
Screening, Growing, and Validation by Catalog: Using Synthetic Intermediates from Natural Product Libraries to Discover Fragments for an Aspartic Protease Through Crystallography
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Fragment Test Set
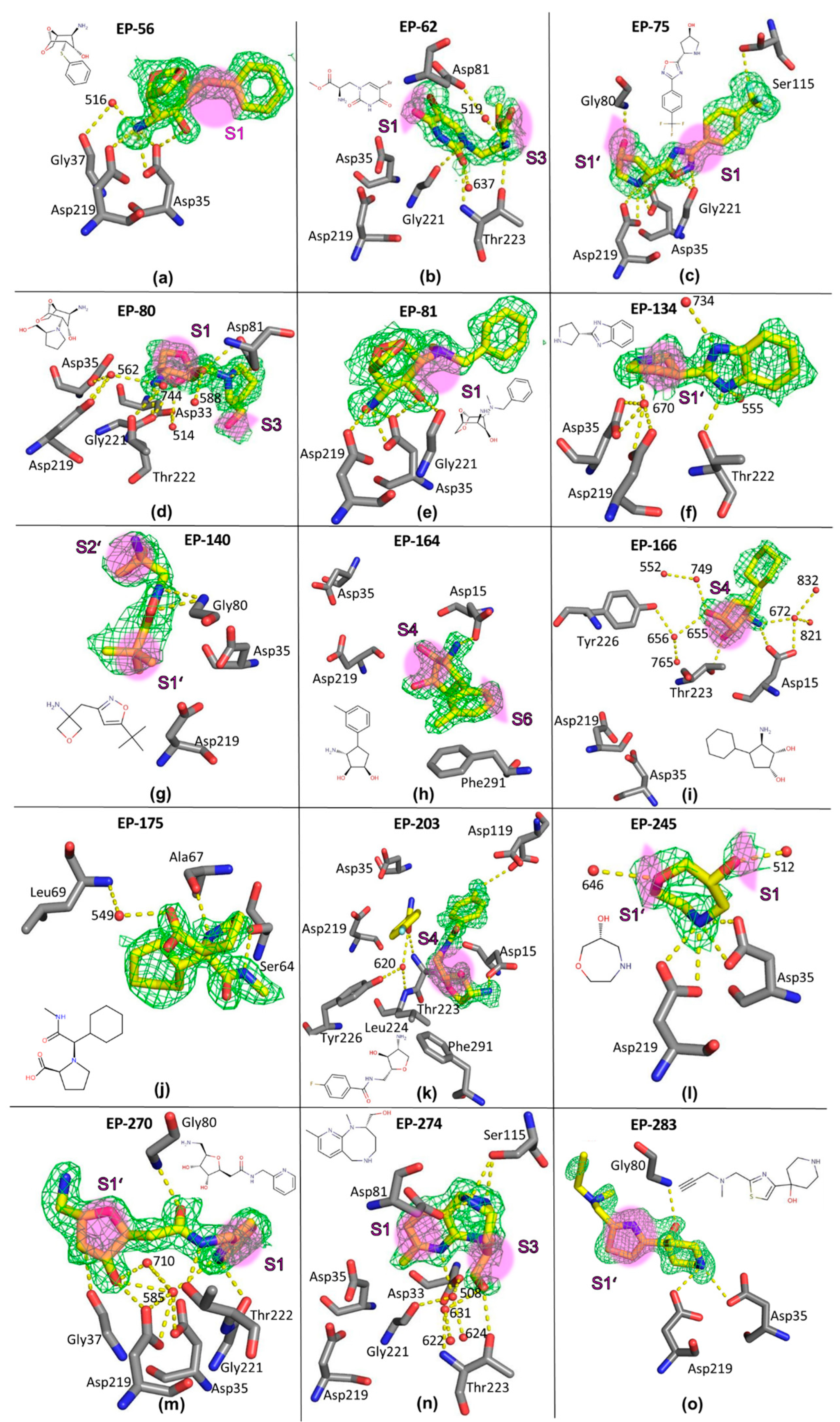
2.2. Crystal Structures of Fragment Complexes
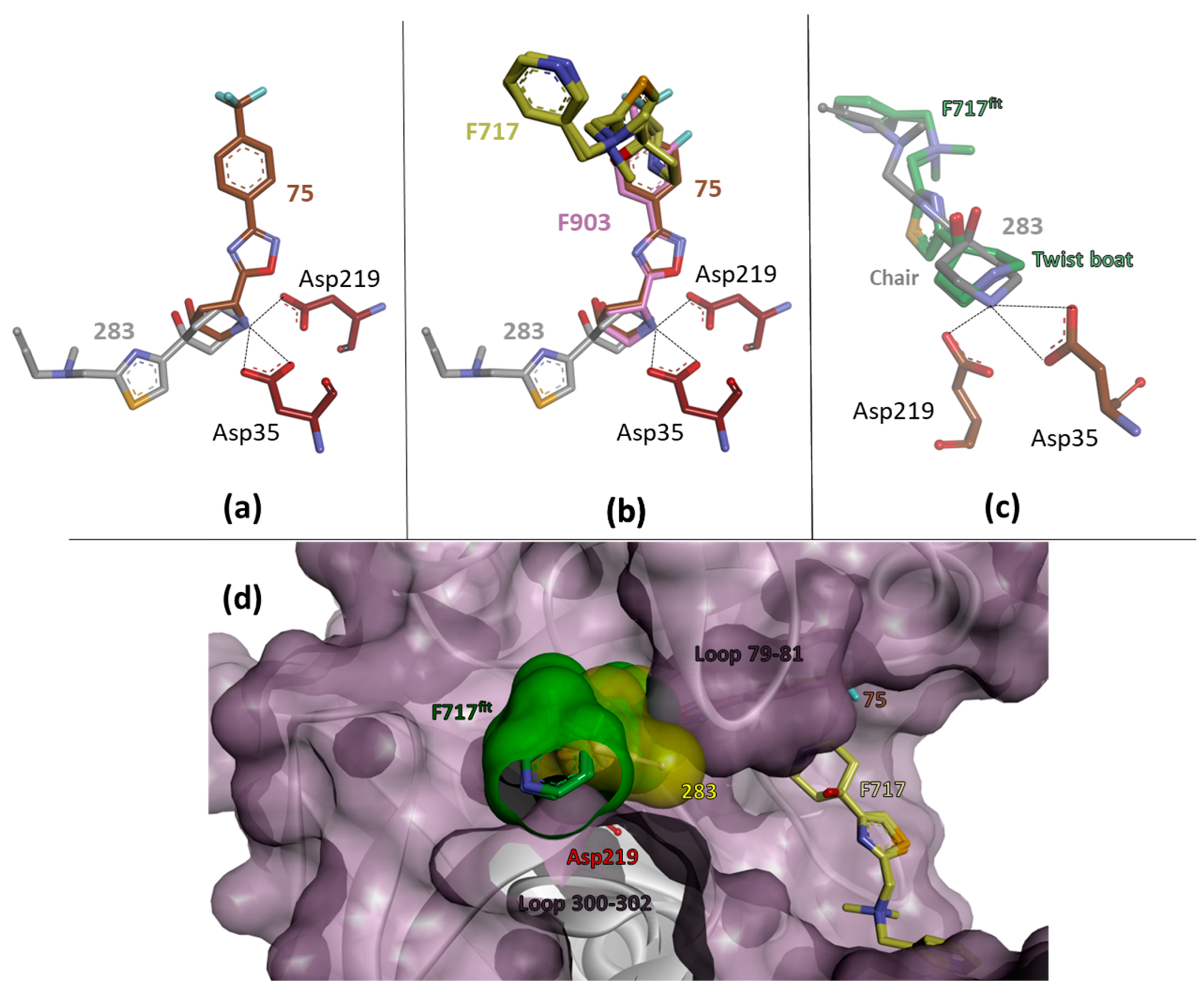
2.3. Comparative Analysis
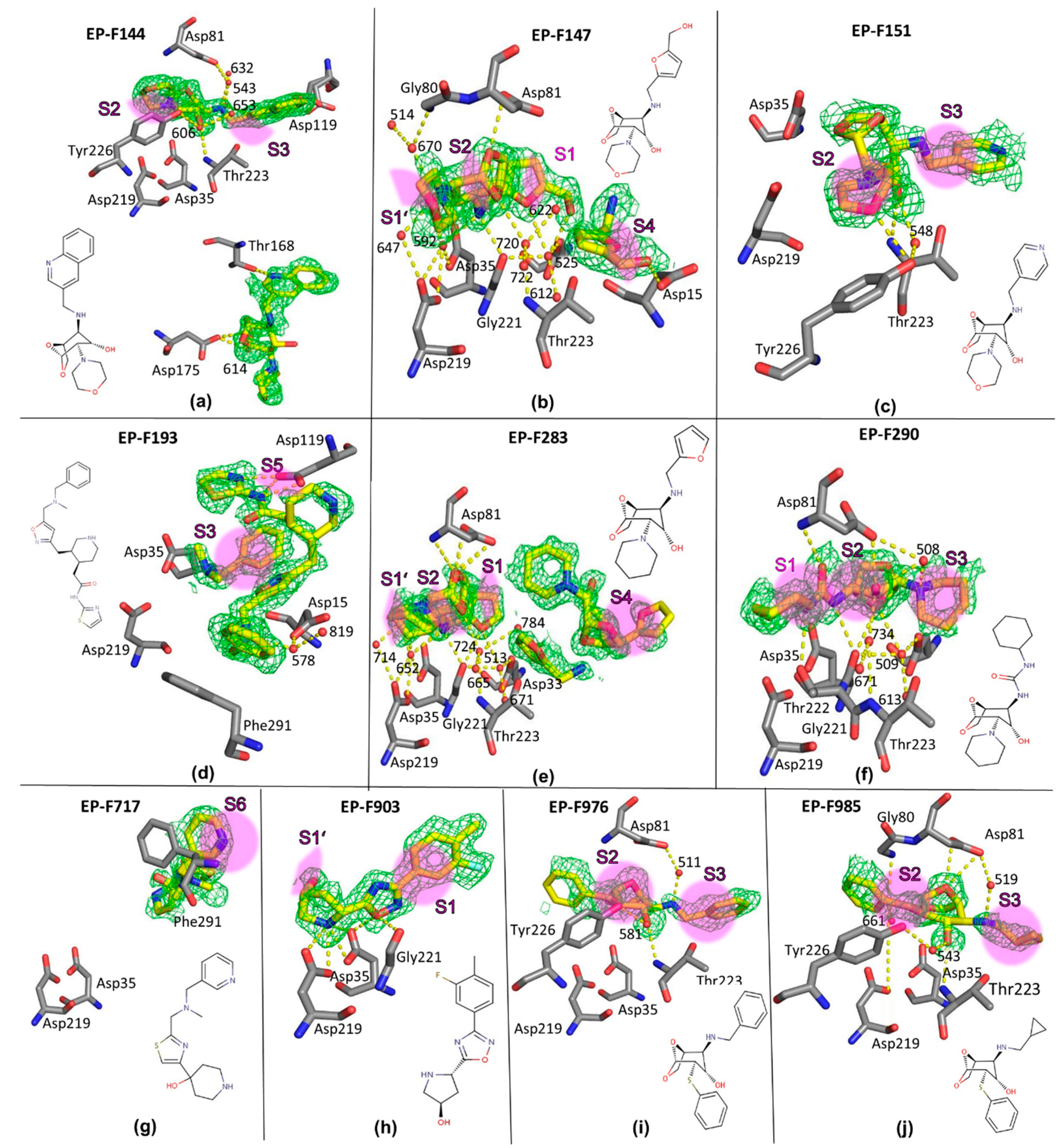
2.4. Crystal Structures and Validation Using Follow-Up Compounds in Complex with EP
2.5. Binding Affinities of AnalytiCon Ligands by Isothermal Titration Calorimetry (ITC)
3. Conclusions and Outlook
4. Materials and Methods
4.1. Ligand Selection for the Initial Crystallographic Fragment Screening
4.2. Crystallization of EP
4.3. Soaking of Crystals
4.4. Data Collection and Processing
4.5. Structure Refinement and Fragment Binding Analysis
4.6. Isothermal Titration Calorimetry (ITC)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hajduk, P.J.; Sheppard, G.; Nettesheim, D.G.; Olejniczak, E.T.; Shuker, S.B.; Meadows, R.P.; Steinman, D.H.; Carrera, G.M., Jr.; Marcotte, P.A.; Severin, J.; et al. Discovery of Potent Nonpeptide Inhibitors of Stromelysin Using SAR by NMR. J. Am. Chem. Soc. 1997, 119, 5818–5827. [Google Scholar] [CrossRef]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-Throughput