843–34850. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Lone, S.H.; Ahmad, I.; Ahangar, A.A.; Ganie, A.A.; Femina, C. Engineering the solid-state luminescence of organic crystals and cocrystals. Mater. Adv. 2024, 5, 1056–1064. [Google Scholar] [CrossRef]
- Duan, W.; Liu, B.; Gong, N.; Famulari, A.; Guo, F. Polymorphs and Transformations of the Solid Forms of Organic Salts of 5-Sulfosalicylic Acid and Isonicotinamide. Cryst. Growth Des. 2020, 20, 7606–7614. [Google Scholar] [CrossRef]
- Ganie, A.A.; Ismail, T.M.; Sajith, P.K.; Dar, A.A. Validation of the supramolecular synthon preference through DFT and physicochemical property investigations of pyridyl salts of organo-sulfonates. New J. Chem. 2021, 45, 4780–4790. [Google Scholar] [CrossRef]
- Ganie, A.A.; Marimuthu, R.; Islam, S.T.; Narang, S.; Dar, A.A. Molecular salts of the isoniazid derivatives. Expanding the scope of sulfonate-pyridinium synthon to design materials. J. Solid State Chem. 2022, 307, 122762. [Google Scholar] [CrossRef]
- Ramesh, K.S.; Saravanabhavan, M.; Muhammad, S.; Edison, D.; Ho, M.-S.; Sekar, M.; Al-Sehemi, A.G. Synthesis, physico-chemical characterization and quantum chemical studies of 2, 3-dimethyl quinoxalinium-5-sulphosalicylate crystal. J. Mol. Struct. 2023, 1285, 135425. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D. Hydrogen bonding in two ammonium salts of 5-sulfosalicylic acid: Ammonium 3-carboxy-4-hydroxybenzenesulfonate monohydrate and triammonium 3-carboxy-4-hydroxybenzenesulfonate 3-carboxylato-4-hydroxybenzenesulfonate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2013, 69, 534–537. [Google Scholar] [CrossRef]
- Selvaraju, D.; Venkatesh, R.; Sundararajan, V. Hydrazine-1,2-diium bis(3-carboxy-4-hydroxybenzenesulfonate) tetrahydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o1236–o1237. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D.; Healy, P.C. Hydrogen bonding in proton-transfer compounds of 5-sulfosalicylic acid with ortho-substituted monocyclic heteroaromatic Lewis bases. J. Chem. Crystallogr. 2006, 36, 841–849. [Google Scholar] [CrossRef]
- Raj, S.B.; Sethuraman, V.; Francis, S.; Hemamalini, M.; Muthiah, P.T.; Bocelli, G.; Cantoni, A.; Rychlewska, U.; Warzajtis, B. Supramolecular organization via hydrogen bonding in trimethoprim sulfonate salts. CrystEngComm 2003, 5, 70–76. [Google Scholar] [CrossRef]
- Hemamalini, M.; Muthiah, P.T.; Sridhar, B.; Rajaram, R.K. Pyrimethaminium sulfosalicylate monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, o1480–o1482. [Google Scholar] [CrossRef]
- Matulkova, I.; Mathauserova, J.; Cisarova, I.; Nemec, I.; Fabry, J. The study of crystal structures and vibrational spectra of inorganic salts of 2,4-diaminopyrimidine. J. Mol. Struct. 2016, 1103, 82–93. [Google Scholar] [CrossRef]
- Bertolasi, V.; Pretto, L.; Gilli, P.; Ferretti, V.; Gilli, G. Hydrogen-bonded supramolecular structures in co-crystals of β- or ζ-diketone enols with 2,6-diaminopyridine or 2,4-diaminopyrimidine. New J. Chem. 2002, 26, 1559–1566. [Google Scholar] [CrossRef]
- Draguta, S.; Fonari, M.S.; Masunov, A.E.; Zazueta, J.; Sullivan, S.; Antipin, M.Y.; Timofeeva, T.V. New acentric materials constructed from aminopyridines and 4-nitrophenol. CrystEngComm 2013, 15, 4700–4710. [Google Scholar] [CrossRef]
- Hutzler, W.M.; Egert, E.; Bolte, M. 6-Propyl-2-thiouracil versus 6-methoxymethyl-2-thiouracil: Enhancing the hydrogen-bonded synthon motif by replacement of a methylene group with an O atom. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2016, 72, 634–646. [Google Scholar] [CrossRef]
