
Co-Crystallization and Structural Studies of Benzophenone Recognized by Positively Shifted ESPs of Perfluorinated β-Diketonate Complexes (M = Cu, Pd, Pt)

Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Crystal Structure Determination
3. Results and Discussion
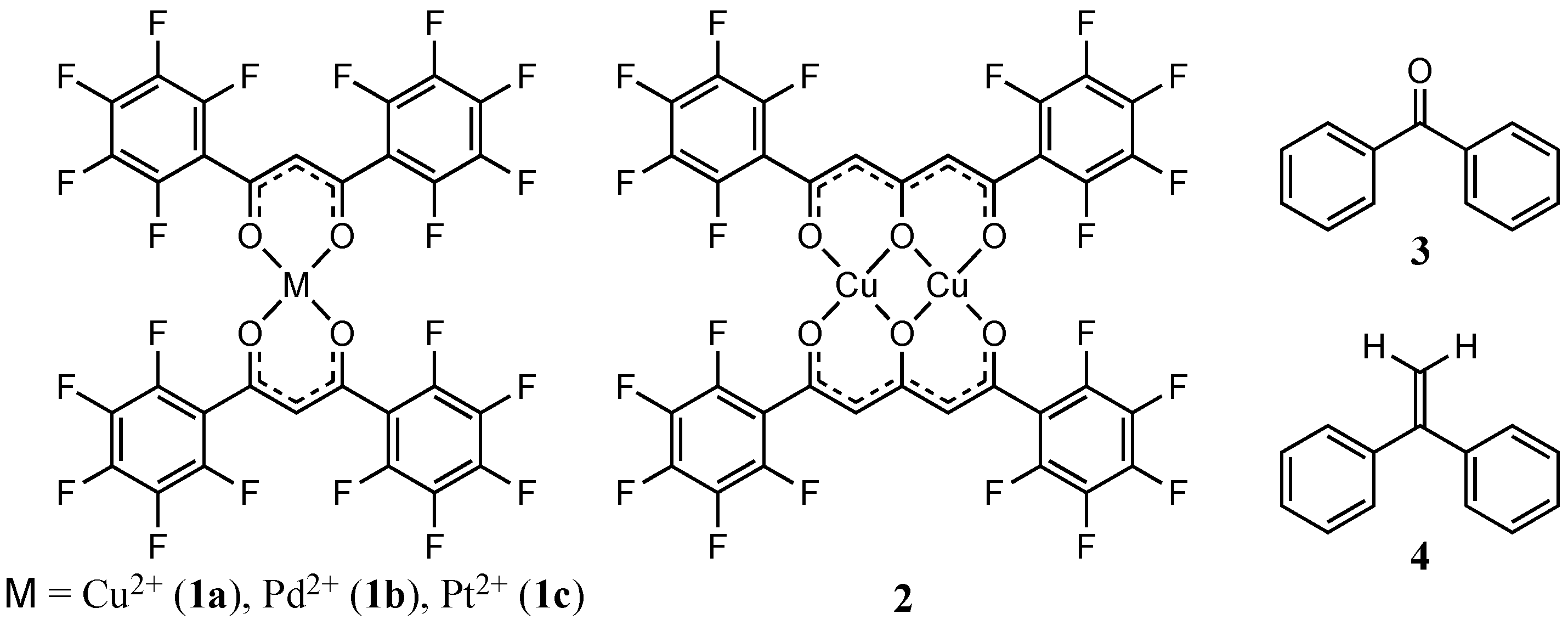
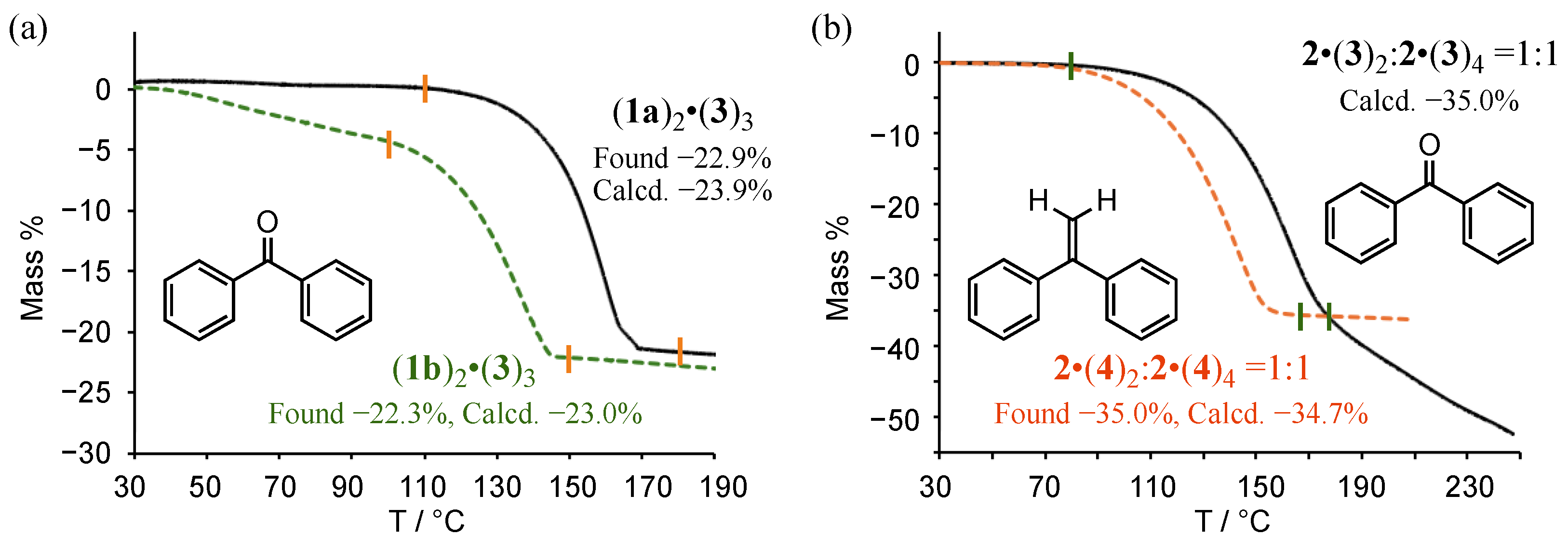
3.1. Preparation and Thermogravimetric Analysis of Co-Crystals
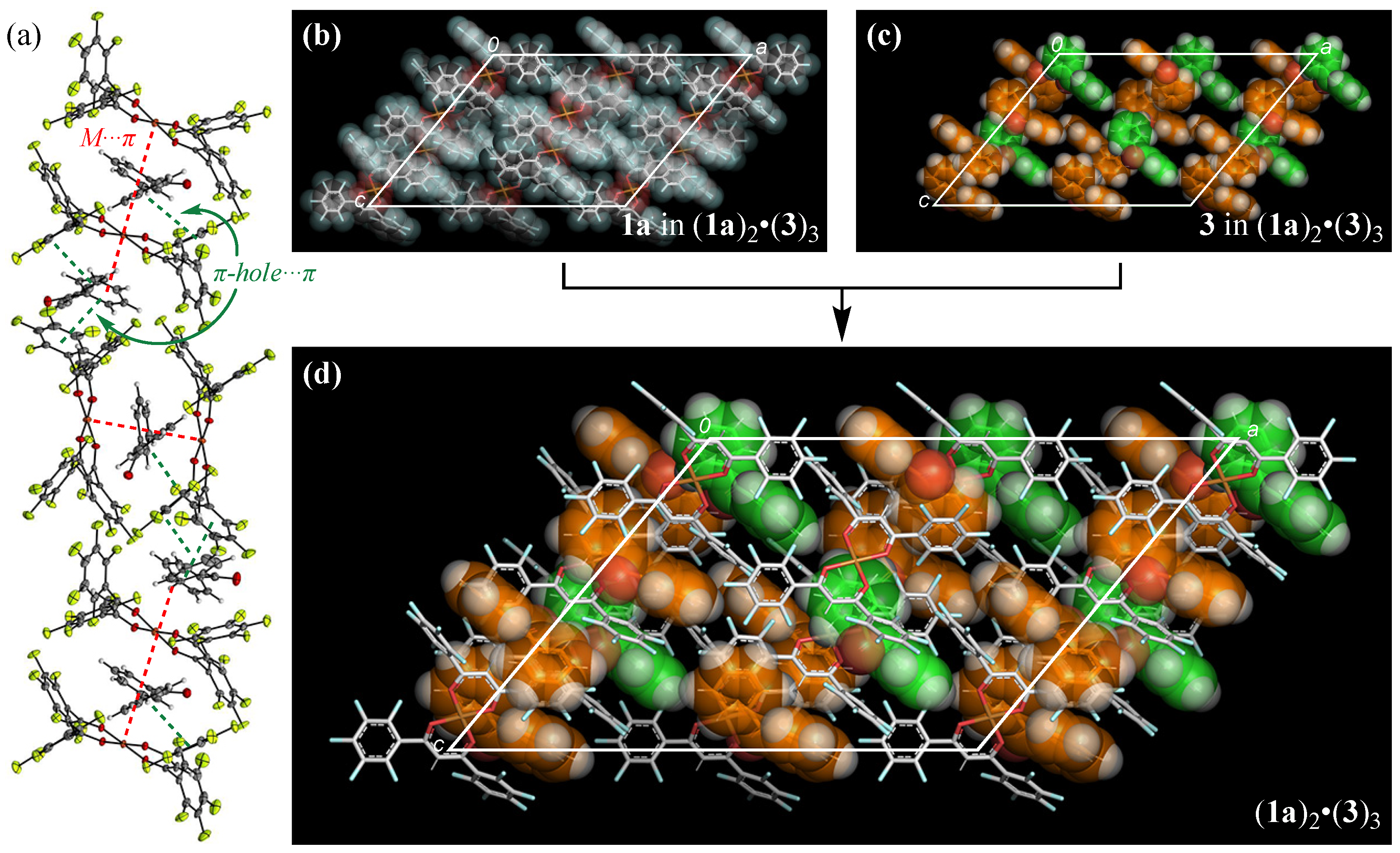
3.2. Crystal Structure of (1)2•(3)3
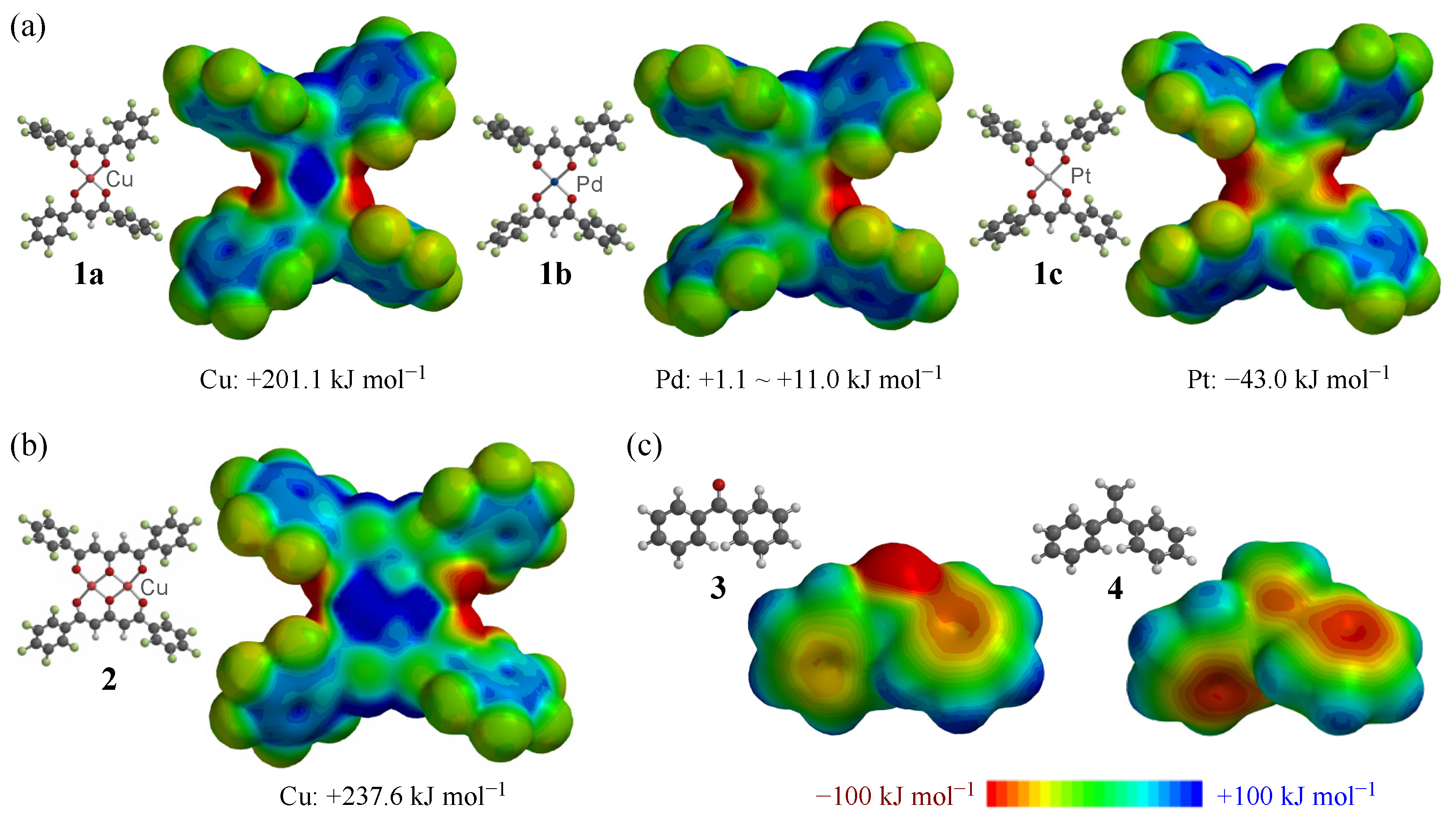
3.3. Density Functional Theory Calculations of the Structures
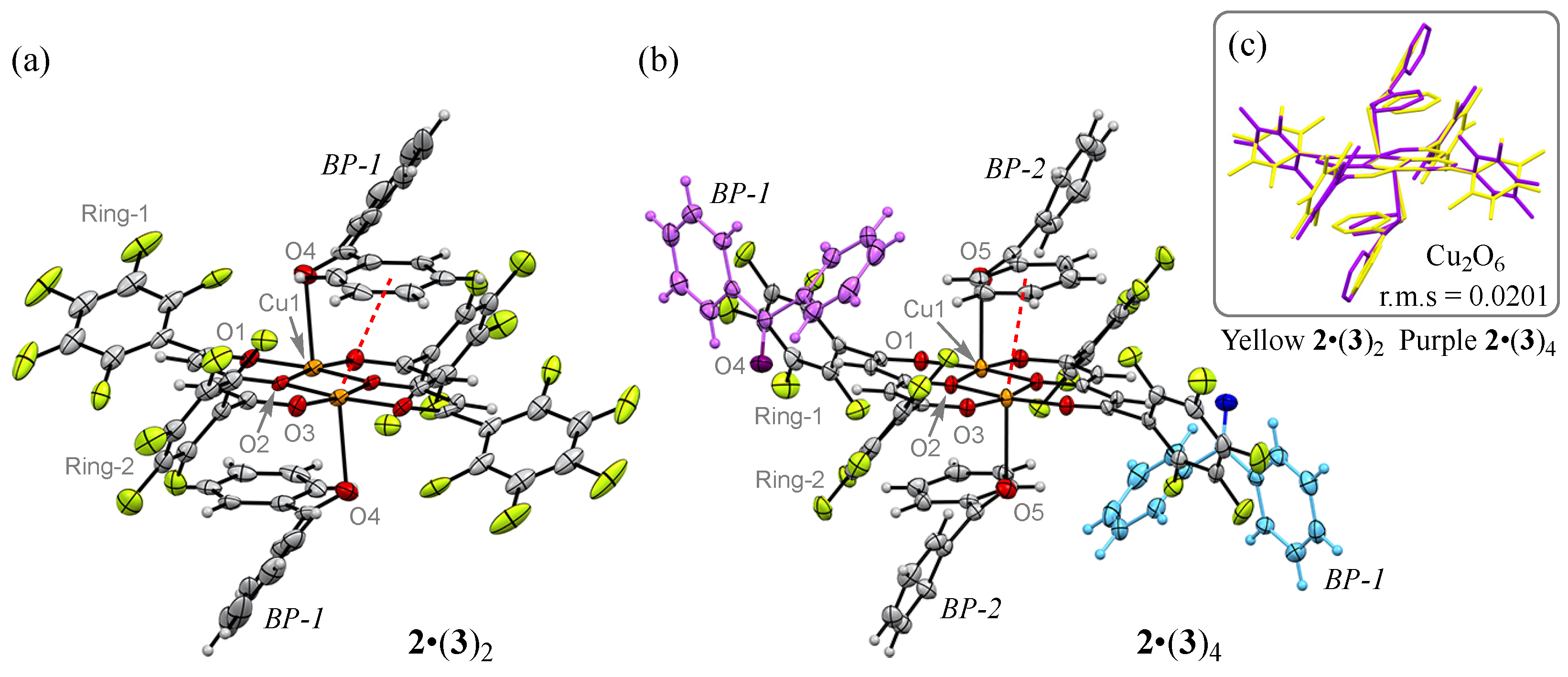
3.4. Pseudo-Polymorph Structures of 2•(3)2 and 2•(3)4
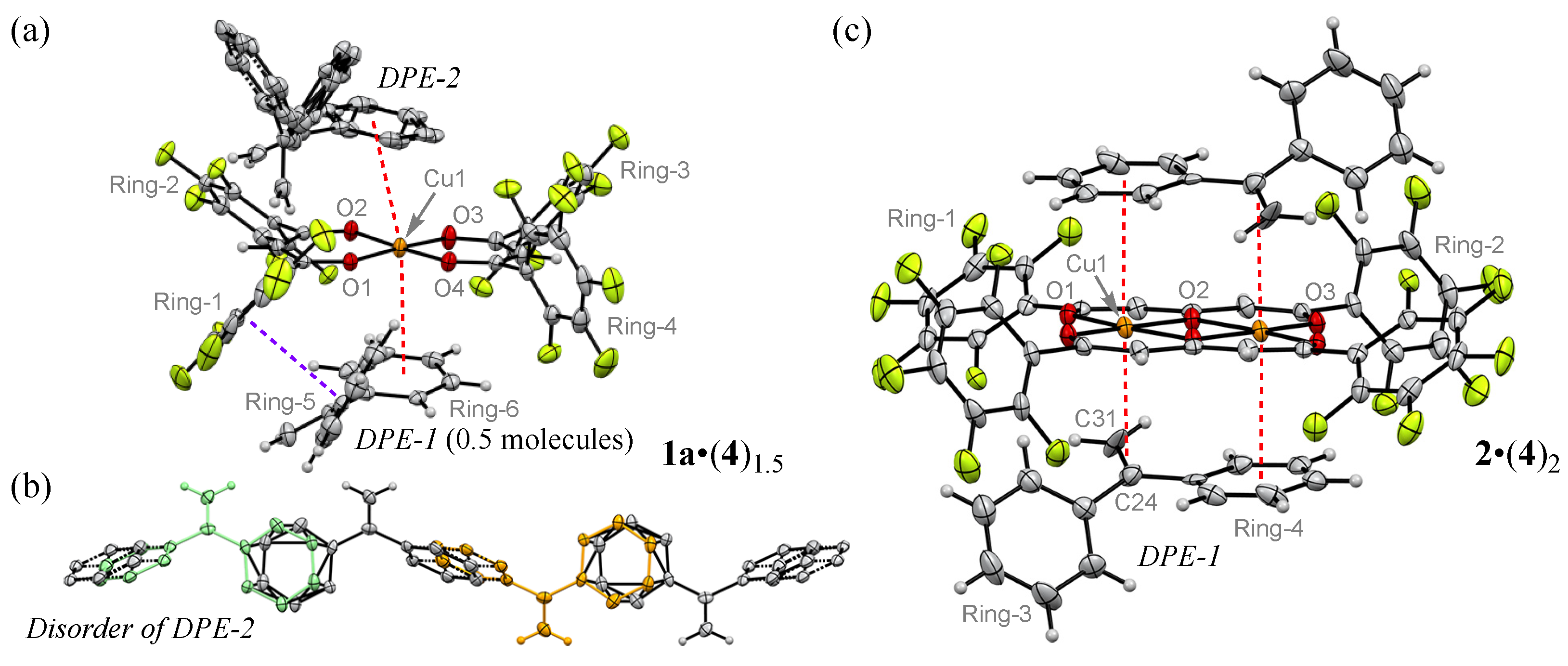
3.5. Crystal Structure with 4
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Fleischer, E.B.; Sung, N.; Hawkinson, S. Crystal structure of benzophenone. J. Phys. Chem. 1968, 72, 4311–4312. [Google Scholar] [CrossRef]
- King, J.B.; Tsunoda, M.; Gabbaï, F.P. Complexation of aldehydes and ketones by trimeric perfluoro-ortho-phenylene Mercury, a tridentate Lewis acid. Organometallics 2002, 21, 4201–4205. [Google Scholar] [CrossRef]
- Demeter, A.; Horváth, K.; Böőr, K.; Molnár, L.; Soós, T.; Lendvay, G. Substituent effect on the photoreduction kinetics of benzophenone. J. Phys. Chem. A 2013, 117, 10196–10210. [Google Scholar] [CrossRef] [PubMed]
- Arivanandhan, M.; Sanjeeviraja, C.; Sankaranarayanan, K.; Das, S.K.; Samanta, G.K.; Datta, P.K. Growth of urea doped benzophenone single crystal for nonlinear optical applications. Opt. Mater. 2006, 28, 324–330. [Google Scholar] [CrossRef]
- Yadav, H.; Sinha, N.; Tyagi, N.; Kumar, B. Enhancement of optical, piezoelectric, and mechanical properties in crystal violet dye-doped benzophenone crystals grown by Czochralski technique. Cryst. Growth Des. 2015, 15, 4908–4917. [Google Scholar] [CrossRef]
- Akrman, J.; Prikryl, J. Application of benzotriazole reactive UV absorbers to cellulose and determining sun protection of treated fabric spectrophotometrically. J. Appl. Polym. Sci. 2007, 108, 334–341. [Google Scholar] [CrossRef]
