
Advances in the Parameter Space Concept towards Picometer Precise Crystal Structure Refinement—A Resolution Study

 , and
, and
Abstract
:1. Introduction
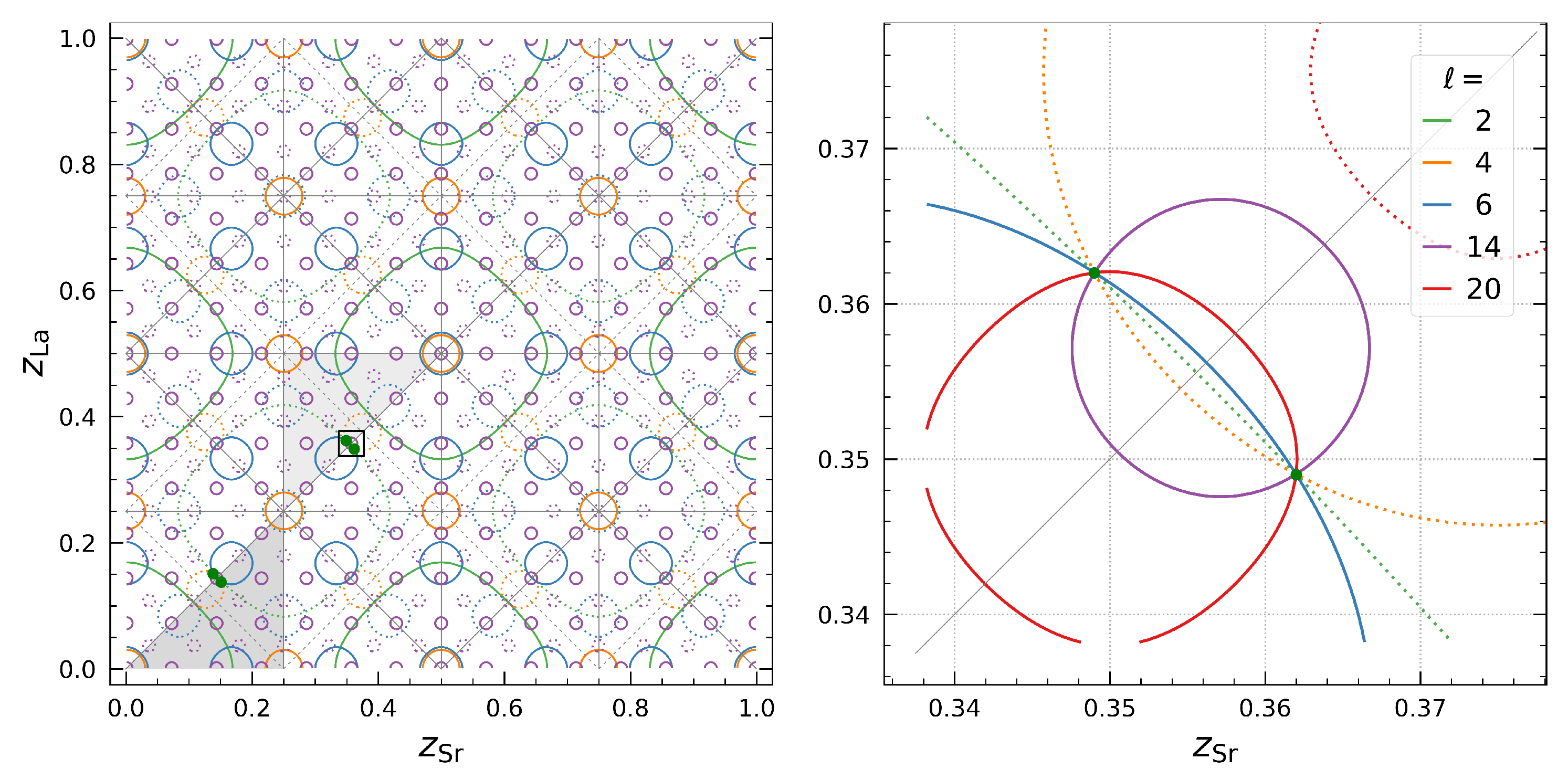
2. The Object and Its PSC-Handling
3. Resolution Studies
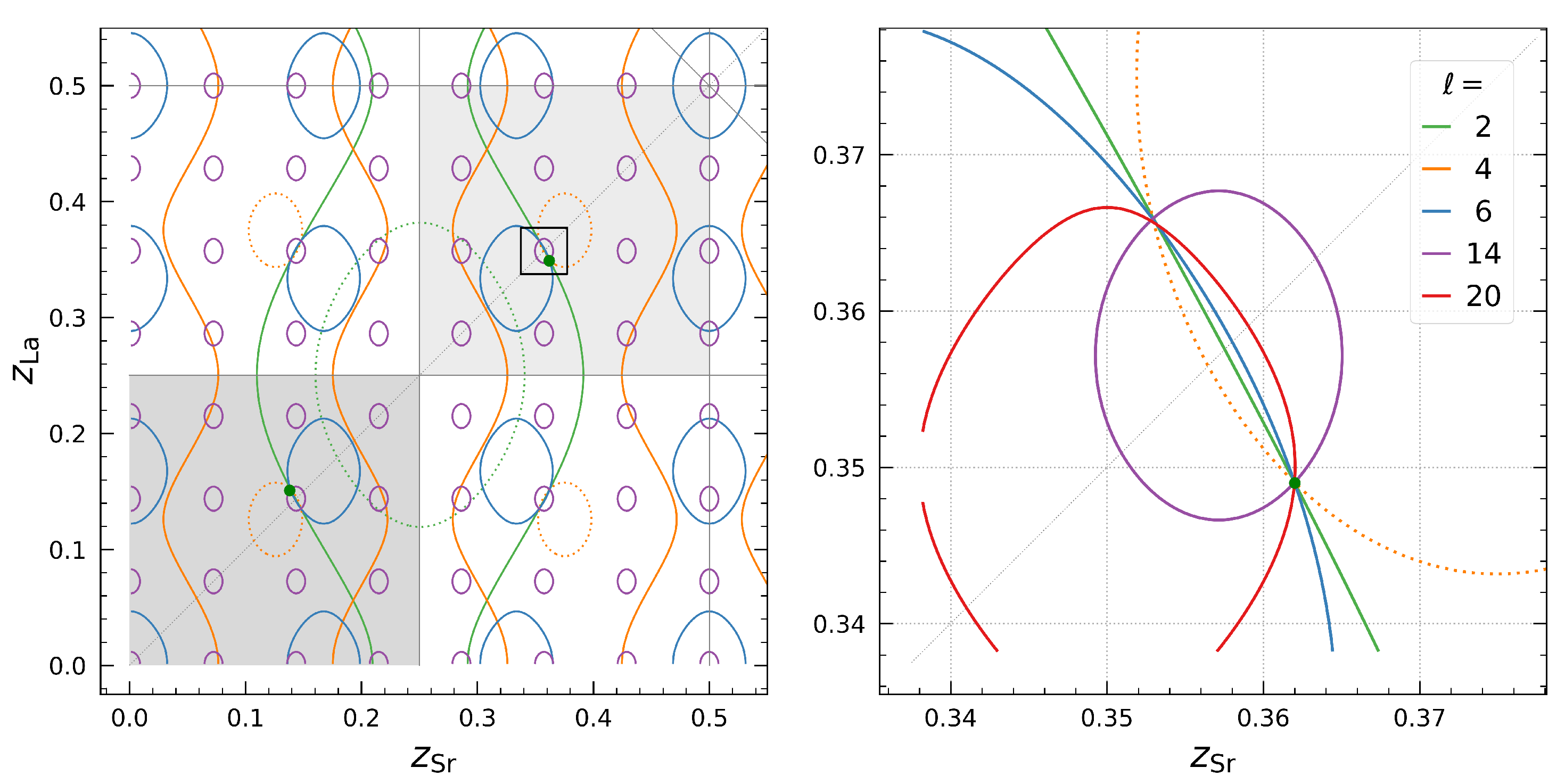
3.1. Step 1: Going beyond the Equal Point-Atom Model
3.2. Step 2: Variations in Atomic Scattering Power
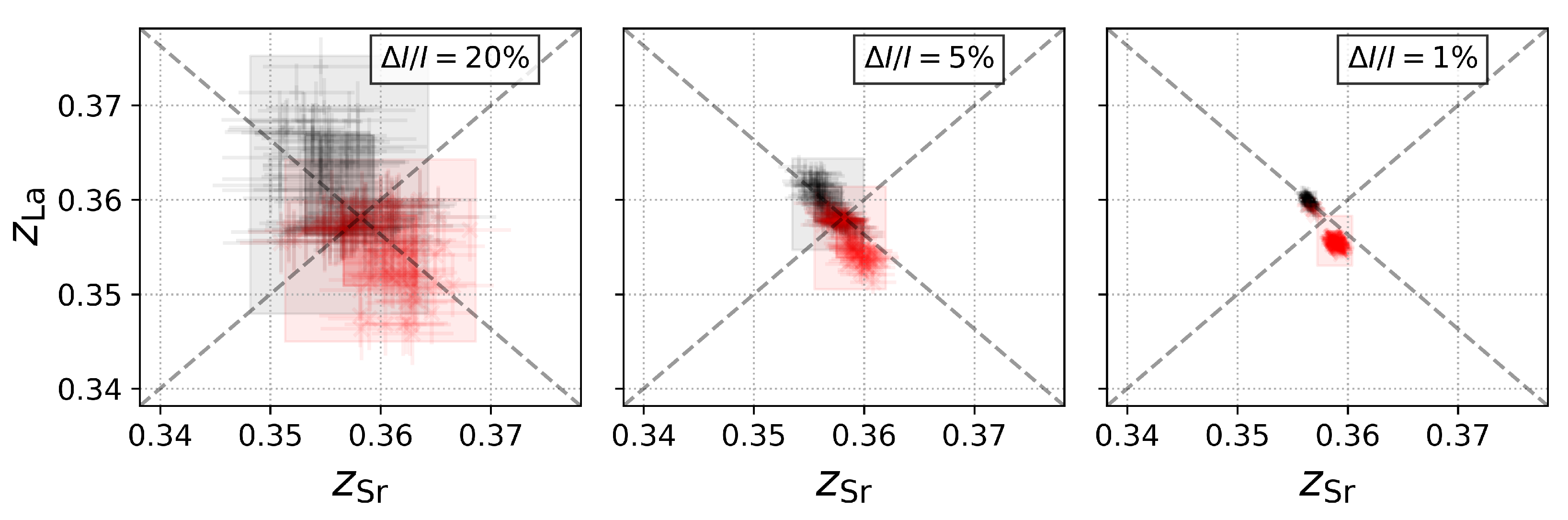
3.3. Step 3: Intensity Errors and Monte-Carlo Calculations
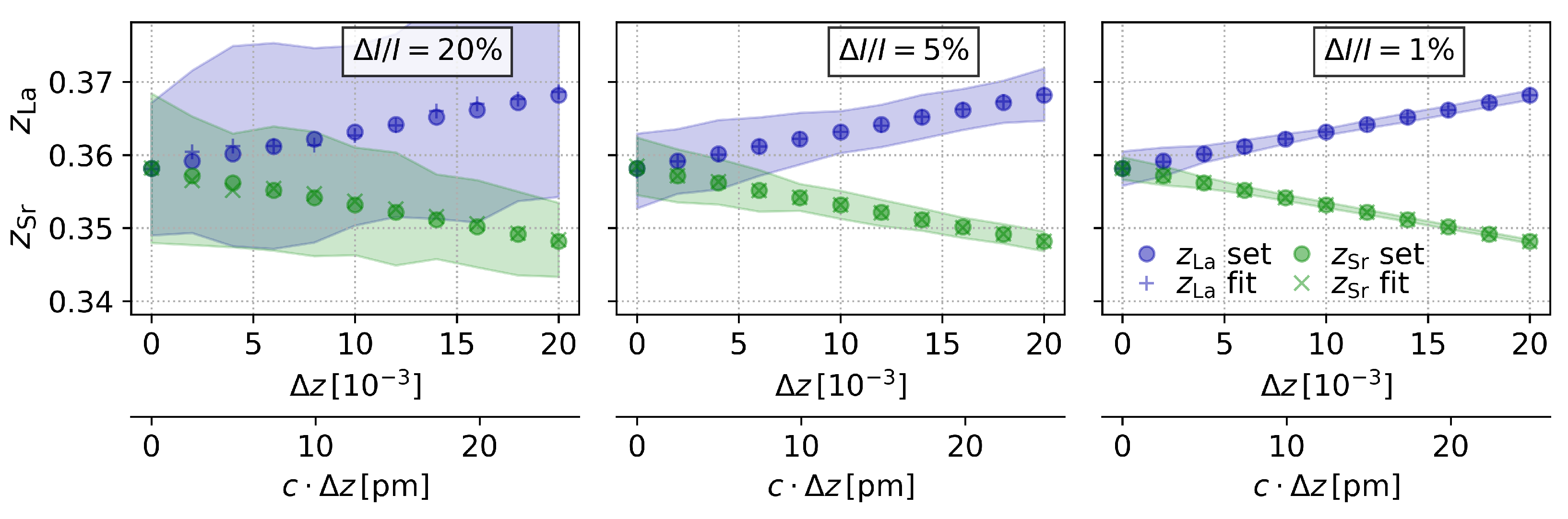
3.4. Step 4: Dependence of the Resolution on the Intensity Error
3.5. Step 5: Pushing the Limits of the PSC
4. Discussion
5. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Simulation Details

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distrontium manganate (IV) | ||
| Sr Mn O La | ||
| 12 8 32 4 | ||
| Direct | ||
| Distrontium manganate (IV) | ||
| Sr Mn O La | ||
| 12 8 32 4 | ||
| Direct | ||
References
- Fischer, K.F.; Kirfel, A.; Zimmermann, H.W. Structure determination without Fourier inversion. Part I. Unique results for centrosymmetric examples. Z. Kristallogr. 2005, 220, 643. [Google Scholar] [CrossRef]
- Kirfel, A.; Fischer, K.F. Structure determination without Fourier inversion. Part VI: High resolution direct space structure information from one-dimensional data obtained with two wavelengths. Z. Kristallogr. 2010, 225, 261. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Fischer, K.F. Structure determination without Fourier inversion. Part V. A Concept based on parameter space. Acta Crystallogr. 2009, A65, 443. [Google Scholar] [CrossRef] [PubMed]