- Hutzler, W.M.; Egert, E.; Bolte, M. One barbiturate and two solvated thiobarbiturates containing the triply hydrogen-bonded ADA/DAD synthon, plus one ansolvate and three solvates of their coformer 2,4-diaminopyrimidine. Acta Cryst. C 2016, 72, 705–715. [Google Scholar] [CrossRef]
- Hall, V.M.; Thornton, A.; Miehls, E.K.; Bertke, J.A.; Swift, J.A. Uric Acid Crystallization Interrupted with Competing Binding Agents. Cryst. Growth Des. 2019, 19, 7363–7371. [Google Scholar] [CrossRef]
- Bojarska, J.; Łyczko, K.; Mieczkowski, A. Synthesis, Crystal Structure and Supramolecular Features of Novel 2,4-Diaminopyrimidine Salts. Crystals 2024, 14, 133. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D.; Healy, P.C. Layered structures in proton-transfer compounds of 5-sulfosalicylic acid with the aromatic polyamines 2,6-diaminopyridine and 1,4-phenylenediamine. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2005, 61, o555–o558. [Google Scholar] [CrossRef] [PubMed]
- Hemamalini, M.; Goh, J.H.; Fun, H.-K. 2,3-Diaminopyridinium 3-carboxy-4-hydroxybenzenesulfonate monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o3122. [Google Scholar] [CrossRef] [PubMed]
- Hemamalini, M.; Fun, H.-K. 2-Amino-5-chloropyridinium 3-carboxy-4-hydroxybenzenesulfonate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o2323–o2324. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Rajakannan, V.; Prabhaharan, M.; Srinivasan, G. 2-Amino-6-chloropyridinium 3-carboxy-4-hydroxybenzenesulfonate. IUCrData 2019, 4, x190566. [Google Scholar] [CrossRef]
- Hemamalini, M.; Fun, H.-K. 2-Amino-5-bromopyridinium 3-carboxy-4-hydroxybenzenesulfonate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o2408–o2409. [Google Scholar] [CrossRef] [PubMed]
- Hemamalini, M.; Fun, H.-K. 2-Amino-5-methylpyridinium 3-carboxy-4-hydroxybenzenesulfonate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o2153–o2154. [Google Scholar] [CrossRef]
- Atria, A.M.; Garland, M.T.; Baggio, R. 2,6-Diamino-9H-purine monohydrate and bis(2,6-diamino-9H-purin-1-ium) 2-(2-carboxylatophenyl)acetate heptahydrate: Two simple structures with very complex hydrogen-bonding schemes. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2010, 66, o547–o552. [Google Scholar] [CrossRef]
- Belletire, J.L.; Schneider, S.; Shackelford, S.A.; Peryshkov, D.V.; Strauss, S.H. Pairing heterocyclic cations with closo-dodecafluorododecaborate (2−): Synthesis of binary heterocyclium(1+) salts and a Ag4(heterocycle)84+ salt of B12F122−. J. Fluor. Chem. 2011, 132, 925–936. [Google Scholar] [CrossRef]
- de Vries, E.J.C.; Kantengwa, S.; Ayamine, A.; Báthori, N.B. Testing the limits of synthon engineering: Salts of salicylic and sulfosalicylic acid with nucleobases and derivatives. CrystEngComm 2016, 18, 7573–7579. [Google Scholar] [CrossRef]
- Xia, M.; Ma, K.-R.; Zhu, Y. Synthesis and Crystal Structure of Hydrate Adduct of 6-Benzylaminopurine and 5-Sulfosalicylic Acid [(C12H12N5)(C7H5O6S)·H2O]. J. Chem. Crystallogr. 2010, 40, 634–638. [Google Scholar] [CrossRef]
- Jones, A.O.F.; Kallay, A.A.; Lloyd, H.; McIntyre, G.J.; Wilson, C.C.; Thomas, L.H. The Effect of Local Crystalline Environment on Hydrogen Atom Behavior in Molecular Complexes of a Proton Sponge. Cryst. Growth Des. 2016, 16, 2123–2129. [Google Scholar] [CrossRef]