- An, Q.F.; Li, L.S.; Lu, D.D. A functional polysiloxane with benzophenone derivative ultraviolet absorbing side groups: Synthesis, morphology, and its performance on fabrics. J. Appl. Polym. Sci. 2007, 104, 680–687. [Google Scholar] [CrossRef]
- Matsumoto, A.; Tsuchiya, S.; Hagiwara, Y.; Ishikawa, K.; Koshima, H.; Asahi, T.; Soai, K. Absolute structure determination of chiral crystals consisting of achiral benzophenone with single-crystal X-ray diffraction and its correlation with solid-state circular dichroism. Chem. Lett. 2016, 45, 526–528. [Google Scholar] [CrossRef]
- Titov, A.A.; Filippov, O.A.; Bilyachenko, A.N.; Smol’yakov, A.F.; Dolgushin, F.M.; Belsky, V.K.; Godovicov, I.A.; Epstein, L.M.; Shubina, E.S. Complexes of trinuclear macrocyclic Copper(I) and Silver(I) 3,5-bis(trifluoromethyl)pyrazolates with ketones. Eur. J. Inorg. Chem. 2012, 33, 5554–5561. [Google Scholar] [CrossRef]
- Geetharani, K.; Bose, S.K.; Sahoo, S.; Varghese, B.; Mobin, S.M.; Ghosh, S. Cluster expansion reactions of group 6 and 8 metallaboranes using transition metal carbonyl compounds of groups 7–9. Inorg. Chem. 2011, 50, 5824–5832. [Google Scholar] [CrossRef]
- Chadwick, K.; Davey, R.J.; Dent, G.; Pritchand, R.G.; Hunter, C.A.; Musumeci, D. Cocrystallization: A solution chemistry perspective and the case of benzophenone and diphenylamine. Cryst. Growth Des. 2009, 9, 1990–1999. [Google Scholar] [CrossRef]
- Ananchenko, G.S.; Udachin, K.A.; Dubes, A.; Ripmeester, J.A.; Perrier, T.; Coleman, A.W. Guest exchange in single crystals of van der Waals nanocapsules. Angew. Chem. Int. Ed. 2006, 45, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.-Q.; Coppens, P. Variable Conformation of benzophenone in a series of resorcinarene-based supramolecular frameworks. Cryst. Growth Des. 2004, 4, 1377–1385. [Google Scholar] [CrossRef]
