
Selective Benzene Recognition in Competitive Solvent System (Cyclohexene, Cyclohexane, Tri- and Hexafluorobenzenes) Using Perfluorinated Dinuclear Cu(II) Complex


Abstract
:1. Introduction
2. Materials and Methods
2.1. General
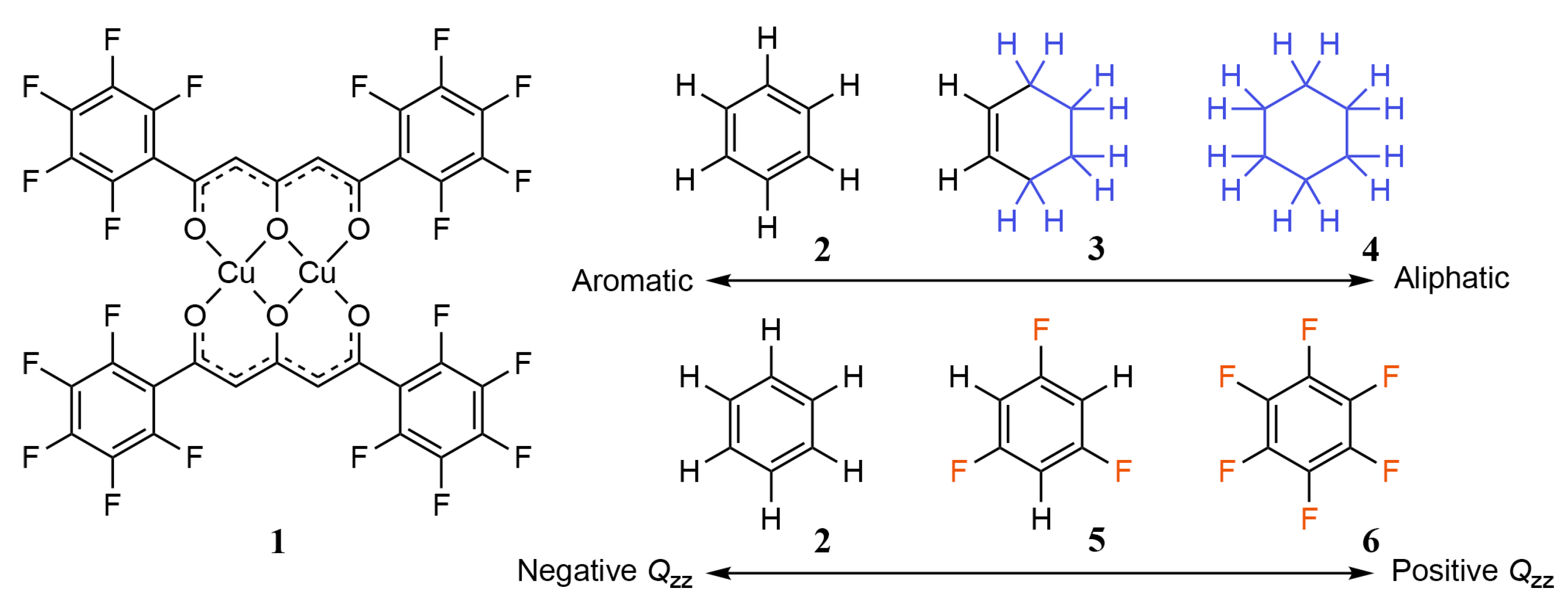
2.2. Quadrupole Moments and Electrostatic Potential of 2–6
2.3. Crystal Structure Determination
3. Results and Discussion
3.1. Preparation and Vapor Adsoption of 1
3.2. Single-Crystal Analysis with Guest-Molecules 3–6
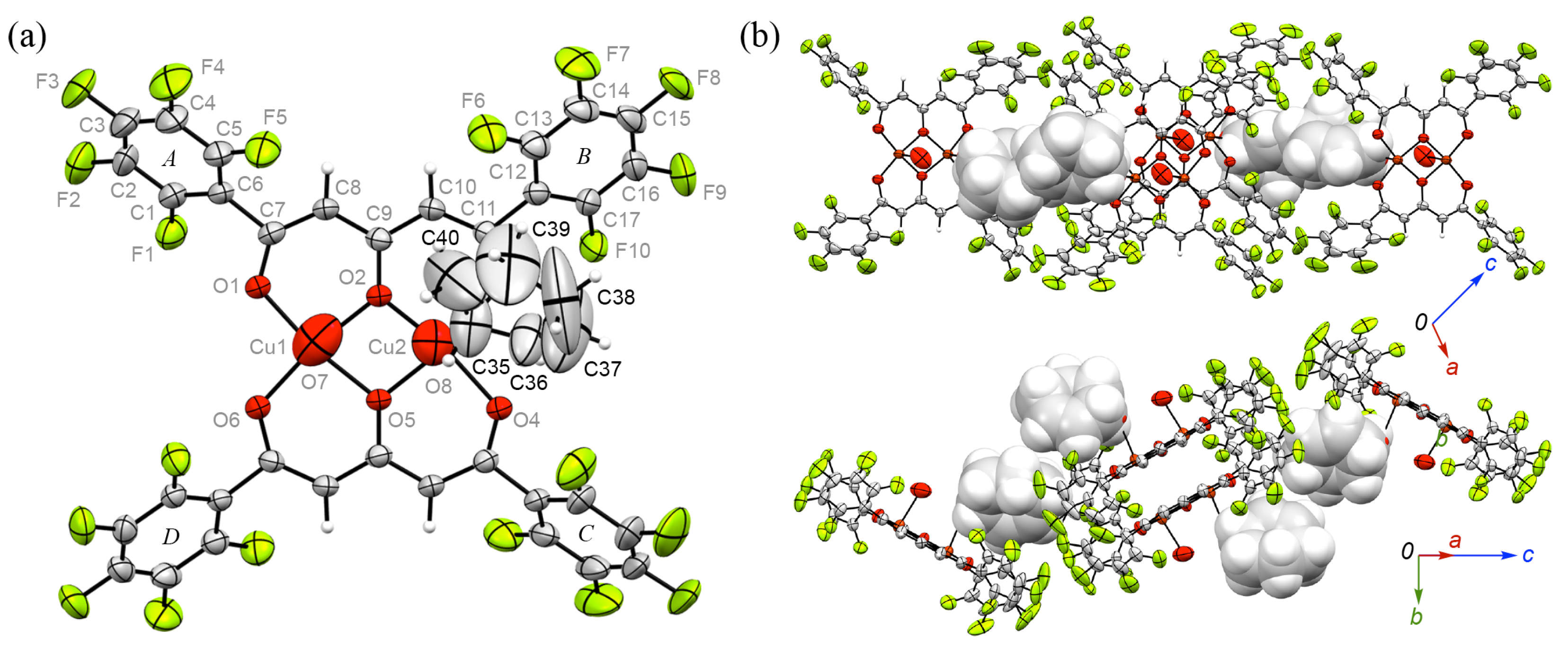
3.3. Crystal Structure of 1
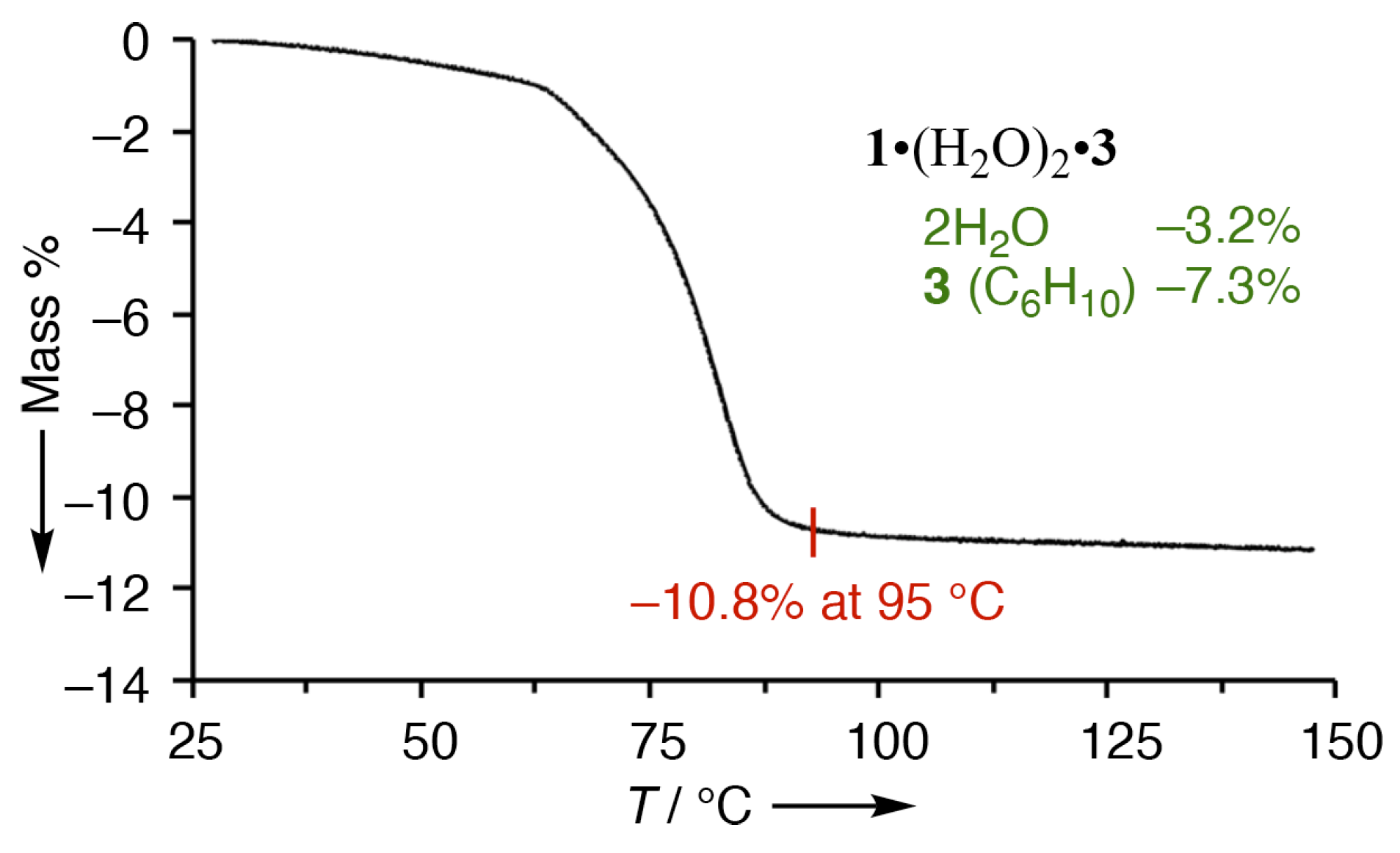
3.4. Crystal Structure of 1•(O)2•3
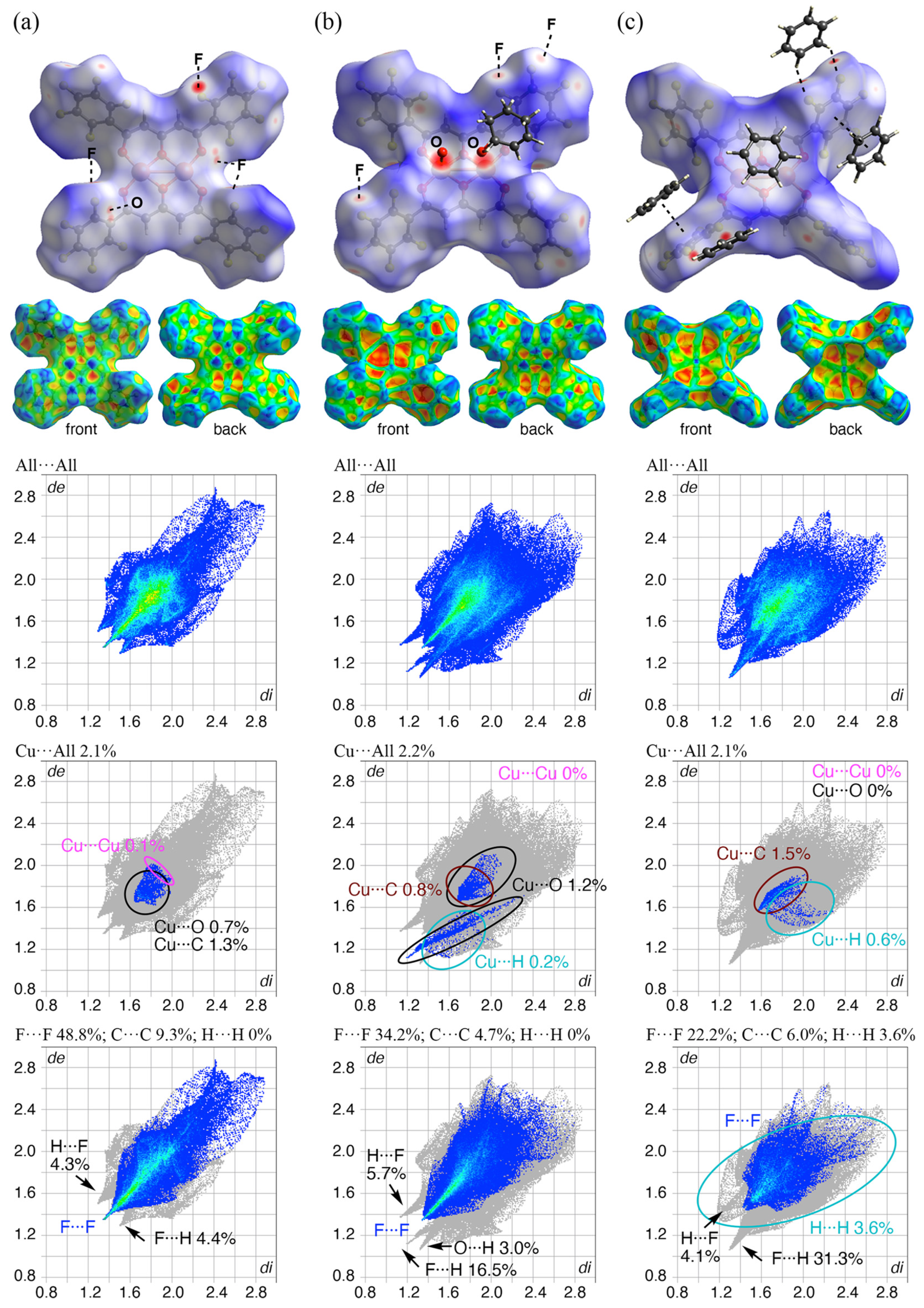
3.5. Hirshfeld Surface Analysis of 1, 1•(O)2•3, and 1•(2)3
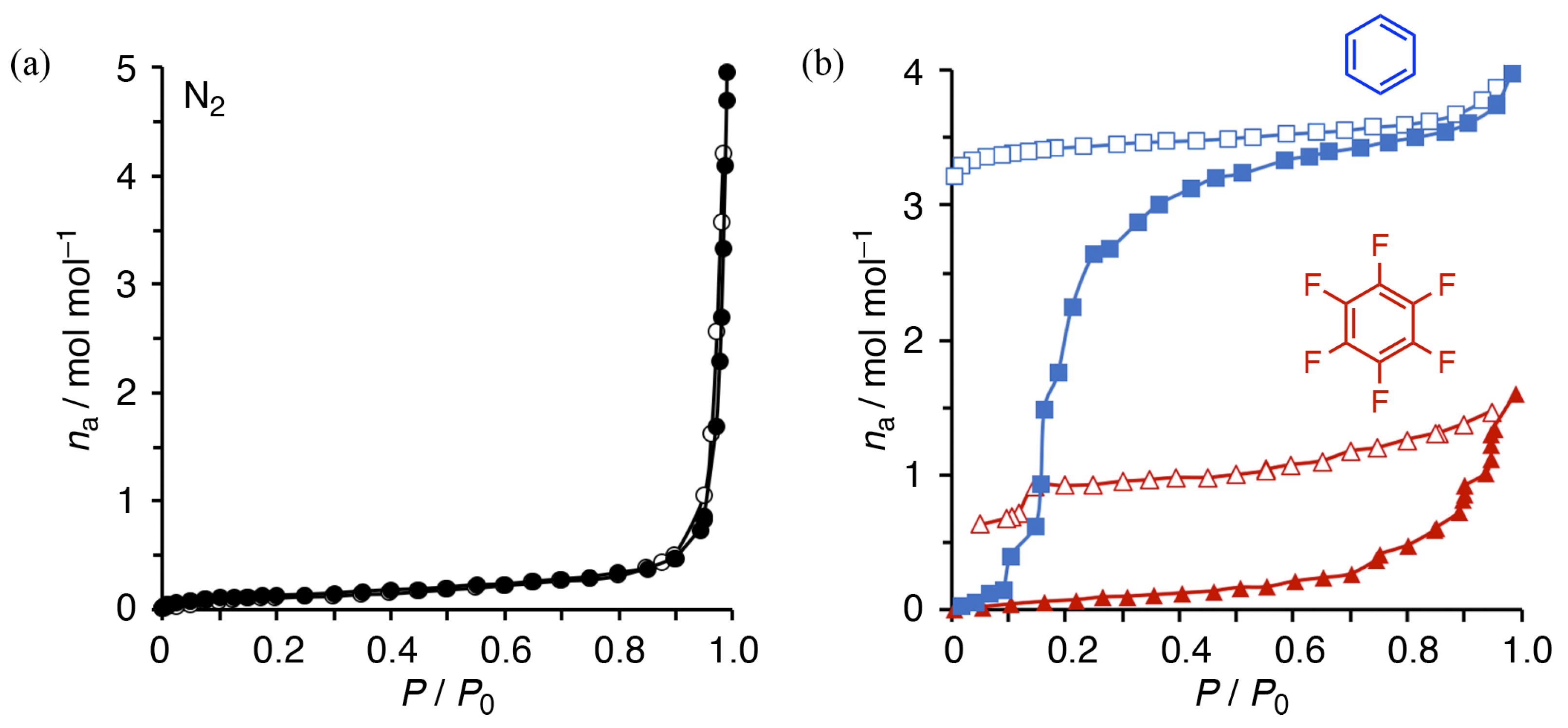
3.6. High Selectivity for Benzene from Mixed Solvents
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
References
- von Ragué Schleyer, P. Introduction: Aromaticity. Chem. Rev. 2001, 101, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Foppa, L.; Dupont, J. Benzene partial hydrogenation: Advances and perspectives. Chem. Soc. Rev. 2015, 44, 1886–1897. [Google Scholar] [CrossRef]
- Aitani, A.; Wang, J.B.; Wang, I.; Al-Khattaf, S.; Tsai, T.-C. Environmental Benign Catalysis for Linear Alkylbenzene Synthesis: A Review. Catal. Surv. Asia 2014, 18, 1–12. [Google Scholar] [CrossRef]
- Mancuso, A.; Sacco, O.; Sannino, D.; Venditto, V.; Vaiano, V. One-Step Catalytic or Photocatalytic Oxidation of Benzene to Phenol: Possible Alternative Routes for Phenol Synthesis? Catalysts 2020, 10, 1424. [Google Scholar] [CrossRef]
