
Lactonization of α-Ferrocenyl Ketocarboxylic Acids via Nucleophilic Attack of Carbonyl Oxygen
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Experimental Procedures
3. Results and Discussion
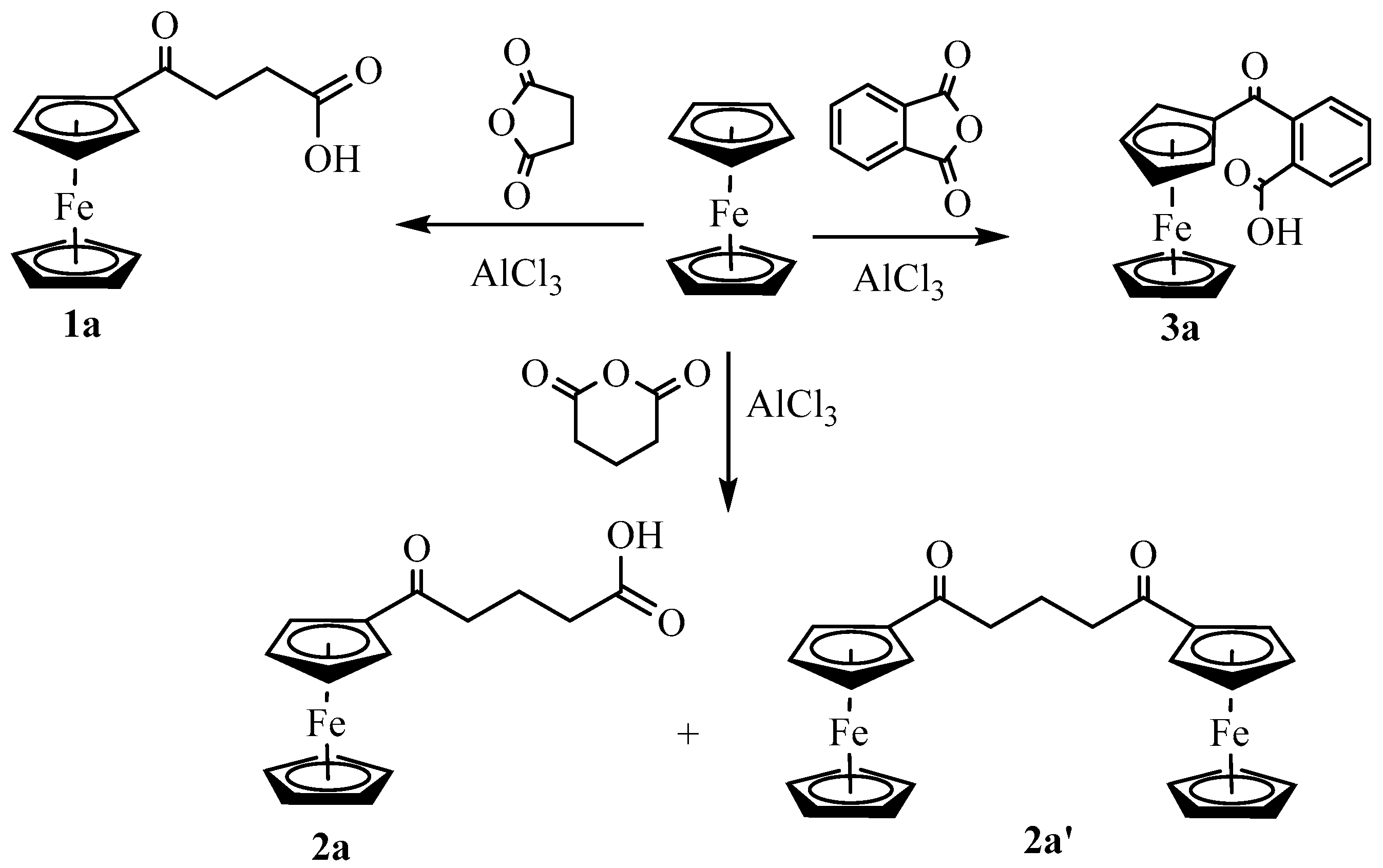
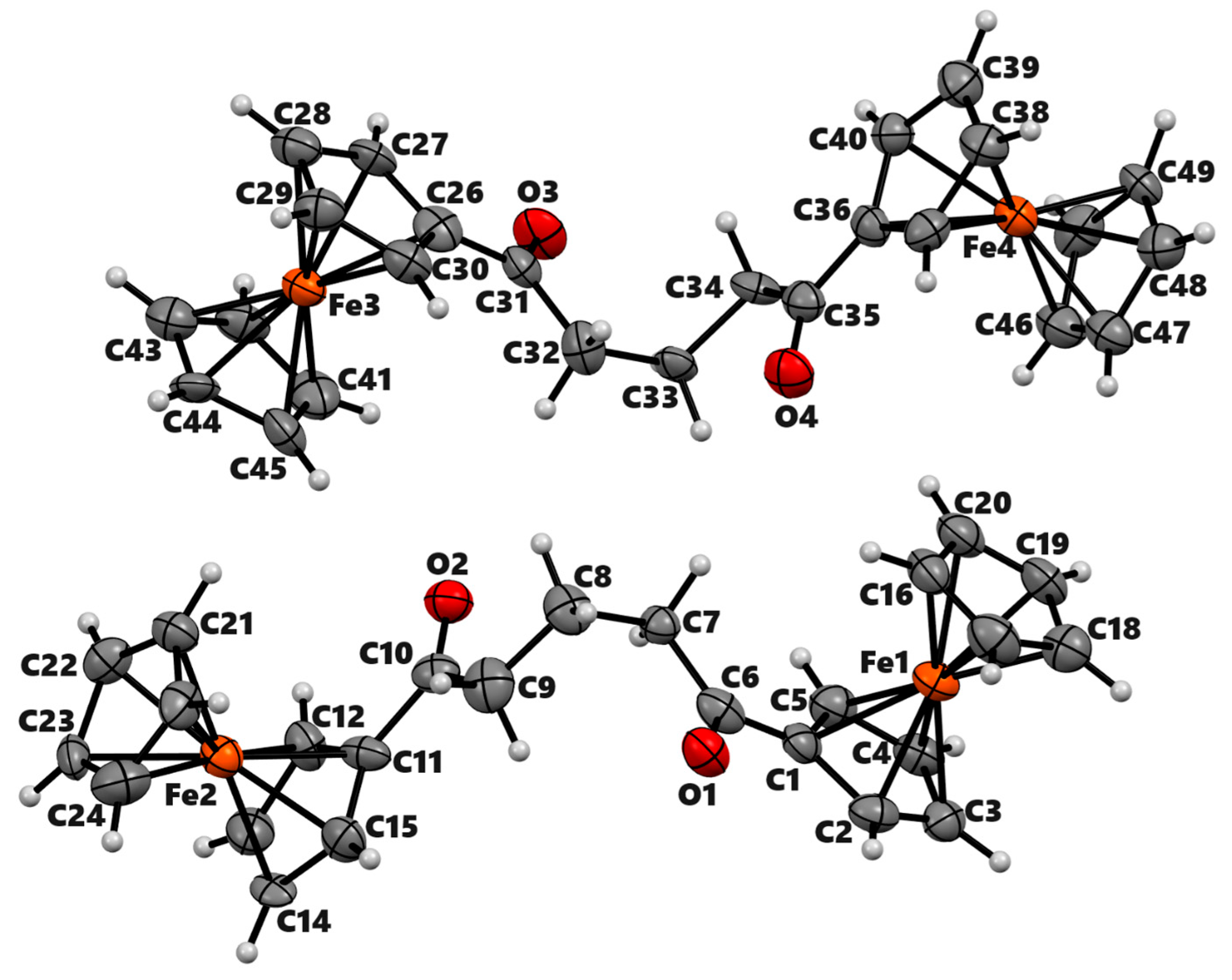
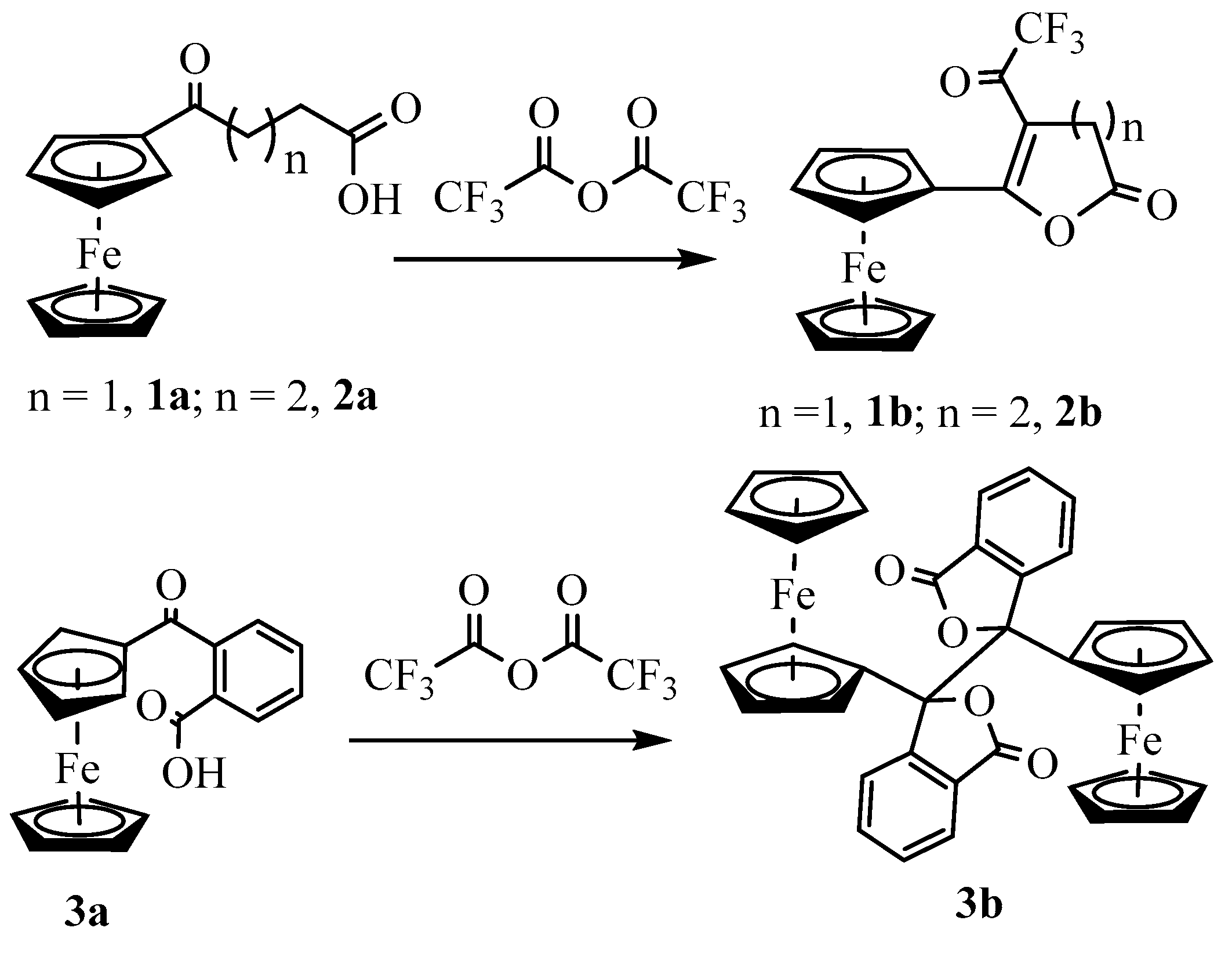
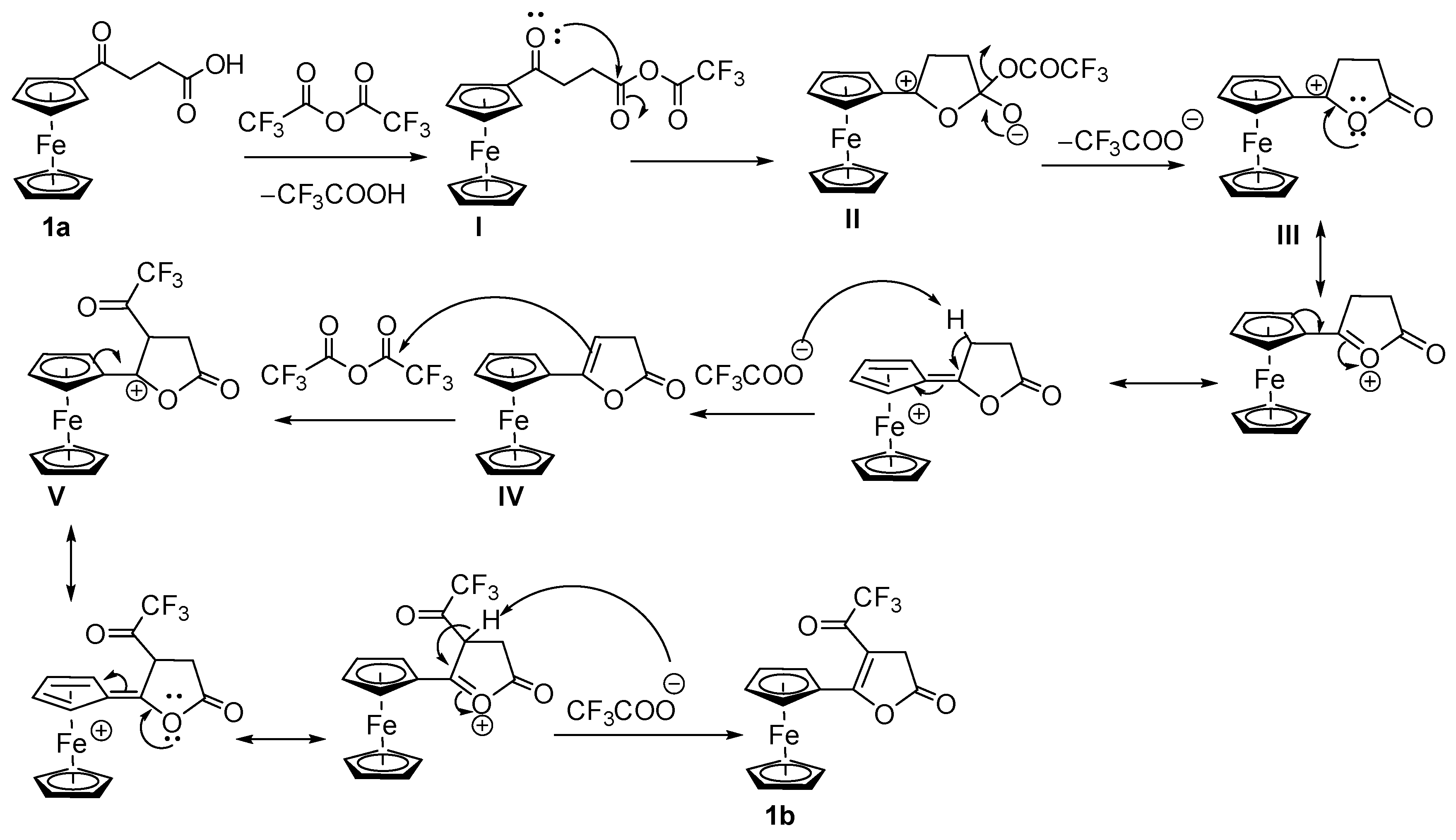
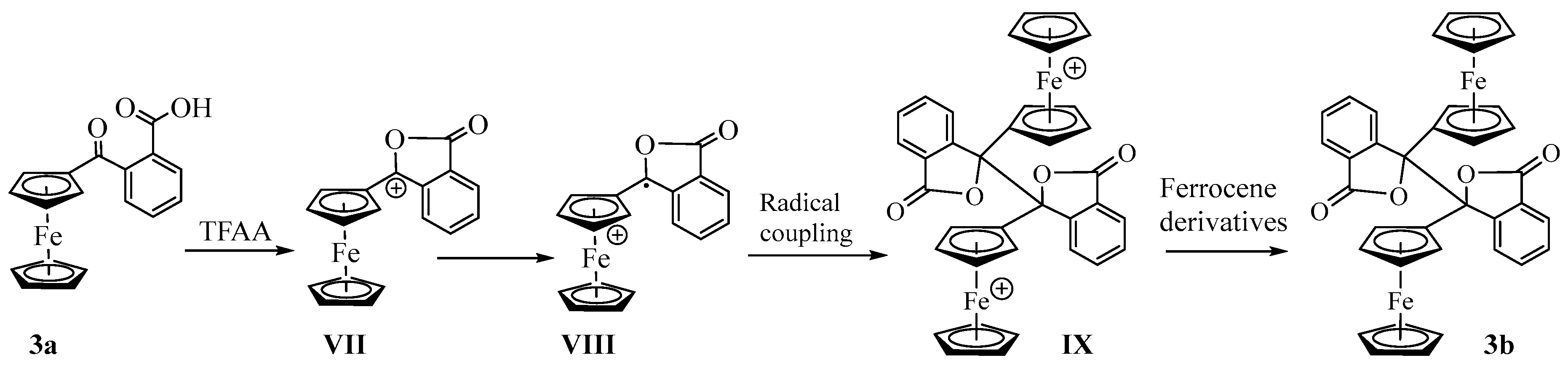
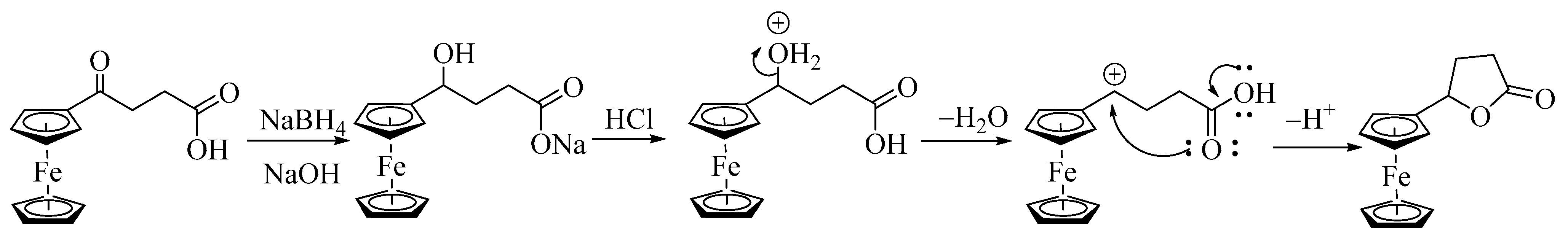
Synthesis and Structural Elucidation of Compounds 2a, 2a′, 1b–5b and Proposed Mechanisms of Their Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kealy, T.J.; Pauson, P.L. A New Type of Organo-Iron Compound. Nature 1951, 168, 1039–1040. [Google Scholar] [CrossRef]
- Richards, J.H.; Hill, A.A. α-Metallocenyl carbonium ions. J. Am. Chem. Soc. 1959, 81, 3484. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Buchholz, H.; Reddy, V.P.; De Meijere, A.; Olah, G.A. Stable carbocations. 285. 1-Ferrocenyl-1-cyclopropyl cation: The first long-lived cyclopropyl cation. J. Am. Chem. Soc. 1992, 114, 1097. [Google Scholar] [CrossRef]
- Cully, N.; Watts, W.E. Stable carbocations. XX. A kinetic study of the SN1 hydrolysis of aryl(ferrocenyl)methyl acetates. J. Organomet. Chem. 1979, 182, 99–103. [Google Scholar] [CrossRef]