- Ma, B.-Q.; Coppens, P. Three-fold interpenetrating three-dimensional networks based on C-methylcalix [4]resorcinarene incorporating benzophenone guest molecules. Chem. Commun. 2003, 3, 412–413. [Google Scholar] [CrossRef]
- Chowdhury, S.; Bridson, J.N.; Georghiou, P.E. Synthesis of calix[4]arene triflates and their unusual chemical reactivity in palladium-catalyzed reactions. J. Org. Chem. 2000, 65, 3299–3302. [Google Scholar] [CrossRef]
- Bas, L.G.; de Rango, C.; Rysanek, N.; Tsoucaris, G. Chiral conformations induced by cyclodextrin. J. Incl. Phenom. 1984, 2, 861–867. [Google Scholar] [CrossRef]
- Toki, S.; Sakurai, H. On the structure of the 2:1-adduct of benzophenone and furan. Tetrahedron Lett. 1967, 8, 4119–4122. [Google Scholar] [CrossRef]
- Lee, H.H.; Warner, J.C. The Systems (I) diphenyl—Diphenylamine, (II) diphenyl—Benzophenone and (III) benzophenone—Diphenylamine. J. Am. Chem. Soc. 1933, 55, 209–214. [Google Scholar] [CrossRef]
- Yuan, W.Z.; Shen, X.Y.; Zhao, H.; Lam, J.W.Y.; Tang, L.; Lu, P.; Wang, C.; Liu, Y.; Wang, Z.; Zheng, Q.; et al. Crystallization-induced phosphorescence of pure organic luminogens at room temperature. J. Phys. Chem. C 2010, 114, 6090–6099. [Google Scholar] [CrossRef]
- He, Z.; Zhao, W.; Lam, J.W.Y.; Peng, Q.; Ma, H.; Liang, G.; Shuai, Z.; Tang, B.Z. White light emission from a single organic molecule with dual phosphorescence at room temperature. Nat. Comm. 2017, 8, 416. [Google Scholar] [CrossRef]
- Hori, A.; Nakajima, K.; Akimoto, Y.; Naganuma, K.; Yuge, H. Guest-adjusted encapsulation and thermal studies of non-porous mononuclear Cu(II) coordination complexes through electrostatic interactions induced by fluorine substitution. CrystEngComm 2014, 16, 8805–8817. [Google Scholar] [CrossRef]
- Ikumura, Y.; Habuka, Y.; Sakai, S.; Shinohara, T.; Yuge, H.; Rzeznicka, I.I.; Hori, A. Enhanced and heteromolecular guest encapsulation in nonporous crystals of a perfluorinated triketonato dinuclear copper complex. Chem. Eur. J. 2020, 26, 5051–5060. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.H. How Molecules Build Solids. In Crystal Engineering; Morgan & Claypool Publishers: Kentfield, CA, USA, 2017. [Google Scholar]