- Rajeev, A.; Balamurugan, M.; Sankaralingam, M. Rational Design of First-Row Transition Metal Complexes as the Catalysts for Oxidation of Arenes: A Homogeneous Approach. ACS Catal. 2022, 12, 9953–9982. [Google Scholar] [CrossRef]
- Wang, H.; Qin, M.; Wu, Q.; Cheng, D.-G.; Meng, X.; Wang, L.; Xiao, F.-S. Zeolite Catalysts for Green Production of Caprolactam. Ind. Eng. Chem. Res. 2023, 62, 2217–2224. [Google Scholar] [CrossRef]
- Hasegawa, S.; Harano, K.; Motokura, K. RhRu Bimetallic Oxide Cluster Catalysts for Cross-Dehydrogenative Coupling of Arenes and Carboxylic Acids. J. Am. Chem. Soc. 2024, 146, 19059–19069. [Google Scholar] [CrossRef]
- Yao, H.; Wang, Y.-M.; Quan, M.; Farooq, M.U.; Yang, L.-P.; Jiang, W. Adsorptive Separation of Benzene, Cyclohexene, and Cyclohexane by Amorphous Nonporous Amide Naphthotube Solids. Angew. Chem. Int. Ed. 2020, 59, 19945–19950. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, H.; Ye, Y.; Yuan, Z.; Wang, J.; Yang, Y.; Lin, S.; Xiang, F.; Xiang, S.; Zhang, Z. Microporous polycarbazole frameworks with large conjugated π systems for cyclohexane separation from cyclohexane-containing mixtures. New J. Chem. 2021, 45, 22437–22443. [Google Scholar] [CrossRef]
- Cui, P.-F.; Liu, X.-R.; Lin, Y.-J.; Li, Z.-H.; Jin, G.-X. Highly Selective Separation of Benzene and Cyclohexane in a Spatially Confined Carborane Metallacage. J. Am. Chem. Soc. 2022, 144, 6558–6565. [Google Scholar] [CrossRef]