- Abram, T.S.; Watts, W.E. Stable carbocations. Part 12. Generation, observation, and properties of ferrocenyl-stabilized vinyl cations. J. Chem. Soc. Perkin Trans. 1 1977, 1, 1522–1526. [Google Scholar] [CrossRef]
- Cerichelli, G.; Floris, B.; Ortaggi, G. Ferrocenyl carbocations. Ionization of ferrocenyl alcohols in aqueous sulfuric acid. J. Organomet. Chem. 1974, 78, 241. [Google Scholar] [CrossRef]
- Natsume, S.; Kurihara, H.; Yamaguchi, T.; Erabi, T.; Wada, M. Stability and reactivity of ferrocenyl(2,4,6-trimethoxyphenyl)carbenium salts. J. Organomet. Chem. 1999, 574, 86–93. [Google Scholar] [CrossRef]
- Korb, M.; Mahrholdt, J.; Liu, X.; Lang, H. Reactivity of Planar-Chiral α-Ferrocenyl Carbocations towards Electron-Rich Aromatics. Eur. J. Inorg. Chem. 2019, 2019, 973–987. [Google Scholar] [CrossRef]
- Rubalcava, H.E.; Thomson, J.B. A spectroscopic study of the protonation of acylferrocenes. Spectrochim. Acta 1962, 18, 449–459. [Google Scholar] [CrossRef]
- Olah, G.A.; Mo, Y.K. Organometallic chemistry: V. Protonation of acylferrocenes under stable ion conditions in fso3h-so2c1f solution. J. Organomet. Chem. 1973, 60, 311–321. [Google Scholar] [CrossRef]