- Patrick, C.R.; Prosser, G.S. A Molecular Complex of Benzene and Hexafluorobenzene. Nature 1960, 187, 1021. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs. π-Hole Bond: A comparison based on halogen bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A. σ- and π-Hole Interactions. Crystals 2020, 10, 721. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. σ/π-Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Vrbancich, J.; Ritchie, G.L.D. Quadrupole moments of benzene, hexafluorobenzene and other non-dipolar aromatic molecules. J. Chem. Soc. Faraday Trans. 2 1980, 76, 648–659. [Google Scholar] [CrossRef]
- Williams, J.H. The molecular electric quadrupole moment and solid-state architecture. Acc. Chem. Res. 1993, 26, 593–598. [Google Scholar] [CrossRef]
- Doerksen, R.J.; Thakkar, A.J. Quadrupole and octopole moments of heteroaromatic rings. J. Phys. Chem. A 1999, 103, 10009–10014. [Google Scholar] [CrossRef]
- Mecozzi, S.; West, A.P.; Dougherty, D.A. Cation-π interactions in simple aromatics: Electrostatics provide a predictive tool. J. Am. Chem. Soc. 1996, 118, 2307–2308. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The cation−π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S. Cation-π interactions in organic crystals. Coord. Chem. Rev. 2020, 415, 213301. [Google Scholar] [CrossRef]
- Ostapenko, N.I.; Sugakov, V.I.; Shpak, M.T. Optical spectra of strained organic crystals. In Spectroscopy of Defects in Organic Crystals; Kluwer: Dordrecht, The Netherlands, 1993; Chapter 3; pp. 103–130. [Google Scholar]
- Nakajima, K.; Hori, A. Dynamic transformation and reversible guest encapsulations of pseudopolymorphs of a fully fluorinated β-diketonate Pd(II) complex. Cryst. Growth Des. 2014, 14, 3169–3173. [Google Scholar] [CrossRef]
- Hori, A.; Gonda, R.; Rzeznicka, I.I. Enhanced adsorption of small gas molecules in metal (Cu2+, Pd2+, Pt2+) complexes induced by ligand fluorination. CrystEngComm 2017, 19, 6263–6266. [Google Scholar] [CrossRef]
