
Effect of Molecular Perturbation on Polymorphism: The Case of 8-Halotheophyllines (8-Cl-Tph and 8-Br-Tph)

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
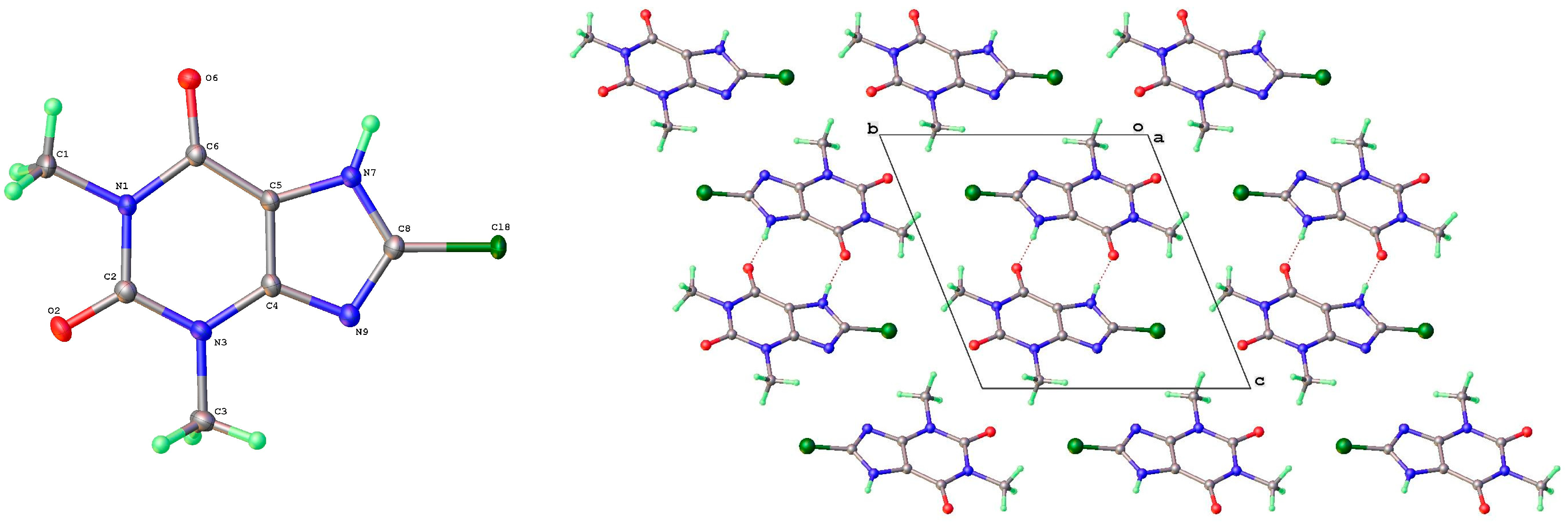
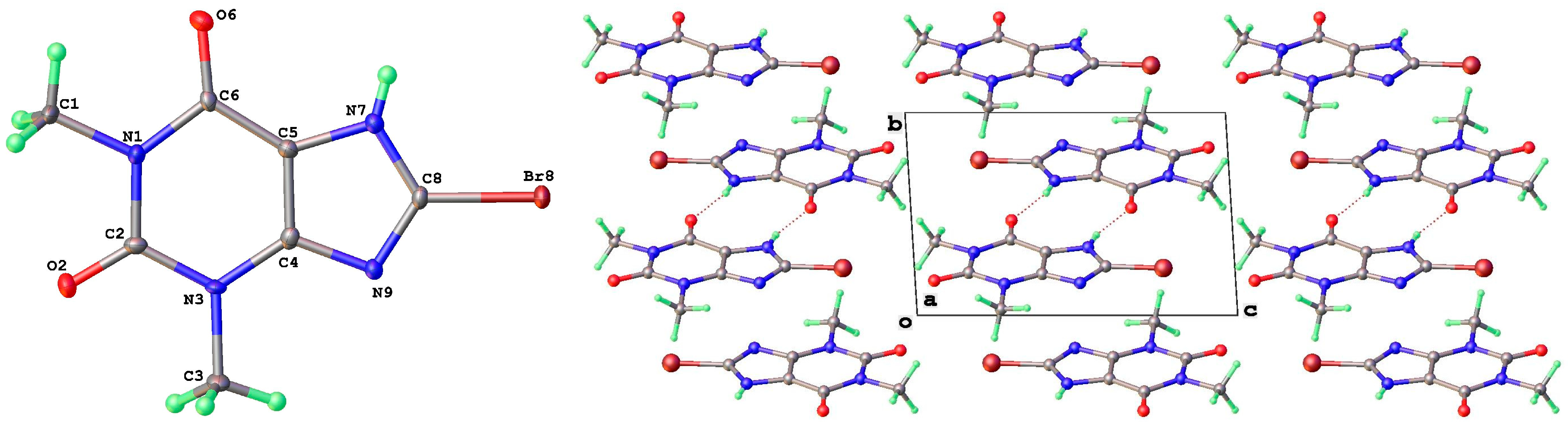
2.1. X-Ray Crystallography
2.2. Computational Methods
3. Results and Discussion
3.1. Polymorphic Forms of Theophylline
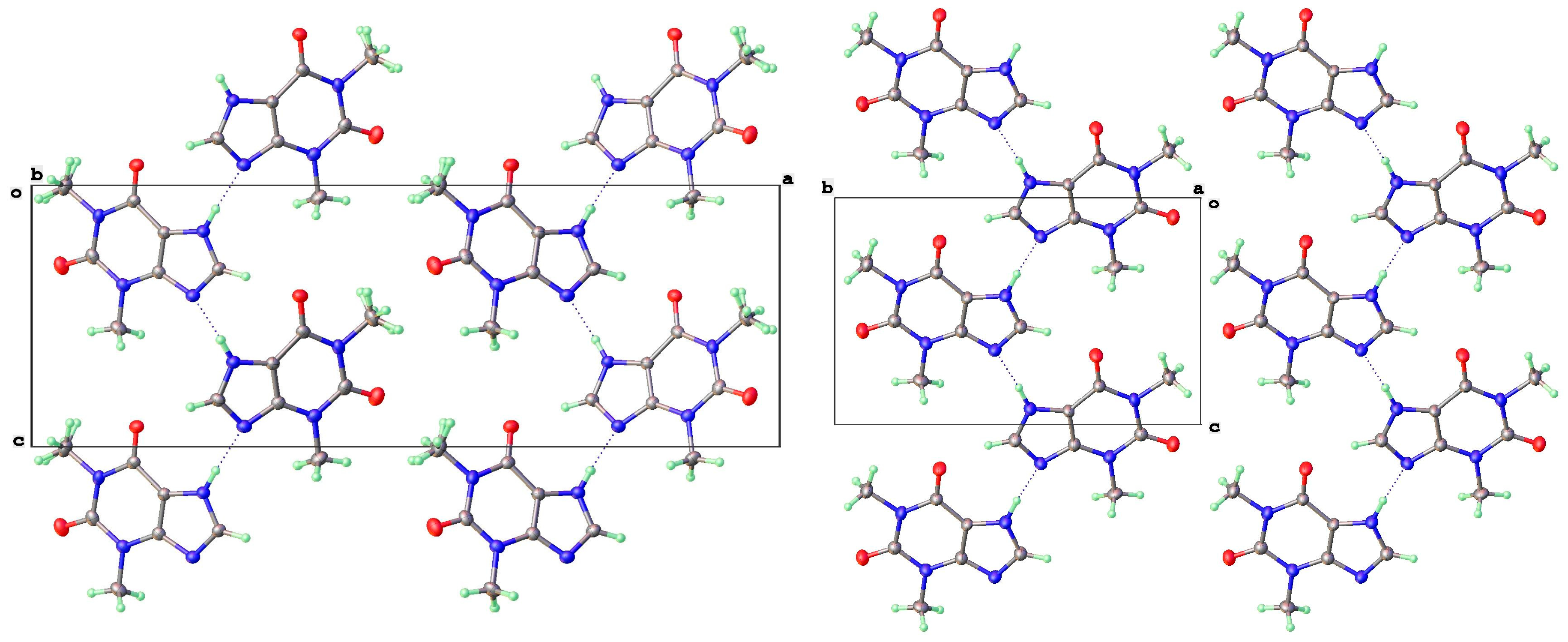
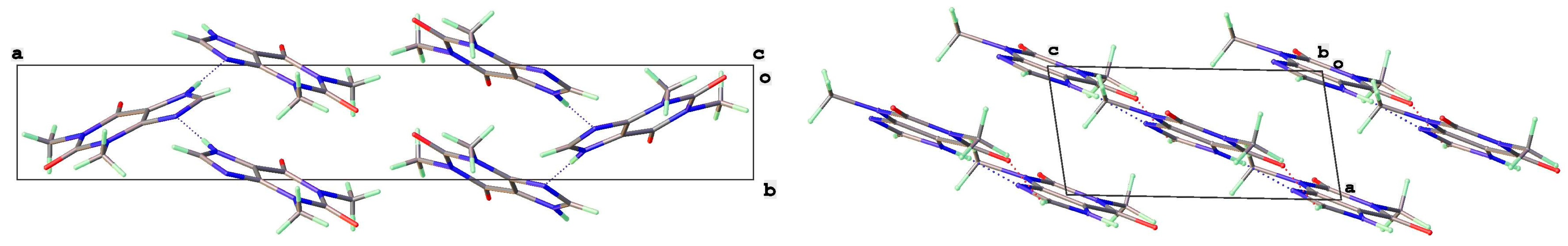
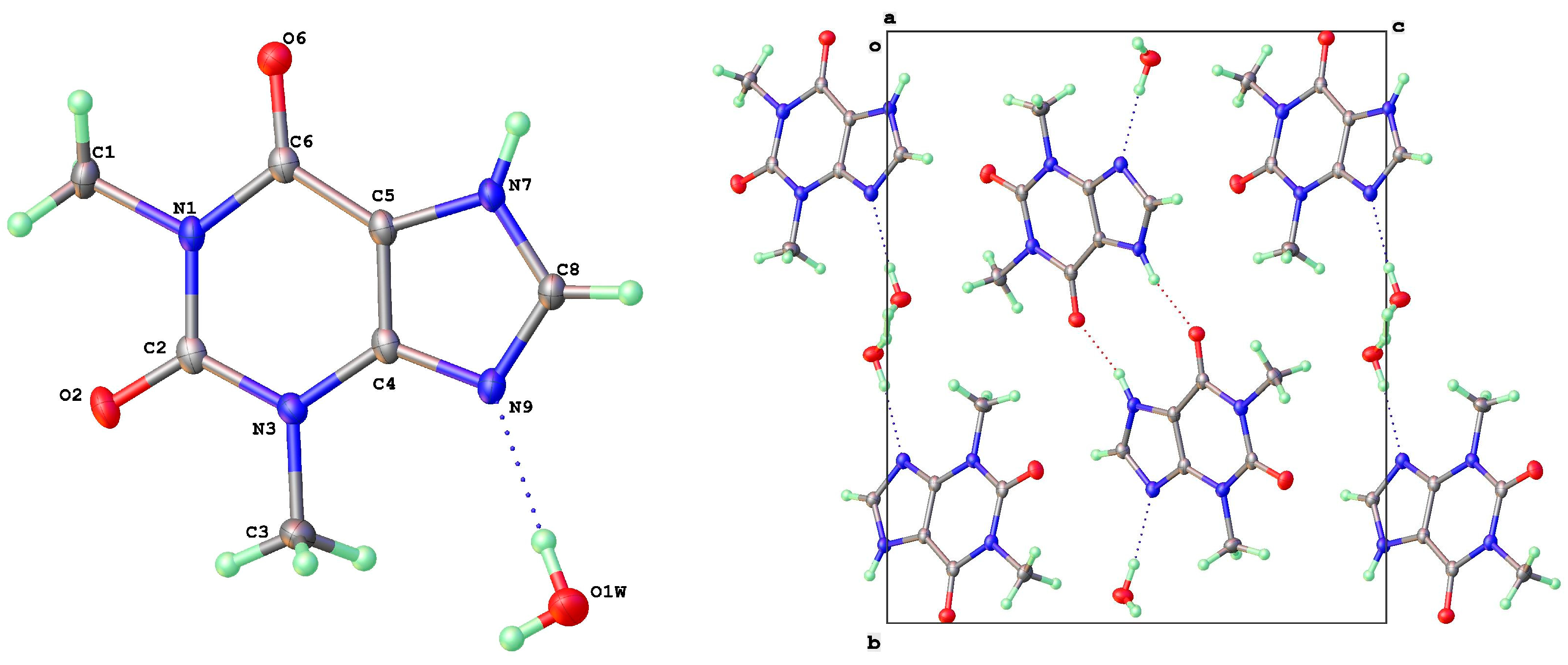
3.2. Polymorphic Forms of 8-Halotheophylline
3.3. Computational Studies of 8-X-Theophyllines (X = H, Cl, Br)
3.4. Dimer Structures of 8-X-Theophyllines (X = H, Cl, Br)
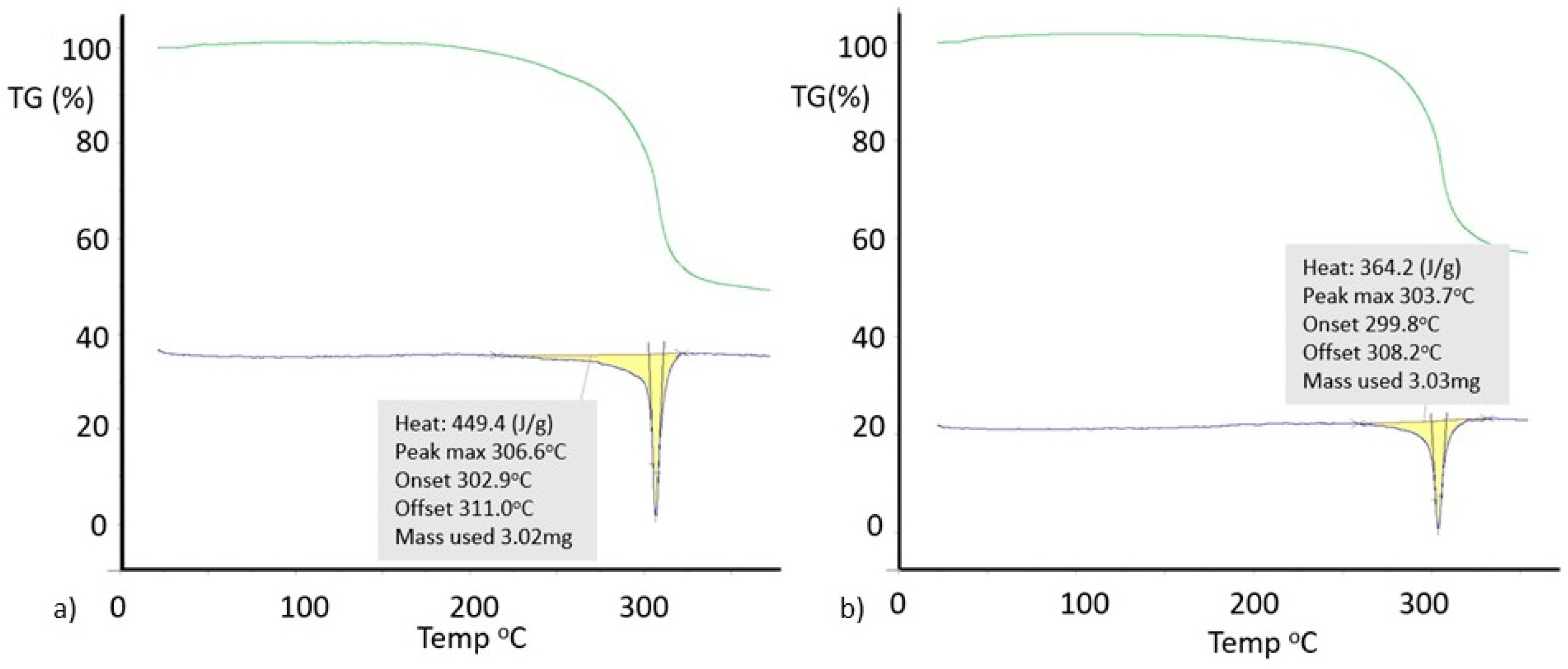
3.5. Powder X-Ray Diffraction of 8-X-Tph Polymorphs
3.6. Further Discussion of 8-X-Tph Polymorphs and Future Prospects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| DSC | differential scanning calorimetry |
| Tph | theophylline |
| LAG | liquid-assisted grinding |
| TGA | thermal gravimetric analysis |
| SS | solid solution |
References