- Hu, L.; Wang, W.; Miao, X.; Hu, M.; Luo, D.; Wu, W.; Lin, D.; Yang, K. A novel Al-based MOF with straight channel and pocket pore structure for trace benzene adsorption and benzene/cyclohexane separation. Chem. Eng. J. 2024, 499, 156376. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, W.; Yang, W.; Xi, F.; He, H.; Liang, M.; Dong, Q.; Hou, J.; Wang, M.; Yu, G.; et al. Separation of benzene and toluene associated with vapochromic behaviors by hybrid[4]arene-based co-crystals. Nat. Commun. 2024, 15, 1260. [Google Scholar] [CrossRef]
- Liang, Z.-J.; Dong, F.-D.; Ye, L.; Zheng, K.; Hu, D.-Y.; Feng, X.; Su, W.-Y.; Wang, Z.-S.; Zhou, M.-Y.; Fang, Z.-L.; et al. Introducing halogen-bonded gates into zeolitic frameworks for efficient benzene/cyclohexene/cyclohexane separation. Chem. Sci. 2025, 16, 3307–3312. [Google Scholar] [CrossRef]
- Shim, W.-G.; Hwang, K.-J.; Chung, J.-T.; Baek, Y.-S.; Yoo, S.-J.; Kim, S.-C.; Moon, H.; Lee, J.-W. Adsorption and thermodesorption characteristics of benzene in nanoporous metal organic framework MOF-5. Adv. Powder Technol. 2012, 23, 615–619. [Google Scholar] [CrossRef]
- Szczęśniak, B.; Choma, J.; Jaroniec, M. Ultrahigh benzene adsorption capacity of graphene-MOF composite fabricated via MOF crystallization in 3D mesoporous graphene. Microporous Mesoporous Mater. 2019, 279, 387–394. [Google Scholar] [CrossRef]