- Kusakawa, T.; Sakai, S.; Nakajima, K.; Yuge, H.; Rzeznicka, I.I.; Hori, A. Synthesis, structures and co-crystallizations of perfluorophenyl substituted β-diketone and triketone compounds. Crystals 2019, 9, 175. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Pang, X.; Wang, H.; Wang, W.; Jin, W.J. Phosphorescent π-hole···π bonding cocrystals of pyrene with halo-perfluorobenzenes (F, Cl, Br, I). Cryst. Growth Des. 2015, 15, 4938–4945. [Google Scholar] [CrossRef]
- Toikka, Y.N.; Mikherdov, A.S.; Ivanov, D.M.; Mooibroek, T.J.; Nadezhda, A.; Bokach, N.A.; Kukushkin, V.Y. Cyanamides as π-hole donor components of structure-directing (cyanamide)···arene noncovalent interactions. Cryst. Growth Des. 2020, 20, 4783–4793. [Google Scholar] [CrossRef]
- Ibrahim, N.A.A.; Rady, A.S.M.; Al-Fahemi, J.H.; Telb, E.M.Z.; Ahmed, S.A.; Shawky, A.M.; Moussa, N.A.M. π-Hole interactions: A comparative investigation based on boron-containing molecules. Chem. Select 2020, 5, 13223–13231. [Google Scholar] [CrossRef]
- Politzer, P.; Murraya, J.S.; Clark, T. The π-hole revisited. Phys. Chem. Chem. Phys. 2021, 23, 16458–16468. [Google Scholar] [CrossRef] [PubMed]
- Shmelev, M.A.; Kuznetsova, G.N.; Dolgushin, F.M.; Voronina Yu, K.; Gogoleva, N.V.; Kiskin, M.A.; Ivanov, V.K.; Sidorov, A.A.; Eremenko, I.L. Influence of the Fluorinated Aromatic Fragments on the Structures of the Cadmium and Zinc Carboxylate Complexes Using Pentafluorobenzoates and 2,3,4,5-Tetrafluorobenzoates as Examples. Russ. J. Coord. Chem. 2021, 47, 127. [Google Scholar] [CrossRef]
- Toikka, Y.N.; Starova, G.L.; Suslonov, V.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Combined σ- and π-hole donor properties of perfluorinated iodo(or bromo)benzenes: Halogen bonding and π-hole Interactions in cocrystals including Cu4I4 Clusters. Cryst. Growth Des. 2023, 23, 5194–5203. [Google Scholar] [CrossRef]
- Mallada, B.; Ondráček, M.; Lamanec, M.; Gallardo, A.; Jiménez-Martín, A.; de la Torre, B.; Hobza, P.; Jelínek, P. Visualization of π-hole in molecules by means of Kelvin probe force microscopy. Nat. Commun. 2023, 14, 4954. [Google Scholar] [CrossRef]