Crystallography for Lead Discovery in Drug Design. Nat. Rev. Drug Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-based drug discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A Decade of Fragment-based Drug Design: Strategic Advances and Lessons Learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef]
- Baker, M. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 2013, 12, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Hou, J.; Zimmerman, M.D.; Wlodawer, A.; Minor, W. The future of crystallography in drug discovery. Expert Opin. Drug Discov. 2013, 9, 125–137. [Google Scholar] [CrossRef]
- Bijak, V.; Szczygiel, M.; Lenkiewicz, J.; Gucwa, M.; Cooper, D.R.; Murzyn, K.; Minor, W. The current role and evolution of X-ray crystallography in drug discovery and development. Expert Opin. Drug Discov. 2023, 18, 1221–1230. [Google Scholar] [CrossRef]
- Woodhead, A.J.; Erlanson, D.A.; de Esch, I.J.P.; Holvey, R.S.; Jahnke, W.; Pathuri, P. Fragment-to-Lead Medicinal Chemistry Publications in 2022. J. Med. Chem. 2024, 67, 2287–2304. [Google Scholar] [CrossRef]
- Schiebel, J.; Radeva, N.; Krimmer, S.G.; Wang, X.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Huschmann, F.U.; Metz, A.; Weiss, M.S.; et al. Six Biophysical Screening Methods Miss a Large Proportion of Crystallographically Discovered Fragment Hits: A Case Study. ACS Chem. Biol. 2016, 11, 1693–1701. [Google Scholar] [CrossRef]
