Abstract
A series of three 1,3-phenylene bis-oxamides 3a–c, structurally related to the GPR35 receptor-agonist drug lodoxamide, has been synthesized by reacting the 1,3-phenylene bis-oxalamates 2a and 2b with amines. The obtained compounds were characterized by 1H and 13C NMR, and IR spectroscopy, they showed characteristic signals for the aromatic, N―H, and C=O groups. Molecular structure was determined using single-crystal X-ray diffraction. The supramolecular architecture is driven by N―H···O=C, N―H···N, C—H···π, and O=C···O=C interactions depicting a supramolecular helix (3a) and tapes (3b–c). Intermolecular interactions were studied using Hirshfeld surface analysis, where N―H∙∙∙X (X = N, O) hydrogen bonding represents 30.2% to the surface of 3a and 17.8–18.8% to the surface of 3b–c. The most energetic interactions involve the amide N—H∙∙∙O hydrogen bonding, contributing in the −113.9 to −97.0 kJ mol−1 range to the crystal energy, being more dispersive than electrostatic in nature. The molecular docking study was performed to evaluate the binding ability of 3a–c compounds to the GPR35 receptor, showing a favorable binding in a similar way to lodoxamide.
1. Introduction
G protein-coupled receptor 35 (GPR35) belongs to the G protein-coupled receptor superfamily. It is a receptor highly expressed in the intestine and immune cells. It has been implicated in pathologies such as asthma, inflammation, hypertension, and diabetes [1]. The role of GPR35 in the inflammatory process as pro-inflammatory or anti-inflammatory response involves the activation of immune cells, production of cytokines, and chemotactic movements [2]. Therefore, the therapeutic potential of the GPR35 receptor can be exploited by developing synthetic ligands for its study [3].