- Liu, A.; Peng, X.; Jin, Q.; Jain, S.K.; Vicent-Luna, J.M.; Calero, S.; Zhao, D. Adsorption and Diffusion of Benzene in Mg-MOF-74 with Open Metal Sites. ACS Appl. Mater. Interfaces 2019, 11, 4686–4700. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yin, H.; Tian, P.; Sun, X.; Pan, X.; Chen, K.; Chen, W.-J.; Wu, Q.-H.; Luo, S. Remarkable adsorption performance of MOF-199 derived porous carbons for benzene vapor. Environ. Res. 2020, 184, 109323. [Google Scholar] [CrossRef]
- Dedecker, K.; Drobek, M.; Rouessac, V.; Julbe, A. A Palladium-Based MOF for the Preferential Sorption of Benzene. ACS Appl. Mater. Interfaces 2023, 15, 6831–6838. [Google Scholar] [CrossRef]
- Weber, E.; Seichter, W.; Goldberg, I. Allenes as clathrate hosts: Formation of a new channel network. J. Chem. Soc. Chem. Commun. 1987, 1426–1428. [Google Scholar] [CrossRef]
- Lavelle, L.; Nassimbeni, L.R. Studies on werner clathrates. Part 131. Selective criteria in werner clathrates. Six crystal structures with bis(isothiocyanato)tetra(4-vinylpyridine)nickel(II) as host. J. Incl. Phenom. Macrocycl. Chem. 1993, 16, 25–54. [Google Scholar] [CrossRef]
- Wang, X.; Eckert, J.; Liu, L.; Jacobson, A.J. Breathing and Twisting: An Investigation of Framework Deformation and Guest Packing in Single Crystals of a Microporous Vanadium Benzenedicarboxylate. Inorg. Chem. 2011, 50, 2028–2036. [Google Scholar] [CrossRef]
- Komiya, N.; Ikeshita, M.; Tosaki, K.; Sato, A.; Itami, N.; Naota, T. Catalytic Enantioselective Rotation of Watermill-Shaped Dinuclear Pd Complexes. Eur. J. Inorg. Chem. 2021, 1929–1940. [Google Scholar] [CrossRef]