- Sigala, P.A.; Ruben, E.A.; Liu, C.W.; Piccoli, P.M.B.; Hohenstein, E.G.; Martinez, T.J.; Schultz, A.J.; Herschlag, D. Determination of Hydrogen Bond Structure in Water versus Aprotic Environments To Test the Relationship Between Length and Stability. J. Am. Chem. Soc. 2015, 137, 5730–5740. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.-Y.; Tang, Y.-Y.; Li, P.-F.; Liao, W.-Q.; Gao, J.-X.; Hua, X.-N.; Cai, H.; Shi, P.-P.; You, Y.-M.; Xiong, R.-G. Metal-free three-dimensional perovskite ferroelectrics. Science 2018, 361, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Duan, H.-B.; Jellen, M.J.; Chen, Y.; Chung, T.S.; Liang, Y.; Garcia-Garibay, M.A. Thermally Activated Transient Dipoles and Rotational Dynamics of Hydrogen-Bonded and Charge-Transferred Diazabicyclo [2.2.2]Octane Molecular Rotors. J. Am. Chem. Soc. 2019, 141, 16802–16809. [Google Scholar] [CrossRef]
- Song, X.; Cui, Q.; Liu, Y.; Xu, Z.; Cohen, H.; Ma, C.; Fan, Y.; Zhang, Y.; Ye, H.; Peng, Z.; et al. Metal-Free Halide Perovskite Single Crystals with Very Long Charge Lifetimes for Efficient X-ray Imaging. Adv. Mater. 2020, 32, 2003353. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.-L.; Zhang, T.; Zhang, W.-Y.; Shi, P.-P.; Gao, J.-X.; Ye, Q.; Fu, D.-W. Three-Dimensional Metal-Free Molecular Perovskite with a Thermally Induced Switchable Dielectric Response. J. Phys. Chem. Lett. 2020, 11, 1668–1674. [Google Scholar] [CrossRef]
- Budzianowski, A.; Petřiček, V.; Katrusiak, A. Metal-free enantiomorphic perovskite [dabcoH2]2+[H3O]+Br3− and its one-dimensional polar polymorph. IUCrJ 2022, 9, 544–550. [Google Scholar] [CrossRef]
- Song, X.; Cohen, H.; Yin, J.; Li, H.; Wang, J.; Yuan, Y.; Huang, R.; Cui, Q.; Ma, C.; Liu (Frank), S.; et al. Low Dimensional, Metal-Free, Hydrazinium Halide Perovskite-Related Single Crystals and Their Use as X-Ray Detectors. Small 2023, 19, 2300892. [Google Scholar] [CrossRef]
- Jin, S.; Guo, J.; Liu, L.; Wang, D. Five organic salts assembled from carboxylic acids and bis-imidazole derivatives through collective noncovalent interactions. J. Mol. Struct. 2011, 1004, 227–236. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, M.-Q.; Famulari, A.; Marti-Rujas, J. Solid state transformations in stoichiometric hydrogen bonded molecular salts: Ionic interconversion and dehydration processes. CrystEngComm 2013, 15, 6237–6243. [Google Scholar] [CrossRef]
- Liu, H.-l.; Xie, Y.-F.; Pan, Z.-G.; Famulari, A.; Guo, F.; Zhou, Z.; Martí-Rujas, J. Cyclic Interconversion among Molecular Salts via Neat Grinding and Related Photoluminescence Properties. Cryst. Growth Des. 2014, 14, 6528–6536. [Google Scholar] [CrossRef]
- Fu, Q.; Xu, X.-K.; Liu, B.-K.; Guo, F. Solid state transformations of different stoichiometric forms of an organic salt formed from 5-sulfosalicylic acid and hexamethylenetetramine upon dehydration and rehydration. CrystEngComm 2018, 20, 1844–1852. [Google Scholar] [CrossRef]
- Yang, D.-J.; Qu, S.-H. Bis(2-aminopyridinium) 5-sulfonatosalicylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, o5127–o5129. [Google Scholar] [CrossRef]
- Jin, S.; Zhao, Y.; Liu, B.; Jin, X.; Zhang, H.; Wen, X.; Liu, H.; Jin, L.; Wang, D. Hydrogen bonded supramolecular structures of eight organic salts based on 2,6-diaminopyridine, and organic acids. J. Mol. Struct. 2015, 1099, 601–615. [Google Scholar] [CrossRef]