The search for synthetic compounds that act as ligands towards the GPR35 receptor has shown that different diacid compounds such as cromolyn, nedocromil, bufrolin, pamoic acid, and lodoxamide exhibit moderate-to-high potency for such receptor [4].
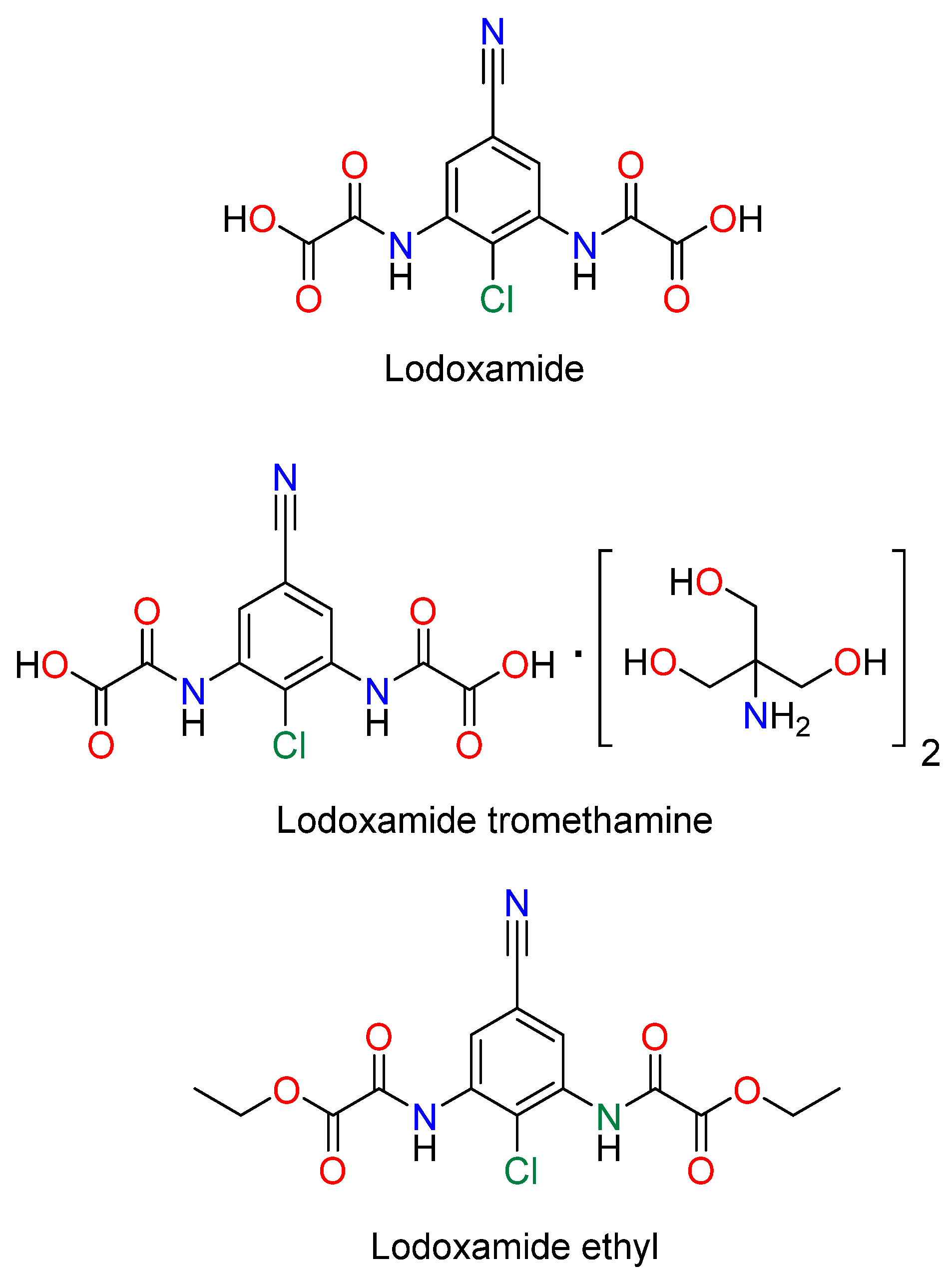
Lodoxamide, commercially available as an eye drop medication, and its derivatives, lodoxamide ethyl and lodoxamide tromethamine, Figure 1, are 1,3-phenylene bis-oxalic acid-derivative agonists of the GPR35 receptor with anti-allergic activity acting as mast cell and eosinophil stabilizers [5,6]. On the other hand, 1,3-phenylene bis-oxalic derivatives (acids, esters, and amides) are versatile molecules used as supramolecular building blocks for crystal engineering and molecular recognition, since they possess N—H and C=O groups able to form intermolecular hydrogen bonds [7]. The 1,3-phenylene bis-oxalamides are compounds structurally related to lodoxamide by the substitution of the –OH of the oxalic acid with a–NHR group. In this context, we report the synthesis, molecular structure, and the in silico molecular docking affinity to the GPR35 receptor of three 1,3-phenylene bis-oxamides, 3a, 3b, and 3c, Scheme 1.
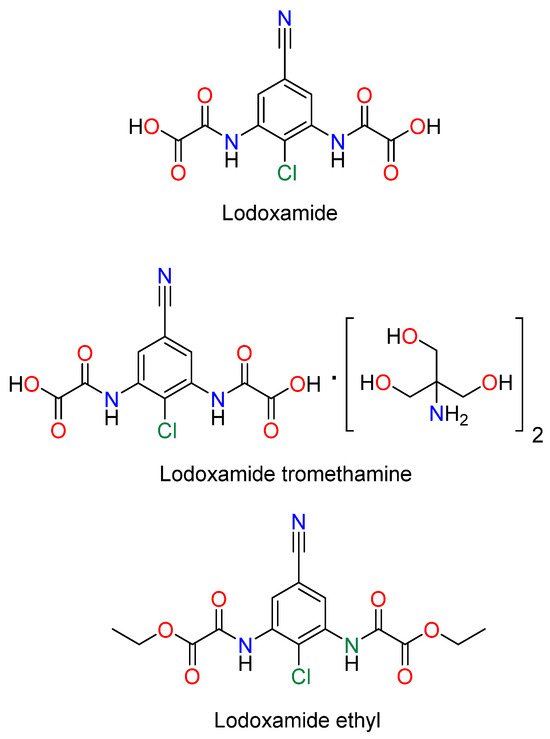
Figure 1.
Lodoxamide derivatives.
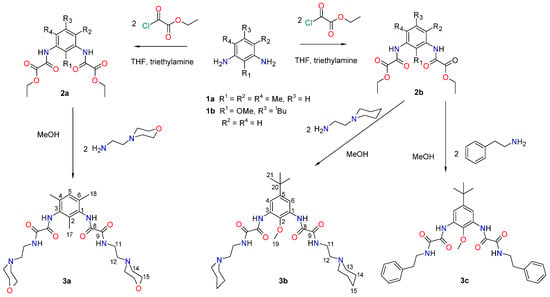
Scheme 1.