- Roberts, R.M.G.; Silver, J.; Wells, A.S. Mössbauer and NMR studies of protonated acyl diphosphaferrocenes. Inorganica Chim. Acta. 1986, 119, 171–176. [Google Scholar] [CrossRef]
- Saric, A.; Vrcek, V.; Buehl, M. Density functional study of protonated formylmetallocenes. Organometallics 2008, 27, 394–401. [Google Scholar] [CrossRef]
- Bunnett, J.F. Nucleophilic Reactivity. Annu. Rev. Phys. Chem. 1963, 14, 271–290. [Google Scholar] [CrossRef]
- Jaramillo, P.; Pérez, P.; Fuentealba, P. Relationship between basicity and nucleophilicity. J. Phys. Org. Chem. 2007, 20, 1050–1057. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, U.R.; Daigle, D.P.; Naquin, S.D.; Engeron, G.S.; Lo, M.A.; Fronczek, F.R. Synthesis, Crystal Structure, and Electrochemistry of Mono- and Bis-Homoannular Ferrocene Derivatives. Crystals 2024, 14, 141. [Google Scholar] [CrossRef]
- Wieczorek, A.; Blauz, A.; Makal, A.; Rychlik, B.; Plazuk, D. Synthesis and evaluation of biological properties of ferrocenyl-podophyllotoxin conjugates. Dalton Trans. 2017, 46, 10847–10858. [Google Scholar] [CrossRef] [PubMed]
- Woltersdorf, M.; Kranich, R.; Schmalz, H.-G. Enantioselective synthesis of new C2-symmetric ferrocenylalkylamines via sonochemical amination of 1-ferrocenylalkyl acetates. Tetrahedron 1997, 53, 7219–7230. [Google Scholar] [CrossRef]