- Alfuth, J.; Kazimierczuk, K.; Połoński, T.; Olszewska, T. Intermolecular hydrogen bonding directed by aryl-perfluoroaryl π–π stacking interactions. Cryst. Growth Des. 2023, 23, 6830–6839. [Google Scholar] [CrossRef]
- Hunter, C.A. Meldola Lecture. The role of aromatic interactions in molecular recognition. Chem. Soc. Rev. 1994, 23, 101–109. [Google Scholar] [CrossRef]
- Cole, J.C.; Taylor, R. Intermolecular Interactions of Organic Fluorine Seen in Perspective. Cryst. Growth Des. 2022, 22, 1352–1364. [Google Scholar] [CrossRef]
- Kutzke, H.; Klapper, H.; Hammond, R.B.; Roberts, K.J. Metastable b-phase of benzophenone: Independentstructure determinations via X-ray powderdiffraction and single crystal studies. Acta Cryst. B 2000, 56, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Kusakawa, T.; Goto, T.; Hori, A. Supramolecular association of M2+···π induced by different electrostatic properties using naphthyl substituted β-diketonate complexes (metal = Cu, Pd, Pt). CrystEngComm 2020, 22, 3090–3094. [Google Scholar] [CrossRef]
- Crowder, J.M.; Han, H.; Wei, Z.; Dikarev, E.V.; Petrukhina, M.A. Unsolvated homo- and heterometallic highly fluorinated β-diketonate complexes of copper(II). Polyhedron 2019, 157, 33–38. [Google Scholar] [CrossRef]
- Habuka, Y.; Takeuchi, E.A.; Hori, A. Co-crystal structure, Hirshfeld surface analysis and DFT studies of 3,4-ethylenedioxythiophene solvated bis[1,3-bis(pentafluorophenyl)propane-1,3-dionato]copper(II). Acta Cryst. 2020, E76, 820–825. [Google Scholar]
- Habuka, Y.; Usui, H.; Okawa, M.; Shinohara, T.; Yuge, H.; Mao, Y.; Gong, J.; Richards, G.J.; Hori, A. Selective recognition and reversible encapsulation of tetrameric alcohol clusters via hydrogen bonds using a perfluorinated dinuclear nickel(II) complex. CrystEngComm 2024, 26, 3014–3020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1a)2•(3)3 | (1b)2•(3)3 | (1c)2•(3)3 | |
|---|---|---|---|