- Jie, K.; Zhou, Y.; Li, E.; Huang, F. Nonporous Adaptive Crystals of Pillararenes. Acc. Chem. Res. 2018, 51, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Wang, Y.; Chena, J.; Zhou, J. Potential of nonporous adaptive crystals for hydrocarbon separation. Chem. Soc. Rev. 2023, 52, 6075–6119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, G.; Li, Q.; Wang, M.; Huang, F. Separation of Benzene and Cyclohexane by Nonporous Adaptive Crystals of a Hybrid[3]arene. J. Am. Chem. Soc. 2020, 142, 2228–2232. [Google Scholar] [CrossRef]
- Williams, J.H. Crystal Engineering: How Molecules Build Solids; Morgan & Claypool Publishers: Saint-Malo, France, 2017. [Google Scholar]
- Wang, W.; Wu, W.X.; Zhang, Y.; Jin, W.J. Perfluoroarylaryl interaction: The most important subset of π-hole⋯π bonding. Chem. Phys. Rev. 2024, 5, 031303. [Google Scholar] [CrossRef]
- Frontera, A. σ- and π-Hole Interactions. Crystals 2020, 10, 721. [Google Scholar] [CrossRef]
- Pang, X.; Wang, H.; Wang, W.; Jin, W.J. Phosphorescent π-Hole···π Bonding Cocrystals of Pyrene with Haloperfluorobenzenes (F, Cl, Br, I). Cryst. Growth Des. 2015, 15, 4938–4945. [Google Scholar] [CrossRef]
- Ikumura, Y.; Kawasaki, T.; Ishida, Y.; Usui, H.; Uchida, S.; Kamata, K.; Nomura, M.; Hori, A. Boosting CO2 and benzene adsorption through π-hole substitution in β-diketonate Cu(II) complex within non-porous adaptive crystals. RSC Adv. 2025, 15, 6184–6190. [Google Scholar] [CrossRef]