- Pilz, K. Weiterentwicklung und Anwendung einer Algebraischen Methode zur Teilstrukturbestimmung, ein Beitrag zur Eindeutigkeit von Strukturanalysen. Ph.D. Thesis, Universität des Saarlandes, Saarbrücken, Germany, 1996. [Google Scholar]
- Herrero-Martín, J.; Mirone, A.; Fernández-Rodríguez, J.; Glatzel, P.; García, J.; Blasco, J.; Geck, J. Hard X-ray probe to study doping-dependent electron redistribution and strong covalency in La1−xSr1+xMnO4. Phys. Rev. B 2010, 82, 075112. [Google Scholar] [CrossRef]
- Senff, D.; Reutler, P.; Braden, M.; Friedt, O.; Bruns, D.; Cousson, A.; Bourée, F.; Merz, M.; Büchner, B.; Revcolevschi, A. Crystal and magnetic structure of La1−xSr1+xMnO4: Role of the orbital degree of freedom. Phys. Rev. B 2005, 71, 024425. [Google Scholar] [CrossRef]
- Lippmann, T.; Kiele, S.; Geck, J.; Reutler, P.; von Zimmermann, M.; Büchner, B. Charge Density Study of La0.5Sr1.5MnO4 at Room Temperature. DESY Annual Report. 2003. Available online: http://hasyweb.desy.de/science/annual_reports/2003_report/part1/contrib/42/9852.pdf (accessed on 1 November 2020).
- Kirfel, A.; Fischer, K.F. High Resolution Structure Determination without Fourier Inversion: Study of a one-dimensional Split Position. In Proceedings of the Annual Conference of the German Society for Crystallography, Cologne, Germany, 28 February–4 March 2005. [Google Scholar]
