
Dissociative Adsorption of O2 on Ag3Au(111) Surface: A Density Functional Theory Study

Abstract
:1. Introduction
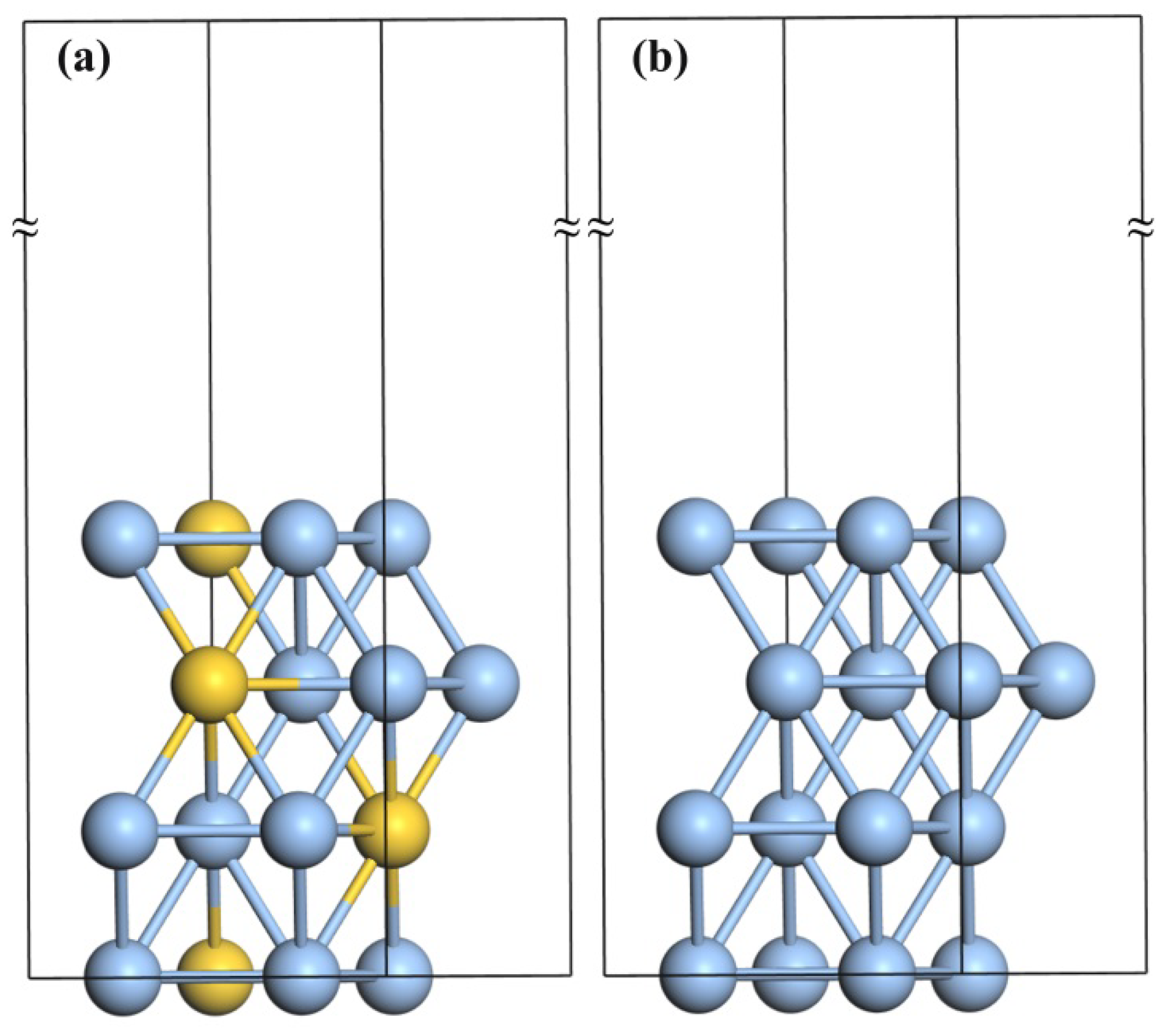
2. Computational Details
3. Results and Discussion
3.1. The Adsorption of O2
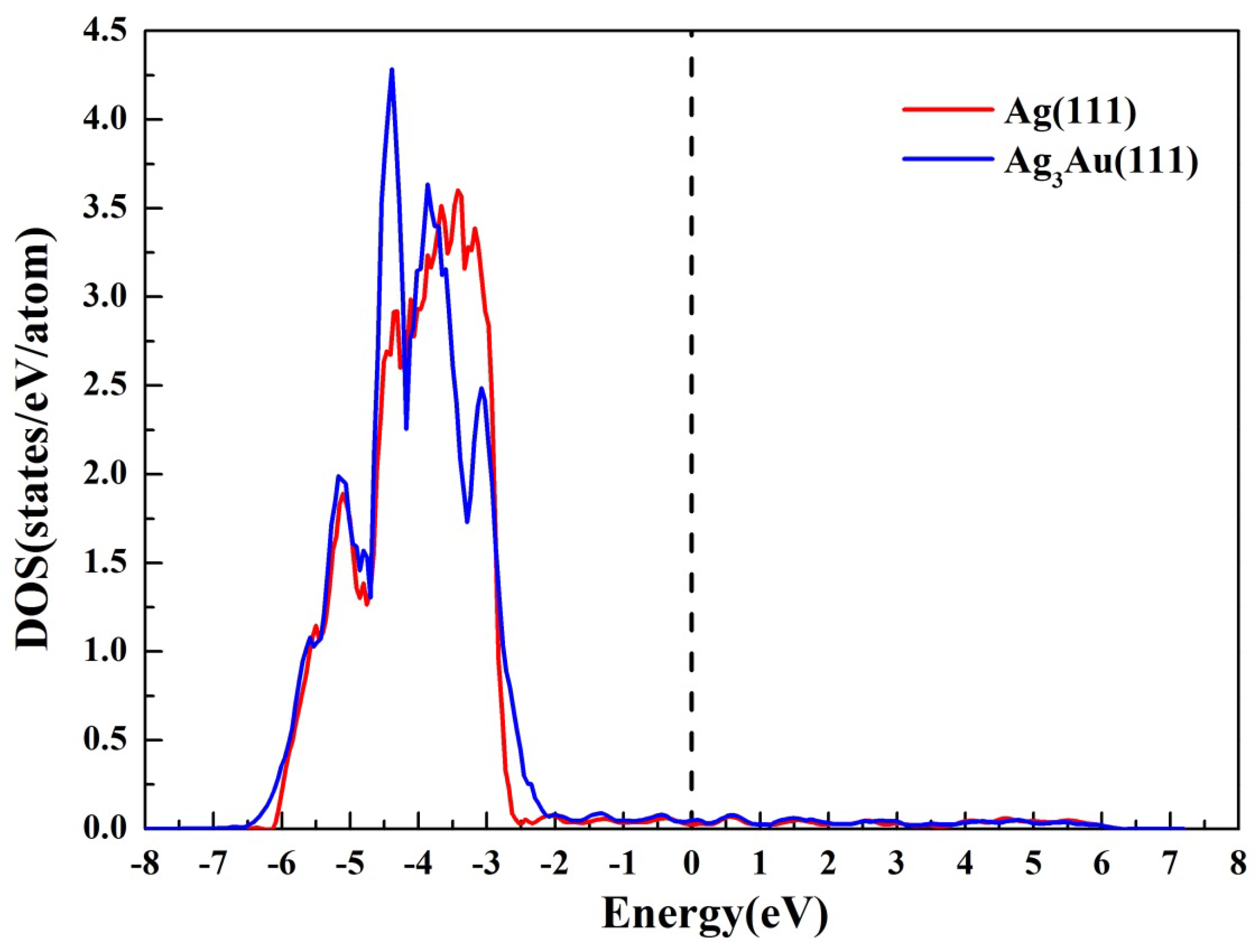
3.2. The Electronic Structure of Ag3Au(111) Surface
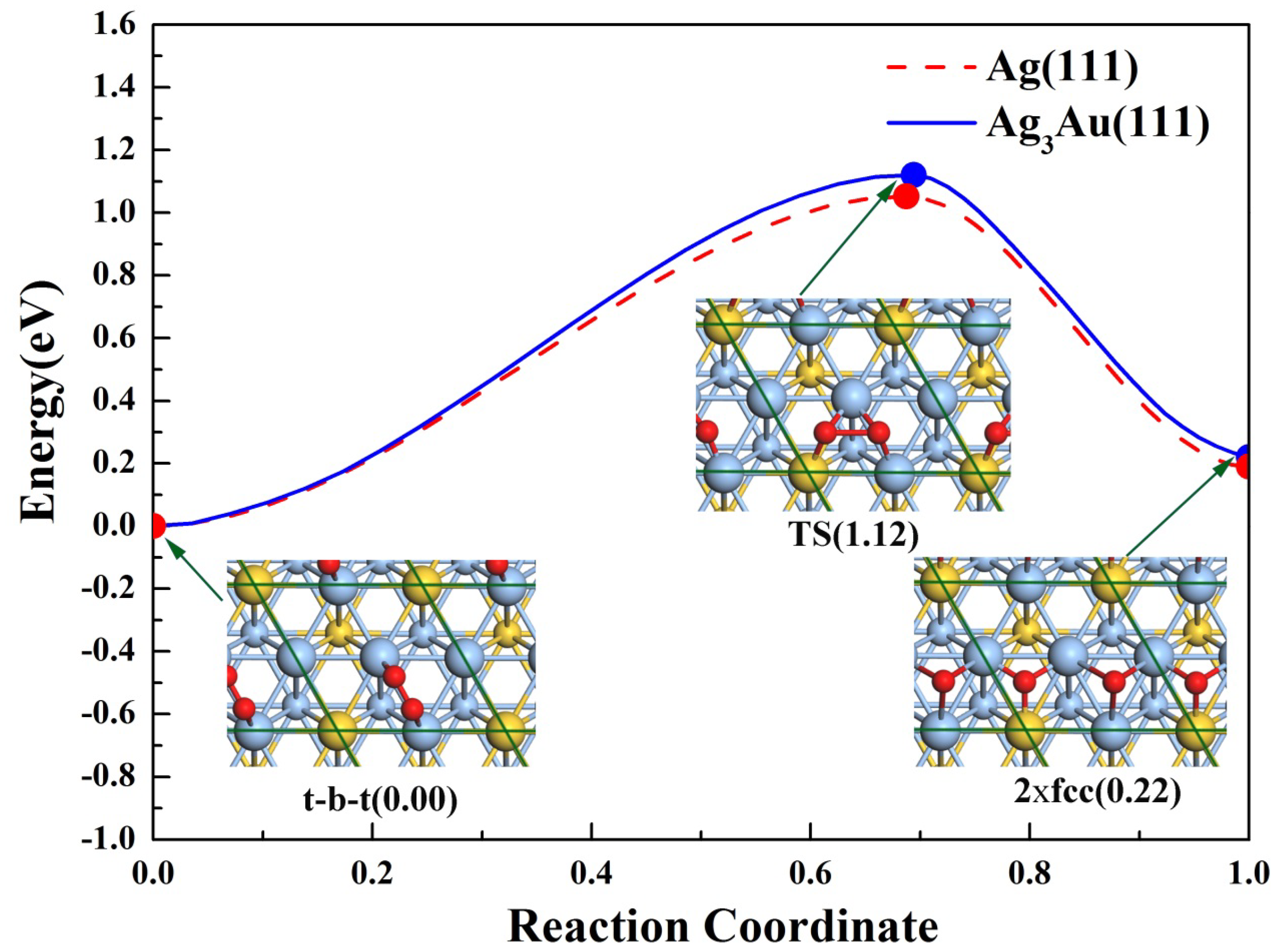
3.3. The Dissociation of O2
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gumuslu, G.; Kondratyuk, P.; Boes, J.R.; Morreale, B.; Miller, J.B.; Kitchin, J.R.; Gellman, A.J. Correlation of Electronic Structure with Catalytic Activity: H2–D2 Exchange across CuxPd1–x Composition Space. ACS Catal. 2015, 5, 3137–3147. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Z.; Huang, W.; Zhou, S.; Hu, Z.; Wang, L. Ab Initio Investigation of the Adsorption and Dissociation of O2 on Cu-Skin Cu3Au (111) Surface. Catalysts 2022, 12, 1407. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, W.; Wang, J.; Wang, L. First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen. Materials 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, J.; Yu, X. The adsorption, diffusion and dissociation of O2 on Pt-skin Pt3Ni (1 1 1): A density functional theory study. Chem. Phys. Lett. 2010, 499, 83–88. [Google Scholar] [CrossRef]
- Qiao, Y.; Xu, L.; Zhang, H.; Luo, H. O2 dissociative adsorption on the Cu-, Ag-, and W-doped Al (111) surfaces from DFT computation. Surf. Interface Anal. 2021, 53, 46–52. [Google Scholar] [CrossRef]
- Yu, Y.; Gu, H.; Wu, G.; Liu, X. Density functional theory study of dissociative adsorption of O2 on Pd-skin Pd3Cu (1 1 1) surface. Comput. Mater. Sci. 2024, 237, 112876. [Google Scholar] [CrossRef]