- Ikumura, Y.; Habuka, Y.; Sakai, S.; Shinohara, T.; Yuge, H.; Rzeznicka, I.I.; Hori, A. Enhanced and heteromolecular guest encapsulation in nonporous crystals of a perfluorinated triketonato dinuclear copper complex. Chem. Eur. J. 2020, 26, 5051–5060. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ikumura, Y.; Lee, C.-H.; Hori, A. Co-Crystallization and Structural Studies of Benzophenone Recognized by Positively Shifted ESPs of Perfluorinated β-Diketonate Complexes (M = Cu, Pd, Pt). Crystals 2024, 14, 593. [Google Scholar] [CrossRef]
- Hernández-Trujillo, J.; Vela, A. Molecular Quadrupole Moments for the Series of Fluoro- and Chlorobenzenes. J. Phys. Chem. 1996, 100, 6524–6530. [Google Scholar] [CrossRef]
- Doerksen, R.J.; Thakkar, A.J. Quadrupole and octopole moments of heteroaromatic rings. J. Phys. Chem. A 1999, 103, 10009–10014. [Google Scholar] [CrossRef]
- Shimizu, K.; Gomes, M.F.C.; Pádua, A.A.H.; Rebelo, L.P.N.; Lopes, J.N.C. On the Role of the Dipole and Quadrupole Moments of Aromatic Compounds in the Solvation by Ionic Liquids. J. Phys. Chem. B 2009, 113, 9894–9900. [Google Scholar] [CrossRef] [PubMed]
- Kusakawa, T.; Sakai, S.; Nakajima, K.; Yuge, H.; Rzeznicka, I.I.; Hori, A. Synthesis, structures and co-crystallizations of perfluorophenyl substituted β-diketone and triketone compounds. Crystals 2019, 9, 175. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and qu antitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1•(2)3 [31] | 1 | 1•(O)2•3 | |