- Huschmann, F.U.; Linnik, J.; Sparta, K.; Ühlein, M.; Wang, X.; Metz, A.; Schiebel, J.; Heine, A.; Klebe, G.; Weiss, M.S.; et al. Structures of endothiapepsin-fragment complexes by crystallographic fragment-screening using a novel, diverse and affordable 96-compound-fragment library. Acta Crystallogr. F 2016, F72, 346–355. [Google Scholar] [CrossRef]
- Füsser, F.T.; Wollenhaupt, J.; Weiss, M.S.; Kümmel, D.; Koch, O. Novel starting points for fragment-based drug design against mycobacterial thioredoxin reductase identified using crystallographic fragment screening. Acta Crystallogr. Sect. D 2023, D79, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Walsh, M.A.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, R.; Groves, M.R. Fragment Screening in the Development of a Novel Anti-Malarial. Crystals 2023, 13, 1610. [Google Scholar] [CrossRef]
- de Souza Neto, L.R.; Montoya, B.O.; Brandão-Neto, J.; Verma, A.; Bowyer, S.; Moreira-Filho, J.T.; Dantas, R.F.; Neves, B.J.; Andrade, C.H.; von Delft, F.; et al. Fragment library screening by X-ray crystallography and binding site analysis on thioredoxin glutathione reductase of Schistosoma mansoni. Sci. Rep. 2024, 14, 1582. [Google Scholar] [CrossRef]