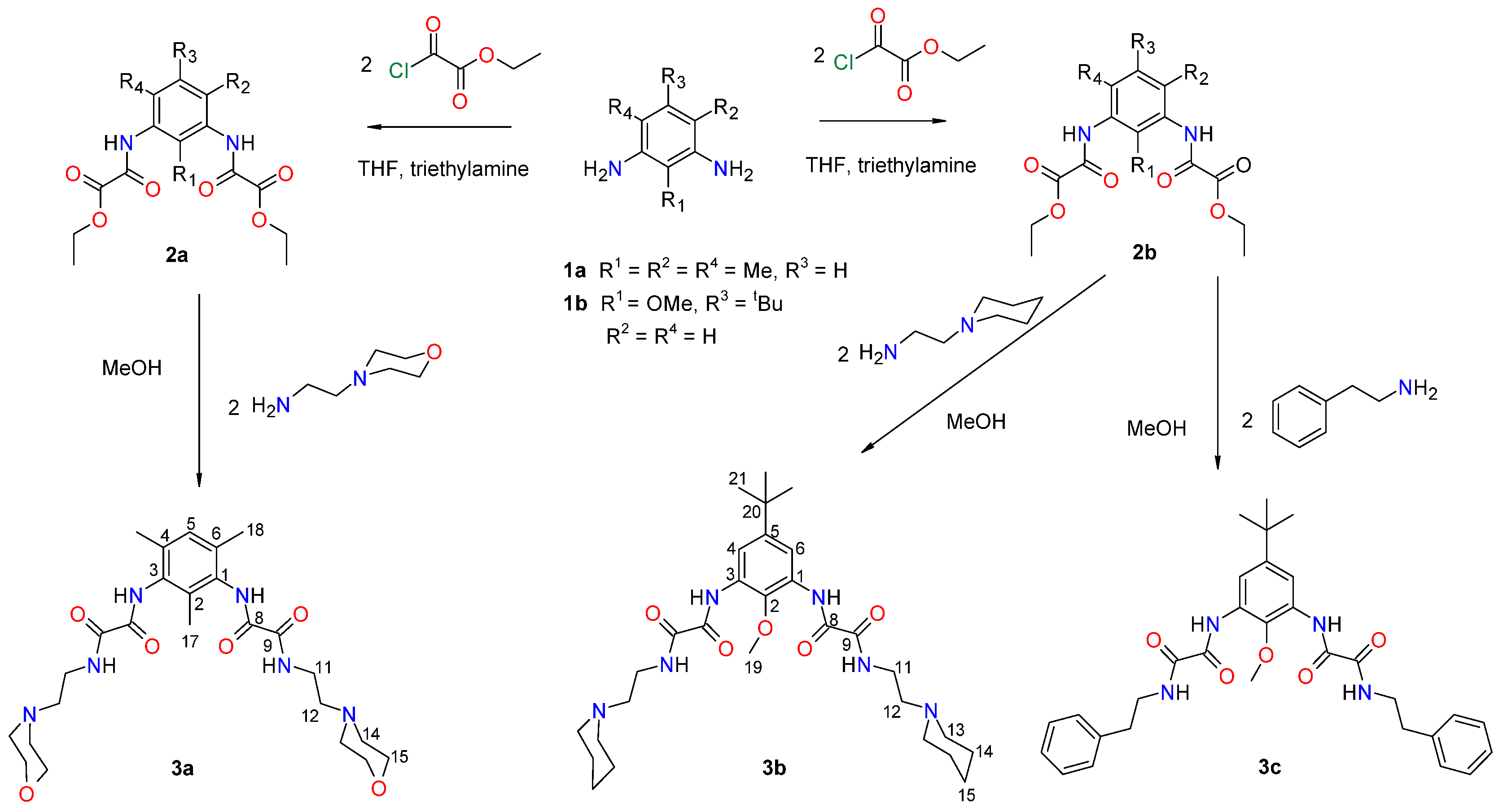
Synthesis of 1,3-phenylene bis-oxamides 3a–c.
2. Materials and Methods
2.1. Materials and Methods
All compounds were purchased from Sigma-Aldrich, 3050 Spruce St., Saint Louis, MO, USA, and were used as received. The 1H and 13C were recorded on a Bruker Ultrashield plus (Ettlingen, Germany) 400 MHz instrument (400 and 100 MHz, respectively) using DMSO-d6 as the solvent and TMS as a reference. Chemical shifts (δ) and coupling constants (J) are expressed in parts per million and Hertz, respectively. Signal multiplicity is expressed as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad singlet). All reactions were monitored by TLC. IR spectra were collected using a Varian 3100 FT-IR EXCALIBUR (Varian Inc., Palo Alto, CA, USA) series spectrophotometer. Melting points were measured on an Electrothermal Mel-Temp 1201D apparatus (Cole-Parmer, Vernon Hills, IL, USA).