| Description | Prismatic | Prismatic | Prismatic |
| Chemical formula | C99H34Cu2F40O11 | C99H34F40O11Pd2 | C99H34F40O11Pt2 |
| Formula weight | 2286.34 | 2372.06 | 2549.44 |
| Temperature [K] | 103 | 103 | 123 |
| Crystal system | monoclinic | Monoclinic | monoclinic |
| Space group | Cc | Cc | Cc |
| a [Å] | 31.4023(17) | 31.269(2) | 31.354(5) |
| b [Å] | 15.0085(7) | 15.1818(10) | 15.220(3) |
| c [Å] | 24.0330(12) | 24.045(3) | 24.470(4) |
| β [°] | 129.932(1) | 130.098(2) | 130.441(4) |
| V [Å3] | 8685.5(8) | 8731.5(13) | 8887(3) |
| Z | 4 | 4 | 4 |
| Dc [Mg m−3] | 1.748 | 1.804 | 1.905 |
| μ [mm−1] | 0.642 | 0.563 | 3.294 |
| F(000) | 4536 | 4672 | 4928 |
| Rint | 0.1012 | 0.1071 | 0.0264 |
| GOF | 1.033 | 1.172 | 1.016 |
| R [(I) > 2σ (I)] | 0.0345 | 0.0712 | 0.0164 |
| wR (Fo2) | 0.0724 | 0.1298 | 0.0355 |
| Flack parameter | 0.271 (8) | 0.47 (4) | 0.214 (3) |
| CCDC No. | 1901579 | 1901580 | 2361541 |
| 2•(3)2 | 2•(3)4 | 1a•(4)1.5 | 2•(4)2 | |
|---|---|---|---|---|
| Description | Plate | Prismatic | Block | Block |
| Chemical formula | C60H24Cu2F20O8 | C43H22CuF10O5 | C51H15CuF20O4 | C62H28Cu2F20O6 |
| Formula weight | 1379.87 | 872.14 | 1135.17 | 1375.92 |
| Temperature [K] | 100 | 150 | 100 | 100 |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic |
| Space group | P21/n | P-1 | C2/c | P21/n |
| a [Å] | 17.900(2) | 11.6773(8) | 31.426(6) | 17.8048(17) |
| b [Å] | 8.0289(9) | 13.0175(9) | 15.545(3) | 8.1625(8) |
| c [Å] | 18.0922(18) | 14.2065(9) | 23.673(5) | 17.9524(18) |
| α [°] | 90 | 63.385(2) | 90 | 90 |
| β [°] | 92.686(3) | 84.163(2) | 129.866(4) | 93.253(3) |
| γ [°] | 90 | 69.029(2) | 90 | 90 |
| V [Å3] | 2597.3(5) | 1798.4(2) | 8877(3) | 2604.8(4) |
| Z | 2 | 2 | 8 | 2 |
| Dc [Mg m−3] | 1.764 | 1.611 | 1.699 | 1.754 |
| μ [mm−1] | 0.951 | 0.708 | 0.625 | 0.946 |
| F(000) | 1372 | 878 | 4496 | 1372 |
| Rint | 0.1815 | 0.0508 | 0.0674 | 0.3277 |
| GOF | 1.204 | 0.918 | 1.094 | 0.815 |
| R [(I) > 2σ (I)] | 0.0973 | 0.0429 | 0.0483 | 0.0339 |
| wR (Fo2) | 0.2018 | 0.1438 | 0.1266 | 0.0658 |