- Carlucci, L.; Gavezzotti, A. Molecular Recognition and Crystal Energy Landscapes: An X-ray and Computational Study of Caffeine and Other Methylxanthines. Chem. Eur. J. 2004, 11, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Physical stability enhancement of theophylline via cocrystallization. Int. J. Pharm. 2006, 320, 114–123. [Google Scholar] [CrossRef]
- Sanphui, P.; Nangia, A. Salts and Co-crystals of Theobromine and their phase transformations in water. J. Chem. Sci. 2014, 126, 1249–1264. [Google Scholar] [CrossRef]
- Enright, G.D.; Terskikh, V.V.; Brouwer, D.H.; Ripmeester, J.A. The Structure of Two Anhydrous Polymorphs of Caffeine from Single-Crystal Diffraction and Ultrahigh-Field Solid-State 13C NMR Spectroscopy. Cryst. Growth Des. 2007, 7, 1406–1410. [Google Scholar] [CrossRef]
- Lehmann, C.W.; Stowasser, F. The crystal structure of anhydrous beta-caffeine as determined from X-ray powder-diffraction data. Chemistry 2007, 13, 2908–2911. [Google Scholar] [CrossRef] [PubMed]
- Ebisuzaki, Y.; Boyle, P.D.; Smith, J.A. Methylxanthines. I. Anhydrous Theophylline. Acta Crystallogr. Sect. C 1997, 53, 777–779. [Google Scholar] [CrossRef]
- Zhang, S.; Fischer, A. A monoclinic polymorph of theophylline. Acta Crystallogr. Sect. E 2011, 67, o3357. [Google Scholar] [CrossRef]