- Balasubramani, K.; Muthiah, P.T.; Lynch, D.E. R22(8) motifs in aminopyrimidine sulfonate/carboxylate interactions: Crystal structures of pyrimethaminium benzenesulfonate monohydrate (2:2:1) and 2-amino-4,6-dimethylpyrimidinium sulfosalicylate dihydrate (4:2:2). Chem. Cent. J. 2007, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Jin, L.; Ye, X.; Li, J.; Jin, B.; Zheng, L.; Wang, D. Crystal and Molecular Structure of Three Organic Salts from N6-Benzyladenine, 5-Sulfosalicylic Acid, Naphthalene-1,5-Disulfonic Acid, and α-Ketoglutaric Acid. J. Chem. Crystallogr. 2015, 45, 9–19. [Google Scholar] [CrossRef]
- Krishnamohan, S.C.V. Crystal engineering––Where do we go from here? Cryst. Growth Des. 2002, 2, 465–474. [Google Scholar] [CrossRef]
- Braga, D.; Brammer, L.; Champness, N.R. New trends in crystal engineering. CrystEngComm 2005, 7, 1–19. [Google Scholar] [CrossRef]
- Bond, A.D.; Jones, W. Supramolecular Organization And Materials Design; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Walsh, R.D.B.; Bradner, M.W.; Fleischman, S.; Morales, L.A.; Moulton, B.; Rodríguez-Hornedo, N.; Zaworotko, M.J. Crystal Engineering of the Composition of Pharmaceutical Phases. Chem. Commun. 2003, 2, 186–187. [Google Scholar] [CrossRef]
- Bojarska, J.; Breza, M.; Remko, M.; Yuan, Y.; Ziora, Z.; Chai, T.-T.; Madura, I.D.; Kaczmarek, K.; Blaskovich, M.A.T.; Wolf, M.W. A supramolecular self-assembly of peptide-derived compounds via 1,5-disubstituted tetrazole-based supramolecular synthons: An experimental and computational study. J. Mol. Struct. 2023, 1288, 135732. [Google Scholar] [CrossRef]
- Bojarska, J.; Breza, M.; Remko, M.; Borowiecki, P.; Fruziński, A.; Madura, I.D.; Kaczmarek, K.; Leśnikowski, Z.; Kraj, A.; Zielenkiewicz, P.; et al. Supramolecular synthon hierarchy in cyclopropyl containing peptide-derived compounds. CrystEngComm 2022, 24, 8372–8389. [Google Scholar] [CrossRef]
- Bojarska, J.; Breza, M.; Remko, M.; Czyz, M.; Gajos-Michniewicz, A.; Zimecki, M.; Kaczmarek, K.; Madura, I.D.; Wojciechowski, J.M.; Wolf, W.M. Structural and Biofunctional Insights into the Cyclo(Pro-Pro-Phe-Phe-)Scaffold from Experimental and In Silico Studies: Melanoma and Beyond. Int. J. Mol. Sci. 2022, 23, 7173. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Madura, I.D.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. Synthesis, experimental and in silico studies of N-fluorenylmethoxycarbonyl-O-tert-butyl-N-methyltyrosine, coupled with CSD data: A survey of interactions in the crystal structures of Fmoc-amino acids. Acta Crystallogr. C 2020, 76, 328–345. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C Struc. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in chemical crystallography. Acta Cryst. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of Molecular Crystal Structures Using Full Interaction Maps. CrystEngComm 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wol, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer, Version 3.1; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Computational Science—ICCS 2003; Sloot, P.M.A., Abramson, D., Bogdanov, A.V., Gorbachev, Y.E., Dongarra, J.J., Zomaya, A.Y., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2660, pp. 142–151. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006. [Google Scholar] [CrossRef] [PubMed]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld Surfaces: A New Tool for Visualizing and Exploring Molecular Crystals. Chem. Eur. J. 1998, 4, 2136. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E Crystallogr. Commun. 2019, 75, 308. [Google Scholar] [CrossRef]