- Henke, B.L.; Gullikson, E.M.; Davis, J.C. X-ray interactions: Photoabsorption, scattering, transmission, and reflection at E = 50–30,000 eV, Z = 1–92. At. Data Nucl. Data Tables 1993, 54, 181–342. [Google Scholar] [CrossRef]
- Materlik, G.; Sparks, C.; Fischer, K. Resonant Anomalous X-ray Scattering: Theory and Applications; North-Holland: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Joly, Y.; Matteo, S.D.; Bunău, O. Resonant X-ray diffraction: Basic theoretical principles. Eur. Phys. J. Spec. Top. 2012, 208, 21. [Google Scholar] [CrossRef]
- Zschornak, M.; Richter, C.; Nentwich, M.; Stöcker, H.; Gemming, S.; Meyer, D.C. Probing a crystal’s short-range structure and local orbitals by Resonant X-ray Diffraction methods. Cryst. Res. Technol. 2014, 49, 43. [Google Scholar] [CrossRef]
- Richter, C.; Zschornak, M.; Novikov, D.; Mehner, E.; Nentwich, M.; Hanzig, J.; Gorfman, S.; Meyer, D.C. Picometer polar atomic displacements in strontium titanate determined by resonant X-ray diffraction. Nat. Commun. 2018, 9, 178. [Google Scholar] [CrossRef]
- Dmitrienko, V. Forbidden reflections due to anisotropic X-ray susceptibility of crystals. Acta Crystallogr. Sect. A 1983, 39, 29–35. [Google Scholar] [CrossRef]
- Richter, C.; Novikov, D.V.; Mukhamedzhanov, E.K.; Borisov, M.M.; Akimova, K.A.; Ovchinnikova, E.N.; Oreshko, A.P.; Strempfer, J.; Zschornak, M.; Mehner, E.; et al. Mechanisms of the paraelectric to ferroelectric phase transition in RbH2PO4 probed by purely resonant X-ray diffraction. Phys. Rev. B 2014, 89, 094110. [Google Scholar] [CrossRef]