- Khamar, D.; Pritchard, R.G.; Bradshaw, I.J.; Hutcheon, G.A.; Seton, L. Polymorphs of anhydrous theophylline: Stable form IV consists of dimer pairs and metastable form I consists of hydrogen-bonded chains. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2011, 67, o496–o499. [Google Scholar] [CrossRef]
- Fucke, K.; McIntyre, G.J.; Wilkinson, C.; Henry, M.; Howard, J.A.K.; Steed, J.W. New insights into an Old Molecule: Interaction Energies of Theophylline Crystal Forms. Cryst. Growth Des. 2012, 12, 1395–1401. [Google Scholar] [CrossRef]
- Larsen, A.S.; Olsen, M.A.; Moustafa, H.; Larsen, F.H.; Sauer, S.P.A.; Rantanen, J.; Madsen, A.Ø. Determining short-lived solid forms during phase transformations using molecular dynamics. CrystEngComm 2019, 21, 4020–4024. [Google Scholar] [CrossRef]
- Seton, L.; Khamar, D.; Bradshaw, I.J.; Hutcheon, G.A. Solid State Forms of Theophylline: Presenting a New Anhydrous Polymorph. Cryst. Growth Des. 2010, 10, 3879–3886. [Google Scholar] [CrossRef]
- Sun, C.; Zhou, D.; Grant, D.J.W.; Young, V.G. Theophylline Monohydrate. Acta Crystallogr. Sect. E 2002, 58, o368–o370. [Google Scholar] [CrossRef]
- Dyulgerov, V.M.; Dimowa, L.T.; Kossev, K.; Nikolova, R.P.; Shivachev, B.L. Solvothermal synthesis of theophylline an N,N ′-(ethane-1,2-diyl)diformamide co-crystals from DMF decomposition and N-formylation trough catalytic effect of 3-carboxyphenylboronic acid and cadmium acetate. Bulgarian Chem. Commun. 2015, 47, 311–316. [Google Scholar]