- Kitaigorodskii, A.I. Organic Chemical Crystallography; Consultants Bureau: New York, NY, USA, 1961; pp. 1–30, 65–112. [Google Scholar]
- Kitaigorodskii, A.I. Molecular Crystals and Molecules; Academic Press: London, UK, 1973; pp. 1–133. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Chemical formula | C11H16N4O8S | C11H13ClN4O7S | C12H15N3O7S | C12H14N6O7S | C13H20N2O7S | C21H24N2O6S |
| Formula weight | 364.34 | 380.76 | 345.33 | 386.35 | 348.37 | 432.48 |
| λ (Cu Kα) (Å) | 1.54184 | 1.54184 | 154184 | 1.54184 | 1.54184 | 1.54184 |
| Crystal system | triclinic | triclinic | monoclinic | triclinic | triclinic | monoclinic |
| Space group | P | P | P 21 | P | P | P 21/c |
| a (Å) | 7.9539(3) | 6.8906(6) | 6.6358(2) | 6.6755(4) | 6.7903(4) | 19.7312(3) |
| b (Å) | 9.5529(4) | 6.9077(6) | 12.9481(4) | 10.6495(5) | 8.7284(8) | 12.33559(11) |
| c (Å) | 11.2394(4) | 16.9140(11) | 8.4812(3) | 10.9153(5) | 13.3336(7) | 18.4437(3) |
| α (°) | 67.904(4) | 96.621(6) | 90 | 84.427(4) | 101.707(6) | 90 |
| β (°) | 85.375(3) | 96.880(6) | 104.703(3) | 87.076(5) | 103.408(5) | 114.7954(16) |
| γ (°) | 78.700(3) | 108.636(8) | 90 | 83.698(5) | 94.585(6) | 90 |
| Volume (Å) | 775.92(6) | 747.20(11) | 704.84(4) | 767.01(7) | 746.02(9) | 4075.28(10) |
| Z | 2 | 2 | 2 | 2 | 2 | 8 |
| Dcalc (g·cm−3) | 1.559 | 1.692 | 1.627 | 1.673 | 1.551 | 1.775 |
| μ (mm−1) | 2.344 | 4.022 | 2.470 | 2.405 | 2.312 | 1.410 |
| F (000) | 380 | 392 | 360 | 400 | 368 | 1824 |
| Crystal size (mm) | 0.18 × 0.16 × 0.08 | 0.15 × 0.08 × 0.03 | 0.18 × 0.10 × 0.05 | 0.22 × 0.04 × 0.03 | 0.18 × 0.14 × 0.10 | 0.20 × 0.18 × 0.10 |
| θ range (°) | 4.245–70.422 | 2.667–70.282 | 5.392–70.328 | 4.073–70.352 | 3.500–70.225 | 2.467–70.187 |
| Reflections collected | 11,295 | 5414 | 4501 | 9276 | 4728 | 28,523 |
| Unique reflections | 2926 | 2792 | 2451 | 2858 | 2776 | 7674 |
| Reflections I > 2σ(I) | 2805 | 2419 | 2365 | 2484 | 2513 | 6949 |
| Rint | 0.0220 | 0.0290 | 0.0211 | 0.0361 | 0.0187 | 0.0222 |
| Restraints/parameters | 1/261 | 1/253 | 1/244 | 0/274 | 1/223 | 2/567 |
| Goodness-of-fit | 1.086 | 1.116 | 1.073 | 1.073 | 1.047 | 1.043 |
| R1, wR2 (I > 2σ(I)) | 0.0261, 0.0735 | 0.0379, 0.0986 | 0.0276, 0.0695 | 0.0432, 0.1096 | 0.0335, 0.0853 | 0.0303, 0.0827 |
| R1, wR2 (all data) | 0.0273, 0.0745 | 0.0443, 0.1047 | 0.0293, 0.0716 | 0.0533, 0.1161 | 0.0373, 0.0881 | 0.0338, 0.0855 |
| Max. peak/hole (e·Å−3) | 0.359/−0.468 | 0.307/−0.602 | 0.202/−0.298 | 0.674/−0.473 | 0.398/−0.427 | 0.302/−0.482 |
| K.P.I. [%] | 70.2 | 72.6 | 72.7 | 74.2 | 70.6 | 74.4 |
| 9 | D-H [Å] | H⋯A [Å] | D⋯A [Å] | D-H⋯A [o] |
|---|---|---|---|---|
| 1 | ||||
| N1-H1⋯O2 i | 0.883(19) | 1.869(19) | 2.7472(14) | 172.7(17) |
| N3-H3A⋯O8 ii | 0.886(19) | 2.154(19) | 2.9655(16) | 152.1(17) |
| N3-H3B⋯O5 iii | 0.853(19) | 2.136(19) | 2.9698(16) | 165.5(19) |
| O4-H4⋯O7 | 0.90(2) | 1.69(2) | 2.5685(13) | 166(3) |
| N4-H4A⋯O6 ii | 0.857(19) | 2.141(19) | 2.9205(17) | 151.1(17) |
| N4-H4B⋯O3 iv | 0.869(19) | 2.082(19) | 2.9313(16) | 165.5(18) |
| *O6-H6⋯O5 | 0.86(2) | 1.87(2) | 2.6397(14) | 149(2) |
| O6-H6⋯N2 iii | 0.86(2) | 2.44(2) | 2.9052(15) | 114.9(17) |
| O7-H7A⋯O1 v | 0.83(2) | 1.96(2) | 2.7803(14) | 172(2) |
| O7-H7B⋯O8 | 0.84(2) | 1.90(2) | 2.7306(15) | 168(2) |
| O8-H8A⋯O3 iv | 0.81(2) | 2.10(2) | 2.8640(14) | 157(2) |
| O8-H8B⋯O1 vi | 0.85(3) | 1.93(3) | 2.7594(15) | 166(2) |
| *C2-H2⋯O3 | 0.95 | 2.53 | 2.9197(17) | 105 |
| C5-H5⋯O1 vii | 0.95 | 2.57 | 3.2961(16) | 134 |
| C10-H10⋯O4 iv | 0.95 | 2.54 | 3.2942(18) | 137 |