- Neumann, P.; Heidemann, J.L.; Wollenhaupt, J.; Dickmanns, A.; Agthe, M.; Weiss, M.S.; Ficner, R. A small step towards an important goal: Fragment screen of the c-di-AMP-synthesizing enzyme Cda. Acta Crystallogr. Sect. D 2024, D80, 350–361. [Google Scholar] [CrossRef]
- Douangamath, A.; Powell, A.; Fearon, D.; Collins, P.M.; Talon, R.; Krojer, T.; Skyner, R.; Brandao-Neto, J.; Dunnett, L.; Dias, A.; et al. Achieving Efficient Fragment Screening at XChem Facility at Diamond Light Source. J. Vis. Exp. 2021, 29, e62414. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, D.P.; Steuber, J.; Fritz, G.; Wojdyla, J.A.; Sharpe, M.E. Fast fragment and compound screening pipeline at the Swiss Light Source. Methods Enzymol. 2023, 690, 235–284. [Google Scholar]
- Metz, A.; Stegmann, D.P.; Panepucci, E.H.; Buehlmann, S.; Huang, C.-Y.; McAuley, K.E.; Wang, M.; Wojdyla, J.A.; Sharpea, M.E.; Smitha, K.M.L. HEIDI: An experiment-management platform enabling high-throughput fragment and compound screening. Acta Crystallogr. Sect. D 2024, D80, 328–335. [Google Scholar] [CrossRef]
- Keserű, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef]
- Troelsen, N.S.; Clausen, M.H. Library Design Strategies To Accelerate Fragment-Based Drug Discovery. Chemistry 2020, 26, 11391–11403. [Google Scholar] [CrossRef]
- Available online: https://practicalfragments.blogspot.com/2023/12/review-of-2023-reviews.html (accessed on 24 August 2024).