- Liu, J.; Fan, X.; Sun, C.Q.; Zhu, W. DFT study on intermetallic Pd–Cu alloy with cover layer Pd as efficient catalyst for oxygen reduction reaction. Materials 2017, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Henkelman, G. Charge redistribution in core-shell nanoparticles to promote oxygen reduction. J. Chem. Phys. 2009, 130, 194504. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Chen, S. Comparative study of oxygen reduction reaction mechanisms on the Pd (111) and Pt (111) surfaces in acid medium by DFT. J. Phys. Chem. C 2013, 117, 1342–1349. [Google Scholar] [CrossRef]
- Maatallah, M.; Guo, M.; Cherqaoui, D.; Jarid, A.; Liebman, J.F. Aluminium clusters for molecular hydrogen storage and the corresponding alanes as fuel alternatives: A structural and energetic analysis. Int. J. Hydrogen Energy 2013, 38, 5758–5767. [Google Scholar] [CrossRef]
- Mortensen, J.J.; Hansen, L.B.; Hammer, B.; Nørskov, J.K. Nitrogen adsorption and dissociation on Fe (111). J. Catal. 1999, 182, 479–488. [Google Scholar] [CrossRef]
- Liu, X.; Wang, A.; Wang, X.; Mou, C.-Y.; Zhang, T. Au–Cu alloy nanoparticles confined in SBA-15 as a highly efficient catalyst for CO oxidation. Chem. Commun. 2008, 27, 3187–3189. [Google Scholar] [CrossRef]
- Lu, F.; Sun, D.; Huang, J.; Du, M.; Yang, F.; Chen, H.; Hong, Y.; Li, Q. Plant-mediated synthesis of Ag–Pd alloy nanoparticles and their application as catalyst toward selective hydrogenation. ACS Sustain. Chem. Eng. 2014, 2, 1212–1218. [Google Scholar] [CrossRef]
- Gao, S.T.; Liu, W.; Feng, C.; Shang, N.Z.; Wang, C. A Ag–Pd alloy supported on an amine-functionalized UiO-66 as an efficient synergetic catalyst for the dehydrogenation of formic acid at room temperature. Catal. Sci. Technol. 2016, 6, 869–874. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J.P. Water gas shift catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Wang, X.; Feng, X.; Liu, J.; Huang, Z.; Zong, S.; Liu, L.; Liu, J.; Fang, Y. Photo-thermo catalytic selective oxidation of cyclohexane by In-situ prepared nonstoichiometric Molybdenum oxide and Silver-palladium alloy composite. J. Colloid Interface Sci. 2022, 607, 954–966. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol steam reforming for hydrogen production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Rong, H.; Tong, X.; Wang, Z. Self-template synthesis of Ag–Pt hollow nanospheres as electrocatalyst for methanol oxidation reaction. Langmuir 2017, 33, 5991–5997. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, H. Platinum-based oxygen reduction electrocatalysts. Acc. Chem. Res. 2013, 46, 1848–1857. [Google Scholar] [CrossRef]
- Wang, J.; Markovic, N.; Adzic, R. Kinetic analysis of oxygen reduction on Pt (111) in acid solutions: Intrinsic kinetic parameters and anion adsorption effects. J. Phys. Chem. B 2004, 108, 4127–4133. [Google Scholar] [CrossRef]
- Vesborg, P.C.; Jaramillo, T.F. Addressing the terawatt challenge: Scalability in the supply of chemical elements for renewable energy. RSC Adv. 2012, 2, 7933–7947. [Google Scholar] [CrossRef]
- Han, B.; Ling, L.; Zhang, R.; Liu, P.; Fan, M.; Wang, B. Dimethyl oxalate synthesis via CO oxidation on Pd-doped Ag (111) surface: A theoretic study. Mol. Catal. 2020, 484, 110731. [Google Scholar] [CrossRef]
- Fu, T.; Huang, J.; Lai, S.; Zhang, S.; Fang, J.; Zhao, J. Pt skin coated hollow Ag-Pt bimetallic nanoparticles with high catalytic activity for oxygen reduction reaction. J. Power Sources 2017, 365, 17–25. [Google Scholar] [CrossRef]
- Fu, T.; Fang, J.; Wang, C.; Zhao, J. Hollow porous nanoparticles with Pt skin on a Ag–Pt alloy structure as a highly active electrocatalyst for the oxygen reduction reaction. J. Mater. Chem. A 2016, 4, 8803–8811. [Google Scholar] [CrossRef]
- Hu, P.; Song, Y.; Chen, L.; Chen, S. Electrocatalytic activity of alkyne-functionalized AgAu alloy nanoparticles for oxygen reduction in alkaline media. Nanoscale 2015, 7, 9627–9636. [Google Scholar] [CrossRef]
- Song, Y.; Liu, K.; Chen, S. AgAu bimetallic Janus nanoparticles and their electrocatalytic activity for oxygen reduction in alkaline media. Langmuir 2012, 28, 17143–17152. [Google Scholar] [CrossRef]