| CCDC No. | 2361542 | 2361543 | 2361544 | 2361545 |
| (1a)2•(3)3 | (1b)2•(3)3 | (1c)2•(3)3 | |
|---|---|---|---|
| Ph in BP-1 ··· Ring-1 | 3.579(3) | 4.073(12) | 3.988(5) |
| Ph in BP-1 ··· Ring-2 | 4.108(4) | 3.542(9) | 3.633(3) |
| Ph in BP-2 ··· Ring-7 | 3.653(3) | 3.887(12) | 3.554(4) |
| Ph in BP-2 ··· Ring-8 | 3.896(4) | 3.601(8) | 3.788(4) |
| Ph in BP-3 ··· Ring-2 [a] | 3.721(4) | 3.728(11) | 3.989(5) |
| Ph in BP-3 ··· M1 | 3.389 | 3.437 | 3.583 |
| Ph in BP-3 ··· M2 | 3.436 | 3.492 | 3.524 |
| Ph in BP-1 ··· M2 [b] | 3.524 | 3.521 | 3.523 |
| Ph in BP-2 ··· M1 [b] | 3.401 | 3.435 | 3.575 |
| 1a | 1b | 1c | 2 | 3 | 4 | |
|---|---|---|---|---|---|---|
| maximum | +201.13 | +137.45 | +140.24 | +237.56 | +96.27 | +77.16 |
| minimum | −152.09 | −139.14 | −134.34 | −148.95 | −182.29 | −92.03 |
| Cg of metal | +201.1 | +11.0 | −38.5 | +237.56 | --- | --- |
| ~ +200.3 | ~ +1.1 | ~ −43.0 | ~ +160.3 | |||
| Cg of aromatic ring | +100.2 | +100.9 | +110.0 | +99.7 | −70.7 | −92.0 |
| ~ +82.0 | ~ +81.0 | ~ +83.0 | ~ +93.1 | ~ −56.6 | ~ −90.7 | |
| others | --- | --- | --- | --- | −182.29 | −78.2 |
| O in CO | Cg in C=C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, H.; Ikumura, Y.; Lee, C.-H.; Hori, A. Co-Crystallization and Structural Studies of Benzophenone Recognized by Positively Shifted ESPs of Perfluorinated β-Diketonate Complexes (M = Cu, Pd, Pt). Crystals 2024, 14, 593. https://doi.org/10.3390/cryst14070593
Kobayashi H, Ikumura Y, Lee C-H, Hori A. Co-Crystallization and Structural Studies of Benzophenone Recognized by Positively Shifted ESPs of Perfluorinated β-Diketonate Complexes (M = Cu, Pd, Pt). Crystals. 2024; 14(7):593. https://doi.org/10.3390/cryst14070593
Chicago/Turabian StyleKobayashi, Hiroyuki, Yoshinori Ikumura, Chang-Hyoun Lee, and Akiko Hori. 2024. "Co-Crystallization and Structural Studies of Benzophenone Recognized by Positively Shifted ESPs of Perfluorinated β-Diketonate Complexes (M = Cu, Pd, Pt)" Crystals 14, no. 7: 593. https://doi.org/10.3390/cryst14070593