- Available online: https://www.cambridgemedchemconsulting.com/ (accessed on 24 August 2024).
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H.A. ‘Rule of Three’ for fragment-based lead discovery. Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The ‘rule of three’ for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644–645. [Google Scholar] [CrossRef]
- Over, B.; Wetzel, S.; Grütter, C.; Nakai, Y.; Renner, S.; Rauh, D.; Waldmann, H. Natural-product-derived fragments for fragment-based ligand discovery. Nat. Chem. 2013, 5, 21–28. [Google Scholar] [CrossRef]
- Prescher, H.; Koch, G.; Schuhmann, T.; Ertl, P.; Bussenault, A.; Glick, M.; Dix, I.; Petersen, F.; Lizos, D.E. Construction of a 3D-shaped, natural product like fragment library by fragmentation and diversification of natural products. Bioorg. Med. Chem. 2017, 25, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Pascolutti, M.; Campitelli, M.; Nguyen, B.; Pham, N.; Gorse, A.-D.; Quinn, R.J. Capturing Nature’s Diversity. PLoS ONE 2015, 10, e0120942. [Google Scholar] [CrossRef]
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytother. Res. 2021, 35, 4660–4702. [Google Scholar] [CrossRef] [PubMed]
- Grigalunas, M.; Burhop, A.; Zinken, S.; Pahl, A.; Gally, J.M.; Wild, N.; Mantel, Y.; Sievers, S.; Foley, D.J.; Scheel, R.; et al. Natural product fragment combination to performance-diverse pseudo-natural products. Nat. Commun. 2021, 12, 1883. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.F.; Hamilton, D.J.; de Esch, I.J.P.; Wijtmans, M.; O’Brien, P. Escape from planarity in fragment-based drug discovery: A synthetic strategy analysis of synthetic 3D fragment libraries. Drug Discov. Today 2022, 27, 2484–2496. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Available online: https://ac-discovery.com/wp-content/uploads/AnalytiCon_Discovery_NATx_Product_Information.pdf (accessed on 24 August 2024).
- Haustedt, L.O.; Siems, K. The Role of Natural Products in Drug Discovery. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W., Hamley, P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Chapter 14; ISBN 97811187716002016. [Google Scholar]
- Available online: https://ac-discovery.com/fragments-nature/ (accessed on 24 August 2024).