- Pires, F.; Villullas, H. Pd-based catalysts: Influence of the second metal on their stability and oxygen reduction activity. Int. J. Hydrogen Energy 2012, 37, 17052–17059. [Google Scholar] [CrossRef]
- Sankarasubramanian, S.; Singh, N.; Mizuno, F.; Prakash, J. Ab initio investigation of the oxygen reduction reaction activity on noble metal (Pt, Au, Pd), Pt3M (M = Fe, Co, Ni, Cu) and Pd3M (M = Fe, Co, Ni, Cu) alloy surfaces, for LiO2 cells. J. Power Sources 2016, 319, 202–209. [Google Scholar] [CrossRef]
- Ramanathan, M.; Li, B.; Greeley, J.; Prakash, J. Microstructure-ORR activity relationships in Pd3M (M = Cu, Ni, Fe) electrocatalysts synthesized at various temperatures. ECS Trans. 2010, 33, 181. [Google Scholar] [CrossRef]
- Ou, L. Design of Pd-Based Bimetallic Catalysts for ORR: A DFT Calculation Study. J. Chem. 2015, 2015, 932616. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1993, 49, 14251–14269. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 2665–2668. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kunisada, Y.; Nakanishi, H.; Kasai, H. A First Principles Study of O2/Ag (111)–Adsorption and Magnetic Properties–. J. Phys. Soc. Jpn. 2011, 80, 084605. [Google Scholar] [CrossRef]
- Li, W.X.; Stampfl, C.; Scheffler, M. Oxygen adsorption on Ag (111): A density-functional theory investigation. Phys. Rev. B 2002, 65, 075407. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Hendrik, J.; James, D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Karatok, M.; Vovk, E.I.; Koc, A.V.; Ozensoy, E. Selective catalytic ammonia oxidation to nitrogen by atomic oxygen species on Ag (111). J. Phys. Chem. C 2017, 121, 22985–22994. [Google Scholar] [CrossRef]
- Sheu, W.-S.; Chang, M.-W. CO oxidation on Ag (111): The catalytic role of H2O. Surf. Sci. 2014, 628, 104–110. [Google Scholar] [CrossRef]