- Putra, O.D.; Yoshida, T.; Umeda, D.; Higashi, K.; Uekusa, H.; Yonemochi, E. Crystal Structure Determination of Dimenhydrinate after More than 60 Years: Solving Salt–Cocrystal Ambiguity via Solid-State Characterizations and Solubility Study. Cryst. Growth Des. 2016, 16, 5223–5229. [Google Scholar] [CrossRef]
- Srirambhatla, V.K.; Guo, R.; Price, S.L.; Florence, A.J. Isomorphous template induced crystallisation: A robust method for the targeted crystallisation of computationally predicted metastable polymorphs. Chem. Commun. 2016, 52, 7384–7386. [Google Scholar] [CrossRef]
- Case, D.H.; Srirambhatla, V.K.; Guo, R.; Watson, R.E.; Price, L.S.; Polyzois, H.; Cockcroft, J.K.; Florence, A.J.; Tocher, D.A.; Price, S.L. Successful Computationally Directed Templating of Metastable Pharmaceutical Polymorphs. Cryst. Growth Des. 2018, 18, 5322–5331. [Google Scholar] [CrossRef]
- Trask, A.V.; Jones, W. Crystal Engineering of Organic Cocrystals by the Solid-State Grinding Approach. Top. Curr. Chem. 2005, 254, 41–70. [Google Scholar] [CrossRef]
- Trask, A.V.; van de Streek, J.; Motherwell, W.D.S.; Jones, W. Achieving Polymorphic and Stoichiometric Diversity in Cocrystal Formation: Importance of Solid-State Grinding, Powder X-ray Structure Determination, and Seeding. Cryst. Growth Des. 2005, 5, 2233–2241. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected. Acta Crystallogr. Sect. A 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comp. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comp. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Etter, M.C.; Macdonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen bond patterns. Acta Crystallogr. Sect. B 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Singh, V.B. Spectroscopic signatures and structural motifs in isolated and hydrated theophylline: A computational study. RSC Adv. 2015, 5, 11433–11444. [Google Scholar] [CrossRef]
- Krivovichev, S.V. Structural complexity and configurational entropy of crystals. Acta Crystallogr. Sect. B 2016, 72, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Jencks, W.P.; Westheimer, F.H. Compilation of pKa Acidity Constants (ACS Organic Division). 2025. Available online: https://www.webqc.org/pkaconstants.php (accessed on 31 January 2025).