- Pearl, L.; Blundell, T. The active site of aspartic proteinases. FEBS Lett. 1984, 174, 96–101. [Google Scholar] [CrossRef]
- Dash, C.; Kulkarni, A.; Dunn, B.; Rao, M. Aspartic Peptidase Inhibitors: Implications in Drug Development. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 89–119. [Google Scholar] [CrossRef]
- Eder, J.; Hommel, U.; Cumin, F.; Martoglio, B.; Gerhartz, B. Aspartic Proteases in Drug Discovery. Curr. Pharm. Des. 2007, 13, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K. (Ed.) Aspartic acid proteases as therapeutic targets. In Methods and Principles in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2010; Volume 45. [Google Scholar]
- Schiebel, J.; Krimmer, S.G.; Sparta, K.; Knörlein, A.; Wang, X.; Park, A.Y.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Radeva, N.; et al. High-throughput Crystallography: Reliable and Efficient Identification of Fragment Hits. Structure 2016, 24, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Radeva, N.; Krimmer, S.G.; Stieler, M.; Schiebel, J.; Fu, K.; Wang, X.; Ehrmann, F.R.; Metz, A.; Huschmann, F.U.; Weiss, M.; et al. Remote Interplay of Small Molecules with Endothiapepsin—Hot spot analysis. J. Med. Chem. 2016, 59, 7561–7575. [Google Scholar] [CrossRef]
- Radeva, N.; Schiebel, J.; Wang, X.; Krimmer, S.G.; Fu, K.; Stieler, M.; Ehrmann, F.R.; Metz, A.; Rickmeyer, T.; Betz, M.; et al. Active Site Mapping of an Aspartic Protease by Multiple Fragment Crystal Structures: Versatile Warheads to Address a Catalytic Dyad. J. Med. Chem. 2016, 59, 9743–9759. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bancet, A.; Raingeval, C.; Lomberget, T.; Le Borgne, M.; Guichou, J.F.; Krimm, I. Fragment Linking Strategies for Structure-Based Drug Design. J. Med. Chem. 2020, 63, 11420–11435. [Google Scholar] [CrossRef]
- Grenier, D.; Audebert, S.; Preto, J.; Guichou, J.F.; Krimm, I. Linkers in fragment-based drug design: An overview of the literature. Expert Opin. Drug Discov. 2023, 18, 987–1009. [Google Scholar] [CrossRef] [PubMed]
- Bedwell, E.V.; McCarthy, W.J.; Coyne, A.G.; Abell, C. Development of potent inhibitors by fragment-linking strategies. Chem. Biol. Drug Des. 2022, 100, 469–486. [Google Scholar] [CrossRef]
- Murray, C.W.; Verdonk, M.L. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J. Comput. Aided Mol. Des. 2002, 16, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Nazaré, M.; Matter, H.; Will, D.W.; Wagner, M.; Urmann, M.; Czech, J.; Schreuder, H.; Bauer, A.; Ritter, K.; Wehner, V. Fragment deconstruction of small, potent factor Xa inhibitors: Exploring the superadditivity energetics of fragment linking in protein-ligand complexes. Angew. Chem. Int. Ed. Engl. 2012, 51, 905–911. [Google Scholar] [CrossRef]
- Borsi, V.; Calderone, V.; Fragai, M.; Luchinat, C.; Sarti, N. Entropic contribution to the linking coefficient in fragment-based drug design: A case study. J. Med. Chem. 2010, 53, 4285–4289. [Google Scholar] [CrossRef]
- Chan, L.; Hutchison, G.R.; Morris, G.M. Understanding Ring Puckering in Small Molecules and Cyclic Peptides. J. Chem. Inf. Model. 2021, 61, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Dragojlovic, V. Conformational analysis of cycloalkanes. ChemTexts 2015, 1, 14. [Google Scholar] [CrossRef]
- Rühmann, E.; Betz, M.; Fricke, M.; Heine, A.; Schäfer, M.; Klebe, G. Thermodynamic Signatures of Fragment Binding: Validation of Direct versus Displacement ITC Titrations. Biochim. Biophys. Acta 2015, 1850, 647–656. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Zhang, Z.-Y. Low-Affinity Binding Determined by Titration Calorimetry Using a High-Affinity Coupling Ligand: A Thermodynamic Study of Ligand Binding to Protein Tyrosine Phosphatase 1B. Anal. Biochem. 1998, 261, 139–148. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Chen, K.; Sharp, K.A.; Kollman, P.A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Blum, A.; Böttcher, J.; Heine, A.; Klebe, G.; Diederich, W.E. Structure-Guided Design of C2-Symmetric HIV-1 Protease Inhibitors Based on a Pyrrolidine Scaffold. J. Med. Chem. 2008, 51, 2078–2087. [Google Scholar] [CrossRef]
- Blum, A.; Böttcher, J.; Sammet, B.; Luksch, T.; Heine, A.; Klebe, G.; Diederich, W.E. Achiral Oligoamines as Versatile Tool for the Development of Aspartic Protease Inhibitors. Bioorg. Med. Chem. 2008, 16, 8574–8586. [Google Scholar]
- Köster, H.; Craan, T.; Brass, S.; Herhaus, C.; Zentgraf, M.; Neumann, L.; Heine, A.; Klebe, G. A Small Nonrule of 3 Compatible Fragment Library Provides High Hit Rate of Endothiapepsin Crystal Structures with Various Fragment Chemotypes. J. Med. Chem. 2011, 54, 7784–7796. [Google Scholar] [CrossRef] [PubMed]
- Köster, H. Endothiapepsin und Proteinkinase A: Komplexstrukturen mit Neuartigen Inhibitoren, Durchmustern einer Fragmentbibliothek Sowie Inhibitordesign Ausgehend von Einer Sonde. Ph.D. Thesis, Philipps-Universität Marburg, Marburg, Germany, 2012. Available online: https://d-nb.info/102718376X/34 (accessed on 24 August 2024).