- Pokharel, U.R.; Bergeron, J.T.; Fronczek, F.R. Synthesis and crystal structures of 2-(ferrocenylcarbonyl)benzoic acid and 3-ferrocenylphthalide. Acta Crystallogr. Sect. E 2020, 76, 1163–1167. [Google Scholar] [CrossRef]
- Goldberg, S.I.; Breland, J.G. Ferrocene studies. XIX. Synthesis of 1,2-terferrocene. J. Org. Chem. 1971, 36, 1499–1503. [Google Scholar] [CrossRef]
- Tombul, M.; Gemici, S.; Bulut, A. Alkyl Lewis Acid Catalyzed Syntheses of Dicarbonyl Ferrocenes. Asian J. Chem. 2010, 22, 7070. [Google Scholar]
- Tombul, M.; Bulut, A.; Guven, K.; Buyukgungor, O. 1,4-Diferrocenylbutane-1,4-dione. Acta Crystallogr. Sect. E 2008, 64, m444–m445. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.t.; Pople, J. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Cais, M.; Modiano, A.; Raveh, A. Organometallic Studies. XVII.1 A Novel Approach to the Synthesis of the Benzopentalene System2. J. Am. Chem. Soc. 1965, 87, 5607–5614. [Google Scholar] [CrossRef]
- Casper, L.A.; Ebel, V.; Linseis, M.; Winter, R.F. Five shades of green: Substituent influence on the (spectro-) electrochemical properties of diferrocenyl(phenyl)methylium dyes. Dalton Trans. 2021, 50, 15336–15351. [Google Scholar] [CrossRef] [PubMed]