| C11-H11⋯O7 iv | 0.95 | 2.42 | 3.3590(18) | 168 |
| (i) 1 − x, 1 − y, 2 − z; (ii) x, y, 1 + z; (iii) −x, 2 − y, 1 − z; (iv) 1 − x, 1 − y, 1 − z; (v) −x, 1 − y, 1 − z; (vi) x, 1 + y, −1 + z; (vii) −x, 1 − y, 2 − z | ||||
| 2 | ||||
| O1-H1⋯O4 i | 0.80(3) | 1.93(3) | 2.666(3) | 154(3) |
| N2-H2⋯O6 | 0.94(4) | 1.84(4) | 2.758(3) | 166(3) |
| *O3-H3⋯O2 | 0.89(4) | 1.85(4) | 2.615(3) | 144(3) |
| N3-H3B⋯N1 ii | 0.87(3) | 2.10(3) | 2.953(3) | 170(3) |
| N3-H3C⋯O5 | 0.89(4) | 1.98(3) | 2.862(3) | 169(3) |
| N4-H4A⋯O2 iii | 0.86(3) | 2.08(3) | 2.925(3) | 168(3) |
| N4-H4B⋯O7 | 0.92(4) | 1.92(4) | 2.813(3) | 163(3) |
| O7-H7A⋯O5 iv | 0.82(3) | 2.02(3) | 2.838(3) | 174(3) |
| O7-H7B⋯O6 v | 0.78(5) | 2.11(5) | 2.867(3) | 164(5) |
| C3-H3A⋯O3 vi | 0.95 | 2.54 | 3.355(4) | 144 |
| *C6-H6⋯O4 | 0.95 | 2.48 | 2.885(3) | 106 |
| C6-H6⋯O1 i | 0.95 | 2.54 | 3.339(3) | 142 |
| (i) 3 − x, 1 − y, 1 − z; (ii) 1 − x, −y, −z; (iii) 2 − x, 1 − y, 1 − z; (iv) −1 + x, 1 + y, z; (v) −1 + x, y, z; (vi) 1 − x, −y, 1 − z | ||||
| 3 | ||||
| N1-H1⋯O7 | 0.86(4) | 1.91(4) | 2.754(4) | 170(4) |
| N2-H2A⋯O3 i | 0.85(5) | 2.38(5) | 3.206(3) | 163(4) |
| N3-H3A⋯O6 ii | 0.85(5) | 2.24(5) | 3.012(3) | 152(4) |
| N3-H3B⋯O2 iii | 0.87(4) | 2.09(4) | 2.924(3) | 162(4) |
| O4-H4⋯O1 iii | 0.88(4) | 1.76(4) | 2.613(2) | 165(4) |
| *O6-H6⋯O5 | 0.80(5) | 1.89(5) | 2.606(3) | 148(5) |
| O7-H7A⋯O3 | 0.81(5) | 2.07(5) | 2.842(4) | 161(5) |
| O7-H7B⋯O1 iv | 0.81(5) | 2.00(5) | 2.768(3) | 160(5) |
| *C2-H2⋯O3 | 0.95 | 2.53 | 2.917(4) | 104 |
| (i) 1 − x, −1/2 + y, 2 − z; (ii) 1 − x, ½ + y, 1 − z; (iii) x, y, −1+z; (iv) 1 + x, y, z | ||||
| 4 | ||||
| N2-H2A⋯O7 | 0.91(3) | 1.84(3) | 2.730(3) | 166(3) |
| O4-H4⋯N3 i | 0.97(4) | 1.63(4) | 2.573(2) | 165(4) |
| N4-H4A⋯N1 ii | 0.87(3) | 2.04(3) | 2.891(3) | 170(3) |
| N5-H5A⋯O7 | 0.88(3) | 2.36(3) | 3.091(3) | 141(3) |
| N5-H5B⋯O2 iii | 0.84(3) | 1.99(3) | 2.817(3) | 170(3) |
| *O6-H6⋯O5 | 0.86(3) | 1.82(3) | 2.594(2) | 150(3) |
| N6-H6B⋯O3 | 0.89(3) | 1.94(3) | 2.781(3) | 159(2) |
| N6-H6C⋯O5 iv | 0.84(3) | 2.02(3) | 2.851(3) | 168(3) |
| O7-H7A⋯O1 v | 0.83(4) | 2.07(4) | 2.827(3) | 153(4) |
| O7-H7B⋯O3 vi | 0.80(4) | 2.09(4) | 2.877(3) | 165(3) |
| *C2-H2⋯O3 | 0.95 | 2.47 | 2.868(3) | 105 |
| C5-H5⋯O6 vii | 0.95 | 2.58 | 3.455(3) | 153 |
| C11-H11⋯O2 viii | 0.95 | 2.37 | 3.098(3) | 133 |
| (i) x, 1 + y, z; (ii) 1 − x, −y, 2 − z; (iii) x, y, 1 + z; (iv) x, −1 + y, z; (v) −x, 1 − y, 1 − z; (vi) 1 − x, 1 − y, 1 − z; (vii) −x, 2 − y, −z; (viii) 1 − x, −y, 1 − z | ||||
| 5 | ||||
| N1-H1⋯O6 i | 0.91(2) | 1.84(2) | 2.7102(18) | 161(2) |
| N2-H2⋯O1 ii | 0.916(17) | 1.632(17) | 2.5451(18) | 174.0(17) |
| N2-H2⋯O2 ii | 0.916(17) | 2.59(2) | 3.1463(19) | 119.9(14) |
| *O3-H3⋯O2 | 0.91(3) | 1.67(3) | 2.5278(19) | 155(2) |
| O7-H7A⋯O4 iii | 0.85(3) | 2.13(2) | 2.9623(19) | 168(2) |
| O7-H7B⋯O5 iv | 0.81(3) | 2.05(3) | 2.841(2) | 167(2) |
| *C6-H6⋯O4 | 0.95 | 2.59 | 2.951(2) | 103 |
| C8-H8A⋯O5 v | 0.99 | 2.47 | 3.423(2) | 161 |
| C8-H8B⋯O7 vi | 0.99 | 2.44 | 3.314(3) | 147 |
| C9-H9B⋯O3 vi | 0.99 | 2.47 | 3.329(2) | 145 |
| C11-H11A⋯O2 vi | 0.99 | 2.59 | 3.363(2) | 135 |
| C13-H13B⋯O7 vi | 0.99 | 2.59 | 3.356(2) | 134 |
| (i) 1 − x, 1 − y, 2 − z; (ii) −x, 1 − y, 1 − z; (iii) 1 + x, y, −1 + z; (iv) 1 − x, −y, 1 − z; (v) 1 + x, 1 + y, z; (vi) 1 − x, 1 − y, 1 − z | ||||
| 6 | ||||
| *N1-H1⋯N2 | 0.930(18) | 1.713(18) | 2.5973(15) | 157.6(17) |
| O1-H1A⋯O5 i | 0.93(2) | 1.63(2) | 2.5468(13) | 169(2) |