- Mueller, U.; Darowski, N.; Fuchs, M.R.; Förster, R.; Hellmig, M.; Paithankar, K.S.; Pühringer, S.; Steffien, M.; Zocher, G.; Weiss, M.S. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Rad. 2012, 19, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.; Förster, R.; Hellmig, M.; Huschmann, F.U.; Kastner, A.; Malecki, P.; Pühringer, S.; Röwer, M.; Sparta, K.; Steffien, M.; et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus 2015, 130, 141–152. [Google Scholar] [CrossRef]
- Geoff, T.; Battye, G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Andrew, G.; Leslie, W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Cryst. 2011, D67, 271–281. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. Sect. 2010, D66, 125–132. [Google Scholar] [CrossRef]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. XDSAPP2.0. J. Appl. Crystallogr. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Cryst. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D 2012, D68, 352–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, D66, 486–501. [Google Scholar] [CrossRef]
- Diederichs, K. Quantifying instrument errors in macromolecular X-ray data sets. Acta Crystallogr. Sect. D 2010, D66, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Tickle, I.J. Statistical quality indicators for electron-density maps. Acta Crystallogr. Sect D 2012, D68, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Rupp, B. Models of protein–ligand crystal structures: Trust, but verify. J. Comput. Aided Mol. Des. 2015, 29, 817–836. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, M.; Köster, H.; Bartholomäus, R.; Park, A.Y.; Shahim, A.; Heine, A.; Steuber, H.; Klebe, G.; Diederich, W.E. Tracing Binding Modes in Hit-to-Lead Optimization: Chameleon-Like Poses of Aspartic Protease Inhibitors. Angew. Chem. Int. Ed. Engl. 2015, 54, 2849–2853. [Google Scholar] [CrossRef]
- Keller, S.; Vargas, C.; Zhao, H.; Piszczek, G.; Brautigam, C.A.; Schuck, P. High-Precision Isothermal Titration Calorimetry with Automated Peak-Shape Analysis. Anal. Chem. 2012, 84, 5066–5073. [Google Scholar] [CrossRef]
- Houtman, J.C.; Brown, P.H.; Bowden, B.; Yamaguchi, H.; Appella, E.; Samelson, L.E.; Schuck, P. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: Application to adaptor protein complexes in cell signaling. Protein Sci. 2007, 16, 30–42. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huschmann, F.U.; Mueller, J.; Metz, A.; Ruf, M.; Senst, J.; Glinca, S.; Schiebel, J.; Heine, A.; Klebe, G. Screening, Growing, and Validation by Catalog: Using Synthetic Intermediates from Natural Product Libraries to Discover Fragments for an Aspartic Protease Through Crystallography. Crystals 2024, 14, 755. https://doi.org/10.3390/cryst14090755
Huschmann FU, Mueller J, Metz A, Ruf M, Senst J, Glinca S, Schiebel J, Heine A, Klebe G. Screening, Growing, and Validation by Catalog: Using Synthetic Intermediates from Natural Product Libraries to Discover Fragments for an Aspartic Protease Through Crystallography. Crystals. 2024; 14(9):755. https://doi.org/10.3390/cryst14090755
Chicago/Turabian StyleHuschmann, Franziska U., Janis Mueller, Alexander Metz, Moritz Ruf, Johanna Senst, Serghei Glinca, Johannes Schiebel, Andreas Heine, and Gerhard Klebe. 2024. "Screening, Growing, and Validation by Catalog: Using Synthetic Intermediates from Natural Product Libraries to Discover Fragments for an Aspartic Protease Through Crystallography" Crystals 14, no. 9: 755. https://doi.org/10.3390/cryst14090755