|---|---|---|---|
| Description | prismatic | prismatic | block |
| Chemical formula | C52H22Cu2F20O6 | C34H4Cu2F20O6 | C40H14Cu2F20O8 |
| Formula weight | 1249.77 | 1015.45 | 1129.59 |
| Crystal system | orthorhombic | monoclinic | monoclinic |
| Space group | Fdd2 | C2/c | P21/n |
| a [Å] | 28.776(4) | 26.567(8) | 14.3647(9) |
| b [Å] | 52.939(7) | 3.8163(12) | 10.1169(8) |
| c [Å] | 6.3912(9) | 32.269(9) | 27.646(2) |
| β [°] | 90 | 101.815(9) | 99.765(2) |
| V [Å3] | 9736(2) | 3202.3(17) | 3959.5(5) |
| Z | 8 | 4 | 4 |
| Dc [Mg m−3] | 1.705 | 2.106 | 1.895 |
| μ [mm−1] | 1.002 | 1.496 | 1.224 |
| F(000) | 4960 | 1976 | 2224 |
| Rint | 0.0299 | 0.0715 | 0.0556 |
| GOF | 1.067 | 1.050 | 1.031 |
| R [(I) > 2σ (I)] | 0.0428 | 0.0410 | 0.0493 |
| wR (Fo2) | 0.1202 | 0.0949 | 0.1484 |
| CCDC No. | 1900742 | 2430009 | 2430010 |
| Guest | 3 | 4 | 5 | 6 |
|---|---|---|---|---|
| Appearance |  |  |  |  |
| Description | block | needle | needle | needle |
| Color | deep green | green | green | green |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| a [Å] | 14.243(6) | 26.56(2) | 26.60(1) | 26.60(4) |
| b [Å] | 10.191(4) | 3.819(3) | 3.816(1) | 3.823(6) |
| c [Å] | 27.736(11) | 32.22(2) | 32.27(1) | 32.30(4) |
| β [°] | 100.103(12) | 101.83(2) | 101.75(2) | 101.87(3) |
| V [Å3] | 3963(5) | 3198(6) | 3207(4) | 3214(13) |
| 2 [=1•(2)3] | 2 + 3 | 2 + 3 + 4 | 2 + 5 + 6 | |
|---|---|---|---|---|
| Appearance |  |  |  |  |
| Description | prismatic | prismatic | prismatic | prismatic |