| *N3-H3⋯N4 | 0.93(2) | 1.707(19) | 2.5907(16) | 158.8(17) |
| *O3-H3A⋯O2 | 0.87(2) | 1.83(2) | 2.6134(17) | 148(2) |
| O7-H7⋯O11 ii | 0.92(2) | 1.66(2) | 2.5714(13) | 169(2) |
| *O9-H9⋯O8 | 0.89(2) | 1.79(2) | 2.6005(16) | 149(2) |
| C25-H25B⋯O4 iii | 0.98 | 2.52 | 3.4370(16) | 157 |
| C25-H25C⋯O2 iv | 0.98 | 2.51 | 3.3061(18) | 138 |
| C26-H26A⋯O5 v | 0.98 | 2.46 | 3.3977(18) | 161 |
| C27-H27C⋯O10 | 0.98 | 2.57 | 3.381(2) | 140 |
| C30-H30⋯O11 vi | 0.95 | 2.49 | 3.4349(16) | 173 |
| C39-H39C⋯O8 iii | 0.98 | 2.51 | 3.0482(17) | 114 |
| C40-H40A⋯O10 vi | 0.98 | 2.39 | 3.3235(18) | 159 |
| C41-H41B⋯O12 | 0.98 | 2.50 | 3.1961(17) | 128 |
| C42-H42C⋯O12 | 0.98 | 2.58 | 3.2782(16) | 128 |
| (i) −x, ½ + y, ½ − z; (ii) 1 − x, −1/2 + y, ½ − z; (iii) x, −1 + y, z; (iv) x, −1 + y, z; (v) −x, −1/2 + y, ½ − z; (vi) 1 − x, 1 − y, −z | ||||
| 1 | 2 | 3 | 4 | 5 | 6 | |
| H⋯H | 0.756 | 0.83 | 0.79 | 0.686 | 0.73 | 0.62 |
| O⋯H | 3.131 | 1.607 | 1.791 | 1.804 | 1.53 | 1.64 |
| C⋯H | 0.962 | 0.407 | 0.42 | 0.512 | 0.81 | 1.64 |
| N⋯H | 0.719 | 1.138 | ||||
| O⋯O | 0.246 | 0.247 | 0.27 | |||
| O⋯C | 0.471 | 1.043 | 0.202 | 0.382 | 0.57 | |
| O⋯N | 0.57 | 0.418 | ||||
| C⋯C | 1.967 | 6.53 | 4.3 | 3.279 | 2.99 | |
| C⋯N | 4.03 | 3.223 |
| N | R | Eele | Epol | Edisp | Erep | Etot | Symmetry Operation | |
|---|---|---|---|---|---|---|---|---|
| 1 | ||||||||
| 1 | 5.98 | 7.1 | 0.0 | −21.2 | 5.8 | −7.1 | −x, −y, −z | |
| 1 | 5.90 | 7.1 | −12.5 | −21.2 | 5.8 | −15.3 | ||
| 1 | 6.55 | −66.6 | 0.0 | −16.5 | 9.3 | −75.2 | −x, −y, −z | |
| 1 | 5.66 | −66.6 | −16.4 | −16.5 | 9.3 | −85.9 | ||
| 1 | 7.85 | 15.5 | −4.0 | −12.9 | 1.3 | 2.6 | −x, −y, −z | |
| 1 | 7.64 | −7.9 | −0.7 | −0.5 | 0.0 | −9.0 | ||
| 1 | 5.68 | −7.9 | −0.7 | −0.5 | 0.0 | −9.0 | ||
| 1 | 5.80 | −66.6 | −16.4 | −16.5 | 9.3 | −85.9 | ||
| 1 | 3.46 | 9.2 | −1.4 | −1.4 | 0.0 | 7.2 | ||
| 1 | 6.38 | 7.1 | −12.5 | −21.2 | 5.8 | −15.3 | ||
| 1 | 6.93 | 15.5 | −4.0 | −12.9 | 1.3 | 2.6 | ||
| 1 | 6.16 | 9.2 | −1.4 | −1.4 | 0.0 | 7.2 | ||
| 1 | 8.58 | −7.4 | −6.1 | −3.5 | 0.3 | −14.5 | −x, −y, −z | |
| 1 | 7.34 | −66.6 | −16.4 | −16.5 | 9.3 | −85.9 | ||
| −218.9 | −92.5 | −162.7 | 57.2 | −383.5 | ||||
| 2 | ||||||||
| 1 | 5.13 | 28.7 | −18.6 | −43.8 | 21.2 | −5.2 | −x, −y, −z | |
| 1 | 8.39 | 6.8 | −4.2 | −10.4 | 8.1 | 1.4 | −x, −y, −z | |
| 0 | 4.07 | 19.2 | −37.6 | −57.8 | 30.3 | −32.4 | −x, −y, −z | |
| 1 | 6.89 | 26.6 | −8.0 | −10.9 | 2.3 | 13.8 | x, y, z | |
| 1 | 7.00 | 0.0 | −4.2 | 0.0 | 0.0 | −2.7 | ||
| 0 | 6.96 | 35.9 | −2.5 | −2.2 | 0.0 | 32.9 | ||
| 1 | 4.79 | 26.6 | −8.0 | −10.9 | 2.3 | 13.8 | ||
| 0 | 6.09 | −176.3 | −61.8 | −22.7 | 79.7 | −175.6 | −x, −y, −z | |
| 0 | 5.65 | 8.7 | −2.3 | −1.8 | 0.0 | 5.7 | ||
| 0 | 6.53 | 8.7 | −2.3 | −1.8 | 0.0 | 5.7 | ||
| 1 | 8.65 | 28.7 | −18.6 | −43.8 | 21.2 | −5.2 | ||
| 1 | 7.90 | 28.7 | −18.6 | −43.8 | 21.2 | −5.2 | ||
| 42.3 | −186.7 | −249.9 | 186.3 | −153 | ||||
| 3 | ||||||||
| 1 | 9.13 | 8.5 | −1.1 | −5.6 | 2.9 | 5.2 | ||
| 1 | 4.05 | −93.4 | −30.6 | −11.1 | 55.8 | −79.8 | ||
| 1 | 3.65 | −46.1 | −18.2 | −50.1 | 32.6 | −77.5 | ||
| 2 | 9.01 | −23.9 | −3.7 | −2.3 | 0.1 | −28.7 | −x, y + 1/2, z | |
| 2 | 8.48 | −93.4 | −30.6 | −11.1 | 55.8 | −79.8 | x, y, z | |
| 1 | 4.14 | −12.0 | −2.4 | −1.9 | 0.0 | −15.5 | ||
| 1 | 6.21 | 0.0 | −0.2 | 0.0 | 0.0 | −0.1 | ||
| 1 | 3.49 | −55.6 | −21.0 | −48.5 | 28.3 | −91.1 | ||
| 1 | 8.13 | 1.4 | −2.5 | −8.0 | 13.5 | 3.6 | ||
| 1 | 6.20 | 0.9 | −8.3 | −14.2 | 8.4 | −10.4 | ||
| 2 | 8.49 | −25.3 | −5.5 | −3.8 | 0.2 | −32.6 | −x, y + 1/2, z | |