- Nesmeyanov, A.N.; Vil’chevskaya, V.D.; Kochetkova, N.S. Reactions of ο-carboxybenzoylferrocene. Dokl. Akad. Nauk SSSR 1965, 165, 835–837. [Google Scholar]
- Gleiter, R.; Seeger, R. The Structure of the Ferrocenyl-Methyl Cation. Preliminary communication. Helv. Chim. Acta 1971, 54, 1217–1220. [Google Scholar] [CrossRef]
- Cais, M.; Eisenstadt, A. Organometallic Studies. X.1a Reductive Dimerization of α-Metallocenylcarbonium Ions. I1b. J. Org. Chem. 1965, 30, 1148–1154. [Google Scholar] [CrossRef]
- Fedin, E.I.; Blumenfeld, A.L.; Petrovskii, P.V.; Kreindlin, A.Z.; Fadeeva, S.S.; Rybinskaya, M.I. Conversion of the diamagnetic nonamethylferrocenylcarbenium salts into the paramagnetic salts of bis(nonamethylferroceniumyl)ethane. J. Organomet. Chem. 1985, 292, 257–268. [Google Scholar] [CrossRef]
- Banide, E.V.; Ortin, Y.; Chamiot, B.; Cassidy, A.; Niehaus, J.; Moore, A.; Seward, C.M.; Müller-Bunz, H.; McGlinchey, M.J. Syntheses, Structures, and Dimerizations of Ferrocenyl- and Fluorenylideneallenes: Push−Pull Multiple Bonds? Organometallics 2008, 27, 4173–4182. [Google Scholar] [CrossRef]
- Casper, L.A.; Deuter, K.L.; Rehse, A.; Winter, R.F. Dimerization of 9-Phenyl-ferroceno[2,3]indenylmethyl Radicals: Electrochemical and Spectroelectrochemical Studies. ACS Org. Inorg. AU 2024. [Google Scholar] [CrossRef]