- Nolasco, M.M.; Amado, A.M.; Ribeiro-Claro, P.J.A. Computationally-Assisted Approach to the Vibrational Spectra of Molecular Crystals: Study of Hydrogen-Bonding and Pseudo-Polymorphism. ChemPhysChem 2006, 7, 2150–2161. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.A.; Du, J.J.; Lai, F.; Stanton, S.A.; Williams, P.A.; Groundwater, P.W.; Platts, J.A.; Overgaard, J.; Hibbs, D.E. An experimental and theoretical charge density study of theophylline and malonic acid cocrystallization. RSC Adv. 2022, 12, 15670–15684. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Comparison of Computational Methods for Atomic Charges. Acta Phys. Chim. Sin. 2012, 28, 1–18. [Google Scholar] [CrossRef]
- Li, K.; Roy, M.; Nisar, M.; Wong, L.W.-Y.; Sung, H.H.-Y.; Haynes, R.K.; Williams, I.D. Control of 11-Aza:4-X-SalA Cocrystal Polymorphs Using Heteroseeds That Switch On/Off Halogen Bonding. Crystals 2022, 12, 1368. [Google Scholar] [CrossRef]
- Xia, Y.; Wei, Y.; Chen, H.; Qian, S.; Zhang, J.; Gao, Y. Competitive cocrystallization and its application in the separation of flavonoids. IUCrJ 2021, 8, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Ye, W. Isolation, Purification and Structural Studies of Natural Product Crystals and Cocrystals. Ph.D. Thesis, Hong Kong University of Science and Technology, Hong Kong, December 2024. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form | T(K) | Sp Gp | a (Å) | b (Å) | c (Å) | β (°) | V (Å3) | Vmol (Å3) | CSD Coden | Ref | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| RT | |||||||||||
| II | 297 | Pna21 | 24.612 | 3.83 | 8.501 | 90 | 801.38 | 200.3 | BAPLOT01 | [6] | |
| III | 298 | P21/c | 4.531 | 11.578 | 15.719 | 93.69 | 822.92 | 205.7 | BAPLOT08 | [10] | Metastable |
| IV | 299 | P21/c | 7.894 | 12.909 | 15.905 | 104.21 | 1571.1 | 196.4 | BAPLOT02 | [7] | |
| V | 290 | Pn | 3.874 | 12.89 | 8.117 | 98.97 | 400.4 | 200.2 | BAPLOT09 | [13] | Solvothermal |
| LT | |||||||||||
| I | 120 | Pna21 | 13.087 | 15.579 | 3.8629 | 90 | 787.6 | 196.9 | BAPLOT05 | [9] | |
| I | 100 | Pna21 | 13.158 | 15.630 | 3.854 | 90 | 792.6 | 198.2 | BAPLOT04 | [8] | Stable HT |
| II | 120 | Pna21 | 24.330 | 3.7707 | 8.4850 | 90 | 778.43 | 194.6 | BAPLOT06 | [9] | |
| II | 100 | Pna21 | 24.3948 | 3.7816 | 8.4779 | 90 | 782.1 | 195.5 | 2,420,759 | This work | Hot cooled |
| IV | 100 | P21/c | 7.705 | 13.001 | 15.7794 | 103.22 | 1538.85 | 192.4 | BAPLOT03 | [8] | Stable RT |
| V | 100 | Pn | 3.8121 | 12.8944 | 8.0643 | 99.56 | 390.9 | 195.5 | 2,420,760 | This work | |
| H2O | 173 | P21/n | 4.468 | 15.355 | 13.121 | 97.79 | 891.9 | 223.0 | THEOPH01 | [12] | |
| H2O | 120 | P21/n | 4.4605 | 15.3207 | 13.0529 | 97.51 | 884.36 | 221.1 | THEOPH02 | [9] | Neutron diff. |
| H2O | 100 | P21/n | 4.453 | 15.3127 | 13.0396 | 97.42 | 881.7 | 220.4 | 2,420,761 | This work | RT growth |
| Form | Sp Gp | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | V (Å3) | Vmol (Å3) | CCDC # | R1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 8-Cl | |||||||||||
| I | P21/c | 3.90604(9) | 16.8562(3) | 12.7664(3) | 90 | 92.031(2) | 90 | 840.02(3) | 210.0 | 2420762 | 3.33 |
| II | P-1 | 8.9581(4) | 8.9956(3) | 11.3519(5) | 80.95(1) | 84.40(1) | 68.80(1) | 841.42(6) | 210.4 | 2420763 | 3.14 |
| III | P-1 | 4.4920(6) | 10.0818(10) | 10.1846(14) | 110.30(1) | 96.88(1) | 99.93(1) | 418.0(1) | 209.0 | 2420764 | 3.32 |