| 1 | 5.82 | −0.9 | −5.1 | −9.2 | 1.2 | −11.5 | ||
| 1 | 7.81 | −11.2 | −8.7 | −12.9 | 15.0 | −16.5 | ||
| 1 | 7.58 | −33.3 | −14.8 | −9.0 | 20.8 | −34.8 | ||
| −384.3 | −152.7 | −187.7 | 234.6 | −469.5 | ||||
| 4 | ||||||||
| 1 | 7.40 | −76.4 | −14.8 | −11.5 | 2.8 | −95.6 | −x, −y, −z | |
| 1 | 8.81 | 6.5 | −3.1 | −8.9 | 5.7 | 1.3 | −x, −y, −z | |
| 1 | 9.38 | 0.0 | −2.4 | 0.0 | 0.0 | −1.6 | ||
| 1 | 9.45 | 15.2 | −2.7 | −5.5 | 0.4 | 9.1 | −x, −y, −z | |
| 1 | 7.04 | 6.5 | −3.1 | −8.9 | 5.7 | 1.3 | ||
| 1 | 8.27 | −76.4 | −14.8 | −11.5 | 2.8 | −95.6 | ||
| 1 | 4.11 | 43.4 | −2.9 | −2.6 | 0.0 | 40.1 | ||
| 1 | 7.88 | 15.2 | −2.7 | −5.5 | 0.4 | 9.1 | ||
| 1 | 8.55 | −10.7 | −0.6 | −0.3 | 0.0 | −11.6 | ||
| 1 | 7.63 | −57.3 | −6.7 | −4.6 | 0.0 | −66.8 | ||
| 1 | 4.30 | −57.3 | −6.7 | −4.6 | 0.0 | −66.8 | ||
| 1 | 3.97 | 43.4 | −2.9 | −2.6 | 0.0 | 40.1 | ||
| −148 | −63.4 | −66.5 | 17.8 | −237 | ||||
| 5 | ||||||||
| 1 | 4.81 | 44.8 | −37.4 | −40.7 | 16.9 | −1.7 | −x, −y, −z | |
| 1 | 7.06 | −3.7 | −0.6 | −1.3 | 0.0 | −5.2 | ||
| 1 | 5.38 | 10.1 | −0.5 | −0.7 | 0.0 | 9.3 | ||
| 1 | 8.75 | 0.0 | −6.4 | 0.0 | 0.0 | −4.1 | −x, −y, −z | |
| 1 | 5.87 | 10.7 | −1.4 | −1.5 | 0.0 | 8.6 | ||
| 1 | 6.64 | −13.6 | −9.5 | −10.7 | 2.6 | −27.6 | ||
| 1 | 7.38 | −39.5 | −4.5 | −7.6 | 0.5 | −49.7 | −x, −y, −z | |
| 1 | 7.29 | 44.8 | −37.4 | −40.7 | 16.9 | −1.7 | ||
| 1 | 4.83 | 4.5 | −0.2 | −0.2 | 0.0 | 4.3 | ||
| 1 | 6.01 | −13.6 | −9.5 | −10.7 | 2.6 | −27.6 | ||
| 1 | 6.96 | 10.7 | −1.4 | −1.5 | 0.0 | 8.6 | ||
| 55.2 | −108.8 | −115.6 | 39.5 | −86.8 | ||||
| 6 | ||||||||
| 2 | 7.91 | −123.7 | −40.9 | −8.7 | 74 | −100.5 | −x, y + 1/2, −z + 1/2 | |
| 1 | 6.17 | −16.6 | −2.4 | −1.7 | 0.0 | −19.9 | ||
| 2 | 9.34 | −16.6 | −2.4 | −1.7 | 0.0 | −19.9 | x, −y + 1/2, z + 1/2 | |
| 1 | 8.06 | 28.9 | −1.4 | −0.5 | 0.0 | 28.1 | ||
| 1 | 6.45 | −123.7 | −40.9 | −8.7 | 74 | −100.5 | ||
| 1 | 6.94 | 0.0 | −0.1 | 0.0 | 0.0 | −0.1 | ||
| 1 | 9.53 | −16.6 | −2.4 | −1.7 | 0.0 | −19.9 | ||
| 1 | 9.60 | 0.0 | −0.3 | 0.0 | 0.0 | −0.2 | ||
| 1 | 9.73 | −123.7 | −40.9 | −8.7 | 74 | −100.5 | ||
| −392 | −131.7 | −31.7 | 222 | −333.4 | ||||
| Energy model | k_ele | k_pol | k_disp | k_rep | ||||
| CE-HF⋯HF/3-21G electron densities | 1.019 | 0.651 | 0.901 | 0.811 | ||||
| CE-B3LYP⋯B3LYP/6-31G(d,p) electron densities | 1.057 | 0.740 | 0.871 | 0.618 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojarska, J.; Łyczko, K.; Mieczkowski, A. Novel Salts of Heterocyclic Polyamines and 5-Sulfosalicylic Acid: Synthesis, Crystal Structure, and Hierarchical Supramolecular Interactions. Crystals 2024, 14, 497. https://doi.org/10.3390/cryst14060497
Bojarska J, Łyczko K, Mieczkowski A. Novel Salts of Heterocyclic Polyamines and 5-Sulfosalicylic Acid: Synthesis, Crystal Structure, and Hierarchical Supramolecular Interactions. Crystals. 2024; 14(6):497. https://doi.org/10.3390/cryst14060497
Chicago/Turabian StyleBojarska, Joanna, Krzysztof Łyczko, and Adam Mieczkowski. 2024. "Novel Salts of Heterocyclic Polyamines and 5-Sulfosalicylic Acid: Synthesis, Crystal Structure, and Hierarchical Supramolecular Interactions" Crystals 14, no. 6: 497. https://doi.org/10.3390/cryst14060497
APA StyleBojarska, J., Łyczko, K., & Mieczkowski, A. (2024). Novel Salts of Heterocyclic Polyamines and 5-Sulfosalicylic Acid: Synthesis, Crystal Structure, and Hierarchical Supramolecular Interactions. Crystals, 14(6), 497. https://doi.org/10.3390/cryst14060497