- Casper, L.A.; Linseis, M.; Demeshko, S.; Azarkh, M.; Drescher, M.; Winter, R.F. Tailoring Valence Tautomerism by Using Redox Potentials: Studies on Ferrocene-Based Triarylmethylium Dyes with Electron-Poor Fluorenylium and Thioxanthylium Acceptors. Chem. Eur. J. 2021, 27, 10854–10868. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.W.; Rabb, D.J. 1,2-(α-Oxotetramethylene)ferrocene. J. Org. Chem. 1961, 26, 3588. [Google Scholar] [CrossRef]
- Sugiyama, N.; Suzuki, H.; Shioura, Y.; Teitei, T. Reaction of ferrocene with acyl chlorides. Bull. Chem. Soc. Jpn. 1962, 35, 767. [Google Scholar] [CrossRef]
- Husain, A.; Khan, S.A.; Iram, F.; Iqbal, M.A.; Asif, M. Insights into the chemistry and therapeutic potential of furanones: A versatile pharmacophore. Eur. J. Med. Chem. 2019, 171, 66–92. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Lah, H.U.; Yousuf, S.K.; Ahmad, Z. α-pyrones: Small molecules with versatile structural diversity reflected in multiple pharmacological activities-an update. Biomed. Pharmacother. 2017, 91, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L., Jr.; Curby, R.J., Jr. Ferrocene bridging and homoannular cyclizations. J. Am. Chem. Soc. 1957, 79, 3290. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Curby, R.J., Jr.; Gustafson, D.H.; Harrison, K.G.; Bozak, R.E.; Bublitz, D.E. Organic chemistry of ferrocene. V. Cyclization of ω-ferrocenylaliphatic acids. J. Am. Chem. Soc. 1962, 84, 3263. [Google Scholar] [CrossRef]


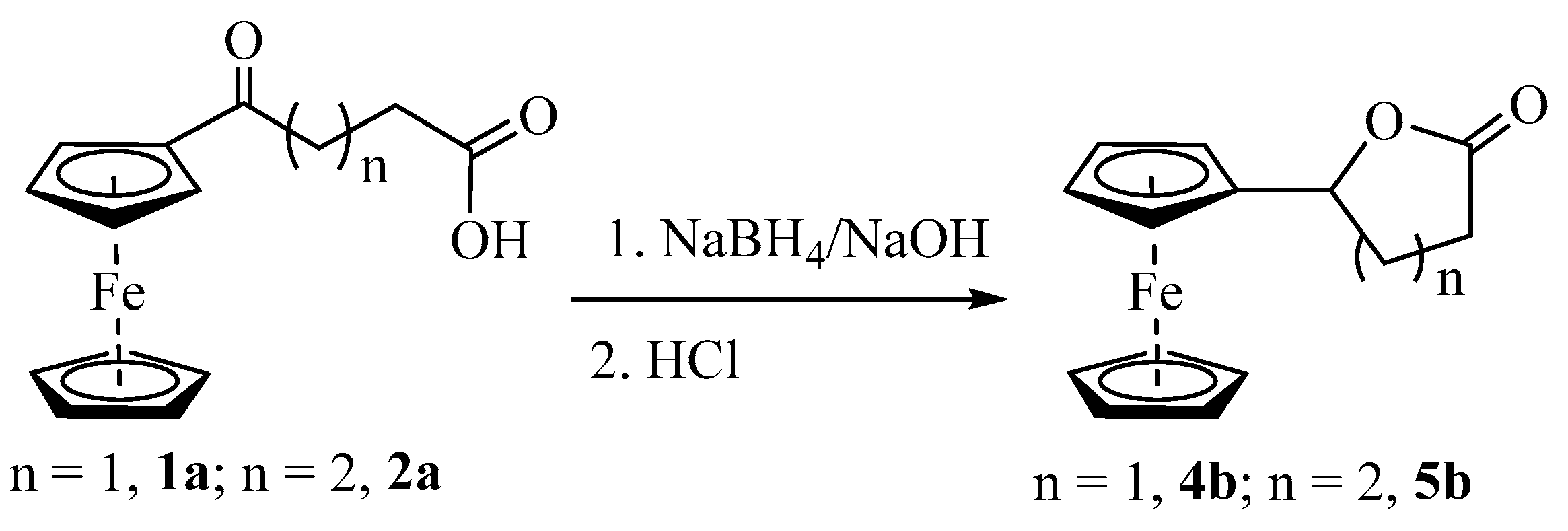

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2a′ | 2b | 3b | |
|---|---|---|---|
| Chemical formula | C25H24Fe2O2 | C17H13F3FeO3 | C36H26Fe2O4 |
| Mr | 468.14 | 378.12 | 634.27 |
| Deposition number | CCDC 2355187 | CCDC 2355188 | CCDC 2355189 |
| Crystal system, space group | Monoclinic, P21 | Monoclinic, P21/n | Monoclinic, P21/c |
| Temperature (K) | 90 | 90 | 90 |
| a, b, c (Å) | 5.7838 (5), 11.8770 (9), 28.071 (2) | 10.6270 (2), 9.7968 (2), 14.9960 (3) | 9.8279 (8), 13.3787 (10), 10.8256 (8) |
| β (°) | 93.773 (6) | 110.1183 (9) | 111.064 (4) |
| V (Å3) | 1924.2 (3) | 1465.98 (5) | 1328.29 (18) |
| Z | 4 | 4 | 2 |
| Radiation type | Cu Kα | Mo Kα | Mo Kα |
| µ (mm−1) | 12.24 | 1.08 | 1.14 |
| Crystal size (mm) | 0.15 × 0.04 × 0.01 | 0.38 × 0.30 × 0.13 | 0.22 × 0.15 × 0.14 |
| Diffractometer | Bruker Kappa APEX-II DUO | Bruker Kappa APEX-II DUO | Bruker Kappa APEX-II DUO |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan |
| Tmin, Tmax | 0.564, 0.887 | 0.764, 0.873 | 0.811, 0.857 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 12,160, 5225, 3851 | 74,201, 13,090, 12,014 | 65,909, 9349, 7963 |
| Rint | 0.074 | 0.019 | 0.031 |
| (sin θ/λ)max (Å−1) | 0.568 | 1.086 | 0.945 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.068, 0.178, 1.04 | 0.021, 0.059, 1.05 | 0.031, 0.086, 1.05 |
| No. of reflections | 5225 | 13090 | 9349 |
| No. of parameters | 524 | 244 | 190 |
| Δρmax, Δρmin (e Å−3) | 1.01, −0.55 | 0.88, −0.48 | 1.02, −0.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokharel, U.R.; Curole, B.J.; Andras, A.M.; LeBlanc, B.P.; Fronczek, F.R. Lactonization of α-Ferrocenyl Ketocarboxylic Acids via Nucleophilic Attack of Carbonyl Oxygen. Crystals 2024, 14, 548. https://doi.org/10.3390/cryst14060548
Pokharel UR, Curole BJ, Andras AM, LeBlanc BP, Fronczek FR. Lactonization of α-Ferrocenyl Ketocarboxylic Acids via Nucleophilic Attack of Carbonyl Oxygen. Crystals. 2024; 14(6):548. https://doi.org/10.3390/cryst14060548
Chicago/Turabian StylePokharel, Uttam R., Brennan J. Curole, Autumn M. Andras, Brandon P. LeBlanc, and Frank R. Fronczek. 2024. "Lactonization of α-Ferrocenyl Ketocarboxylic Acids via Nucleophilic Attack of Carbonyl Oxygen" Crystals 14, no. 6: 548. https://doi.org/10.3390/cryst14060548