| 8-Br | |||||||||||
| I | P21/c | 3.9463(1) | 17.0024(3) | 12.8384(2) | 90 | 91.933(2) | 90 | 860.92(3) | 215.2 | 2420765 | 2.23 |
| IV | P-1 | 6.5878(6) | 6.8562(4) | 10.3366(10) | 92.754(6) | 90.961(7) | 108.06(1) | 443.11(6) | 221.6 | 2420766 | 3.42 |
| 8-Cl/Br | 72:28 * | ||||||||||
| I | P21/c | 3.9204(2) | 16.9274(10) | 12.8080(7) | 90 | 91.97(1) | 90 | 849.46(8) | 212.4 * | 2420767 | 2.16 |
| 8-Cl-Tph | Form I | Form II | Form III |
|---|---|---|---|
| CCDC Deposition | 2420762 | 2420763 | 2420764 |
| Empirical formula | C7H7ClN4O2 | C7H7ClN4O2 | C7H7ClN4O2 |
| Formula weight | 214.62 | 214.62 | 214.62 |
| Temperature/K | 99.99(10) | 100.15 | 100.01(10) |
| Crystal system | Monoclinic | triclinic | triclinic |
| Space group | P21/c | P-1 | P-1 |
| a/Å | 3.90604(9) | 8.9581(4) | 4.4920(6) |
| b/Å | 16.8562(3) | 8.9956(3) | 10.0818(10) |
| c/Å | 12.7664(3) | 11.3519(5) | 10.1846(14) |
| α/° | 90 | 80.95(1) | 110.304(10) |
| β/° | 92.031(2) | 84.40(1) | 96.877(11) |
| γ/° | 90 | 68.80(1) | 99.931(10) |
| Volume/Å3 | 840.02(3) | 841.42(6) | 417.97(10) |
| Z, Z’ | 4, 1 | 4, 2 | 2, 1 |
| ρcalcg/cm3 μ/mm-1 | 1.697, 3.892 | 1.694, 3.886 | 1.705, 3.911 |
| F(000) | 440.0 | 440.0 | 220.0 |
| Crystal size/mm3 | 0.15 × 0.10 × 0.08 | 0.15 × 0.08 × 0.08 | 0.06 × 0.04 × 0.04 |
| Radiation | CuKα (λ = 1.54184) | CuKα (λ = 1.54184) | CuKα (λ = 1.54184) |
| 2Θ range/° | 8.7 to 154.0 | 8.0 to 149.0 | 9.4 to 149.5 |
| Index ranges | −3 ≤ h ≤ 4, −21 ≤ k ≤ 20, −11 ≤ l ≤ 16 | −10 ≤ h ≤ 11, −11 ≤ k ≤ 11 −14 ≤ l ≤ 14 | −5 ≤ h ≤ 5, −6 ≤ k ≤ 12 −12 ≤ l ≤ 9 |
| Reflections collected | 5186 | 12856 | 2307 |
| Independent reflections Rint, Rsigma | 1732 0.0247, 0.0247 | 3347 0.0351, 0.0270 | 1615 0.0187, 0.0273 |
| Data/restraints/parameters | 1732/0/133 | 3347/0/265 | 1615/0/134 |
| Goodness-of-fit on F2 | 1.036 | 1.041 | 1.040 |
| Final R1, wR2 [I ≥ 2σ (I)] | 0.0333, 0.0838 | 0.0314, 0.0801 | 0.0329, 0.0881 |
| Final R indexes [all data] | 0.0395, 0.0882 | 0.0367, 0.0840 | 0.0347, 0.0904 |
| Res. peak/hole/e Å−3 | 0.33/−0.27 | 0.31/−0.33 | 0.43/−0.56 |
| D | H | A | d(D-H)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° | |
|---|---|---|---|---|---|---|---|
| 8-Cl-Tph Form I | N7 | H7 | O2 1 | 0.87(3) | 1.86(3) | 2.7346(19) | 178(3) |
| 8-Cl-Tph Form II | N7 | H7 | O2A | 0.92(2) | 1.81(2) | 2.7185(18) | 174(2) |
| N7A | H7A | O2 1 | 0.91(3) | 1.81(3) | 2.7203(18) | 177(2) | |
| 8-Cl-Tph Form III | N7 | H7 | O6 1 | 0.84(2) | 1.89(2) | 2.7297(18) | 177(2) |
| 8-Br-Tph Form I | N7 | H7 | O2 1 | 0.84(3) | 1.93(3) | 2.760(2) | 174(3) |
| 8-Br-Tph Form IV | N7 | H7 | O2 1 | 0.77(5) | 1.97(5) | 2.723(4) | 166(5) |
| 8-Br-Tph | Form I | Form IV | Form I (8-Cl/8-Br-Tph SS) |
|---|---|---|---|
| CCDC Deposition | 2420765 | 2420766 | 2420767 |
| Empirical formula | C7H7BrN4O2 | C7H7BrN4O2 | C7H7Br0.28Cl0.72N4O2 |
| Formula weight | 259.08 | 259.08 | 227.06 |
| Temperature/K | 100.01(10) | 99.96(12) | 100.01(10) |
| Crystal system | Monoclinic | triclinic | monoclinic |
| Space group | P21/c | P-1 | P21/c |
| a/Å | 3.94630(10) | 6.5461(3) | 3.9204(2) |
| b/Å | 17.0024(3) | 6.8780(5) | 16.9274(10) |
| c/Å | 12.8384(2) | 10.3419(5) | 12.8080(7) |
| α/° | 90 | 93.114(5) | 90 |
| β/° | 91.933(2) | 90.726(4) | 91.972(2) |
| γ/° | 90 | 107.664(5) | 90 |