- Chen, B.W.; Kirvassilis, D.; Bai, Y.; Mavrikakis, M. Atomic and molecular adsorption on Ag (111). J. Phys. Chem. C 2018, 123, 7551–7566. [Google Scholar] [CrossRef]
- Langmuir, I. Modelisation of adsorption. Phys. Rev 1915, 6, 79–80. [Google Scholar]
- Neugebauer, J.; Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al(111). Phys. Rev. B Condens. Matter 1992, 46, 16067–16080. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Xu, Y.; Greeley, J.; Mavrikakis, M. Effect of subsurface oxygen on the reactivity of the Ag (111) surface. J. Am. Chem. Soc. 2005, 127, 12823–12827. [Google Scholar] [CrossRef]
- Dhouib, A.; Guesmi, H. DFT study of the M segregation on MAu alloys (M = Ni, Pd, Pt) in presence of adsorbed oxygen O and O2. Chem. Phys. Lett. 2012, 521, 98–103. [Google Scholar] [CrossRef]
- Hansen, H.A.; Viswanathan, V.; Nørskov, J.K. Unifying kinetic and thermodynamic analysis of 2e– and 4e–reduction of oxygen on metal surfaces. J. Phys. Chem. C 2014, 118, 6706–6718. [Google Scholar] [CrossRef]
- Viswanathan, V.; Hansen, H.A.; Rossmeisl, J.; Nørskov, J.K. Universality in oxygen reduction electrocatalysis on metal surfaces. ACS Catal. 2012, 2, 1654–1660. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Mahata, A.; Pathak, B. Bimetallic core-based cuboctahedral core–shell nanoclusters for the formation of hydrogen peroxide (2e− reduction) over water (4e− reduction): Role of core metals. Nanoscale 2017, 9, 9537–9547. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Kon, K.; Satsuma, A.; Shimizu, K.I. Volcano-Curves for Dehydrogenation of 2-Propanol and Hydrogenation of Nitrobenzene by SiO2-Supported Metal Nanoparticles Catalysts As Described in Terms of a d-Band Model. ACS Catal. 2012, 2, 1904–1909. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J. Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces. Phys. Rev. Lett. 2004, 93, 156801. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, J.; Nørskov, J.K.; Barteau, M.; Chen, J. Modification of the surface electronic and chemical properties of Pt (111) by subsurface 3d transition metals. J. Chem. Phys. 2004, 120, 10240–10246. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of strain on the reactivity of metal surfaces. Phys. Rev. Lett. 1998, 81, 2819. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Liao, S.; Li, B. DFT study of high performance Pt3Sn alloy catalyst in oxygen reduction reaction. Comput. Mater. Sci. 2018, 149, 107–114. [Google Scholar] [CrossRef]
- Goikoetxea, I.; Beltrán, J.; Meyer, J.; Juaristi, J.I.; Alducin, M.; Reuter, K. Non-adiabatic effects during the dissociative adsorption of O2 at Ag (111)? A first-principles divide and conquer study. New J. Phys. 2012, 14, 013050. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ag3Au(111) | Ag (111) | ||||||
|---|---|---|---|---|---|---|---|
| Site | Eads | dO–O | Nchg | Site | Eads | dO–O | Nchg |
| t-f-b1 | −0.095 | 1.308 | 0.503 | t-f-b | −0.196 | 1.316 | 0.544 |
| t-f-b2 | −0.088 | 1.307 | 0.492 | t-h-b | −0.189 | 1.317 | 0.545 |
| t-h-b1 | −0.118 | 1.305 | 0.488 | t-b-t | −0.243 | 1.307 | 0.496 |
| t-h-b2 | −0.052 | 1.304 | 0.482 | ||||
| t-b-t | −0.139 | 1.299 | 0.455 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Fu, M.; Gu, H.; Wang, L.; Liu, W.; Xie, Q.; Wu, G. Dissociative Adsorption of O2 on Ag3Au(111) Surface: A Density Functional Theory Study. Crystals 2024, 14, 504. https://doi.org/10.3390/cryst14060504
Yu Y, Fu M, Gu H, Wang L, Liu W, Xie Q, Wu G. Dissociative Adsorption of O2 on Ag3Au(111) Surface: A Density Functional Theory Study. Crystals. 2024; 14(6):504. https://doi.org/10.3390/cryst14060504
Chicago/Turabian StyleYu, Yanlin, Mingan Fu, Huaizhang Gu, Lei Wang, Wanxiu Liu, Qian Xie, and Guojiang Wu. 2024. "Dissociative Adsorption of O2 on Ag3Au(111) Surface: A Density Functional Theory Study" Crystals 14, no. 6: 504. https://doi.org/10.3390/cryst14060504