- Ovchinnikova, E.; Novikov, D.; Zschornak, M.; Kulikov, A.; Kozlovskaya, K.; Dmitrienko, V.; Oreshko, A.; Blagov, A.; Mukhamedzhanov, E.; Marchenkov, N.; et al. Forbidden Reflections in TeO2 in the Vicinity of the Te L1 Absorption Edge. Crystals 2020, 10, 719. [Google Scholar] [CrossRef]
- Wilson, A.J.C. (Ed.) International Tables for Crystallography. Volume C: Mathematical, Physical and Chemical Tables; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Zschornak, M.; Wagner, C.; Nentwich, M.; Meyer, D.C.; Fischer, K.F. Advances in the Parameter Space Concept for Crystal Structure Determination—A maximum Resolution Study. Acta Crystallogr. 2021, A77, C1254. [Google Scholar] [CrossRef]
- Lindley, P.F. Preparation, selection, and investigation of specimens. In International Tables for Crystallography, Online MRW; John Wiley and Sons, Ltd.: Hoboken, NJ, USA, 2006; Chapter 3.1; pp. 148–155. [Google Scholar] [CrossRef]
- Weigel, T.; Funke, C.; Zschornak, M.; Behm, T.; Stöcker, H.; Leisegang, T.; Meyer, D.C. X-ray diffraction using focused-ion-beam-prepared single crystals. J. Appl. Crystallogr. 2020, 53, 614–622. [Google Scholar] [CrossRef]
- Prince, E. International Tables for Crystallography Volume C: Mathematical, Physical and Chemical Tables; John Wiley and Sons, Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Spiess, L.; Teichert, G.; Schwarzer, R.; Behnken, H.; Genzel, C. Moderne Röntgenbeugung: Röntgendiffraktometrie für Materialwissenschaftler, Physiker und Chemiker; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Weigel, T.; Leisegang, T.; Zschornak, M.; Herrmann, M.; Rothenberger, M.; Wünsche, A.; Stöcker, H.; Meyer, D.C. Influence of environmental parameter variations on X-ray beam intensities: A time-dependent absorption correction. J. Appl. Crystallogr. 2015, 48, 1870–1882. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zschornak, M.; Wagner, C.; Nentwich, M.; Vallinayagam, M.; Fischer, K.F. Advances in the Parameter Space Concept towards Picometer Precise Crystal Structure Refinement—A Resolution Study. Crystals 2024, 14, 684. https://doi.org/10.3390/cryst14080684
Zschornak M, Wagner C, Nentwich M, Vallinayagam M, Fischer KF. Advances in the Parameter Space Concept towards Picometer Precise Crystal Structure Refinement—A Resolution Study. Crystals. 2024; 14(8):684. https://doi.org/10.3390/cryst14080684
Chicago/Turabian StyleZschornak, Matthias, Christian Wagner, Melanie Nentwich, Muthu Vallinayagam, and Karl F. Fischer. 2024. "Advances in the Parameter Space Concept towards Picometer Precise Crystal Structure Refinement—A Resolution Study" Crystals 14, no. 8: 684. https://doi.org/10.3390/cryst14080684
APA StyleZschornak, M., Wagner, C., Nentwich, M., Vallinayagam, M., & Fischer, K. F. (2024). Advances in the Parameter Space Concept towards Picometer Precise Crystal Structure Refinement—A Resolution Study. Crystals, 14(8), 684. https://doi.org/10.3390/cryst14080684