| Volume/Å3 | 860.92(3) | 442.82(5) | 849.46(8) |
| Z, Z’ | 4, 1 | 2, 1 | 2, 1 |
| ρcalcg/cm3 μ/mm−1 | 1.999, 6.381 | 1.943, 4.62 | 1.775, 3.024 |
| F(000) | 512.0 | 256.0 | 460.0 |
| Crystal size/mm3 | 0.10 × 0.10 × 0.10 | 0.12 × 0.10 × 0.10 | 0.11 × 0.07 × 0.06 |
| Radiation | CuKα (λ = 1.54184) | MoKα (λ = 0.71073) | GaKα (λ = 1.34139) |
| 2Θ range /° | 8.6 to 148.9 | 4.0 to 57.5 | 7.5 to 115.0 |
| Index ranges | −4 ≤ h ≤ 4, −20 ≤ k ≤ 19, −15 ≤ l ≤ 11 | −6 ≤ h ≤ 8, −9 ≤ k ≤ 5, −13 ≤ l ≤ 12 | −4 ≤ h ≤ 4, −21 ≤ k ≤ 21, −15 ≤ l ≤ 16 |
| Reflections collected | 4627 | 2629 | 9048 |
| Independent reflections Rint, Rsigma | 1699 0.0311, 0.0673 | 1947 0.0180, 0.0513 | 1675 0.0272, 0.0232 |
| Data/restraints/parameters | 1593/0/133 | 1947/0/133 | 1675/0/144 |
| Goodness-of-fit on F2 | 1.003 | 1.094 | 1.049 |
| Final R1, wR2 [I ≥ 2σ (I)] | 0.0223, 0.0556 | 0.0342, 0.0763 | 0.0216, 0.0577 |
| Final R indexes [all data] | 0.0230, 0.0560 | 0.0397, 0.0796 | 0.0221, 0.0580 |
| Res. peak/hole/e Å−3 | 0.52/−0.47 | 0.95/−0.65 | 0.26/−0.23 |
| X | O | d(X---O)/Å | C-X---O/° | X---O = C/° | |
|---|---|---|---|---|---|
| 8-Cl-Tph Form I | Cl8 | O6 1 | 2.982(3) | 159.6(2) | 155.2(2) |
| 8-Cl-Tph Form II | Cl8 | O6A 1 | 3.011(3) | 148.5(2) | 138.9(2) |
| Cl8A | O6 2 | 2.961(3) | 153.0(2) | 148.8(2) | |
| 8-Cl-Tph Form III | Cl8 | O2 1 | 3.203(3) | 168.3(2) | 138.9(2) |
| 8-Br-Tph Form I | Br8 | O6 1 | 2.947(2) | 160.8(2) | 156.4(2) |
| 8-Br-Tph Form IV | Br8 | O2 1 | 3.031(2) | 172.1(2) | 142.3(2) |
| NBO Charges | ESP Charges | ||||||
|---|---|---|---|---|---|---|---|
| Atom | Tph | 8-Cl-Tph | 8-Br-Tph | Atom | Tph | 8-Cl-Tph | 8-Br-Tph |
| N(1) | −0.436 | −0.434 | −0.434 | N(1) | −0.227 | −0.238 | −0.239 |
| O(2) | −0.622 | −0.615 | −0.614 | O(2) | −0.532 | −0.545 | −0.549 |
| N(3) | −0.394 | −0.391 | −0.391 | N(3) | −0.247 | −0.335 | −0.398 |
| O(6) | −0.603 | −0.600 | −0.600 | O(6) | −0.512 | −0.498 | −0.504 |
| N(7) | −0.472 | −0.483 | −0.488 | N(7) | −0.400 | −0.263 | −0.089 |
| H(7) | 0.434 | 0.443 | 0.444 | H(7) | 0.379 | 0.334 | 0.298 |
| X(8) | H: 0.206 | Cl: 0.068 | Br: 0.121 | X(8) | H: 0.116 | Cl: −0.025 | Br: 0.029 |
| N(9) | −0.482 | −0.488 | −0.494 | N(9) | −0.616 | −0.516 | −0.461 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, W.; Zhang, C.; Sung, H.H.-Y.; Wong, L.W.-Y.; Sheong, F.K.; Williams, I.D. Effect of Molecular Perturbation on Polymorphism: The Case of 8-Halotheophyllines (8-Cl-Tph and 8-Br-Tph). Crystals 2025, 15, 340. https://doi.org/10.3390/cryst15040340
Ye W, Zhang C, Sung HH-Y, Wong LW-Y, Sheong FK, Williams ID. Effect of Molecular Perturbation on Polymorphism: The Case of 8-Halotheophyllines (8-Cl-Tph and 8-Br-Tph). Crystals. 2025; 15(4):340. https://doi.org/10.3390/cryst15040340
Chicago/Turabian StyleYe, Weijian, Chao Zhang, Herman H-Y. Sung, Lawrence W-Y. Wong, Fu Kit Sheong, and Ian D. Williams. 2025. "Effect of Molecular Perturbation on Polymorphism: The Case of 8-Halotheophyllines (8-Cl-Tph and 8-Br-Tph)" Crystals 15, no. 4: 340. https://doi.org/10.3390/cryst15040340
APA StyleYe, W., Zhang, C., Sung, H. H.-Y., Wong, L. W.-Y., Sheong, F. K., & Williams, I. D. (2025). Effect of Molecular Perturbation on Polymorphism: The Case of 8-Halotheophyllines (8-Cl-Tph and 8-Br-Tph). Crystals, 15(4), 340. https://doi.org/10.3390/cryst15040340