| Color | green | green | green | green |
| a [Å] | 28.776(4) | 28.724(4) | 28.787(12) | 28.728(3) |
| b [Å] | 52.939(7) | 53.068(8) | 53.20(2) | 53.149(6) |
| c [Å] | 6.3912(9) | 6.4344(10) | 6.451(3) | 6.4356(7) |
| β [°] | 90 | 90 | 90 | 90 |
| V [Å3] | 9736(2) | 9808(3) | 9879(7) | 9826(2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiomoto, K.; Oimatsu, N.; Hirano, S.; Hori, A. Selective Benzene Recognition in Competitive Solvent System (Cyclohexene, Cyclohexane, Tri- and Hexafluorobenzenes) Using Perfluorinated Dinuclear Cu(II) Complex. Crystals 2025, 15, 322. https://doi.org/10.3390/cryst15040322
Shiomoto K, Oimatsu N, Hirano S, Hori A. Selective Benzene Recognition in Competitive Solvent System (Cyclohexene, Cyclohexane, Tri- and Hexafluorobenzenes) Using Perfluorinated Dinuclear Cu(II) Complex. Crystals. 2025; 15(4):322. https://doi.org/10.3390/cryst15040322
Chicago/Turabian StyleShiomoto, Kazuki, Nanako Oimatsu, Satoshi Hirano, and Akiko Hori. 2025. "Selective Benzene Recognition in Competitive Solvent System (Cyclohexene, Cyclohexane, Tri- and Hexafluorobenzenes) Using Perfluorinated Dinuclear Cu(II) Complex" Crystals 15, no. 4: 322. https://doi.org/10.3390/cryst15040322
APA StyleShiomoto, K., Oimatsu, N., Hirano, S., & Hori, A. (2025). Selective Benzene Recognition in Competitive Solvent System (Cyclohexene, Cyclohexane, Tri- and Hexafluorobenzenes) Using Perfluorinated Dinuclear Cu(II) Complex. Crystals, 15(4), 322. https://doi.org/10.3390/cryst15040322