2.2. Synthesis of Compounds
The 1,3-phenylene bis-oxalamates 2a and 2b were prepared as previously reported [7].
2.2.1. N1,N1′-(2,4,6-Trimethyl-1,3-phenylene)bis(N2-(2-morpholinoethyl)oxalamide) 3a
To a solution of 2a (0.500 g, 1.43 mmol) in methanol (30 mL), 0.416 mL (2.85 mmol) of 4-(2-aminoethyl), morpholine was added. The mixture was left at reflux for 24 h and then filtered. A white solid was obtained, which was washed with acetone to yield 0.443 g (59.9%), m.p. 234–236 °C, I.R. ν (cm−1): 3253 (N-H), 1654 (C=O). 1H NMR (DMSO-d6, δ): 10.18 (s, 2H, NH), 8.69 (t, 2H, 3J = 5.8 Hz), 7.00 (s, 1H, Ph), 1.91 (s, 3H, CH3), 2.09 (s, 6H, CH3), aminoethylmorpholine: NH-CH2 3.55 (m, 4H), CH2-N 2.48 (m, 4H), N-CH2 2.43 (m, 8H), CH2-O 3.57 (m, 8H). 13C NMR (DMSO-d6, δ): 160.5 (C9), 159.6 (C8), 133.4 (C1, C3), 132.9 (C5), 129.3 (C2), 134.5 (C4, C6), 14.2 (C17), 18.5 (C18), aminoethylmorpholine: NH-CH2 36.7, CH2-N 57.3, N-CH2 53.7, CH2-O 66.8. Anal. calcd. % (found %) for C25H38N6O6: C 57.90 (57.23), H 7.39 (7.54), N 16.21 (14.29).
2.2.2. N1,N1′-(5-(Tert-butyl)-2-methoxy-1,3-phenylene)bis(N2-(2-(piperidin-1yl)ethyl)oxalamide) 3b
To a solution of 2b (0.500 g, 1.43 mmol) in methanol (30 mL), 0.416 mL (2.85 mmol) of 2-aminoethylpiperidine was added. The mixture was left to reflux for 24 h and then filtered. A white solid was obtained to yield 0.485 g (90.1%), m.p. 234–237 °C, I.R. ν (cm−1): 3349 (N-H), 1670 (C=O). 1H NMR (DMSO-d6, δ): 9.87 (s, 2H, N7H), 8.94 (t, 2H, N10H, 3J = 5.6 Hz), 7.93 (s, 2H, Ph), 3.75 (s, 3H, O-CH3), 1.27 (s, 9H, t-but), aminoethylpiperidine: NHb-CH2 3.35 (m, 4H), CH2-N 2.48 (m, 4H), N-CH2 2.51 (m, 8H), N-CH2-CH2 1.52 (m, 8H), N-CH2-CH2-CH2 1.41 (m, 4H). 13C NMR (DMSO-d6, δ): 160.1 (C9), 158.5 (C8), 147.5 (C2), 142.6 (C5), 130.0 (C4, C6), 116.0 (C1, C3), 61.6 (C19), 35.1 (C20), 31.6 (C21), aminoethylpiperidine: N10H-CH2 39.6, CH2-N 57.1, N-CH2 54.2, N-CH2-CH2 25.9, N-CH2-CH2-CH2 24.3. Anal. calcd. % (found %) for C29H46N6O5: C 62.34 (61.47), H 8.30 (8.60), N 15.04 (14.03).
2.2.3. N1,N1′-(5-(Tert-butyl)-2-methoxy-1,3-phenylene)bis(N2-phenethyloxalamide) 3c
To a solution of 2b (0.500 g, 1.26 mmol) in methanol (30 mL), 0.318 mL (2.52 mmol) of phenethylamine was added. The mixture was left to reflux for 24 h and then filtered. A white solid was obtained to yield 0.536 g (77.6%), m.p. 156–158 °C, I.R. ν (cm−1): 3390, 3346 (N-H), 1668 (C=O). 1H NMR (DMSO-d6, δ): 9.87 (s, 2H, N7H), 9.20 (t, 2H, N10H, 3J = 5.9 Hz), 7.93 (s, 1H, Ph), 3.74 (s, 3H, O-CH3), 1.27 (s, 9H, t-but), phenethylamine: N10H-CH2 3.44 (t, 4H, 3J = 6.5 Hz), CH2-Ph 2.85 (t, 4H, 3J = 7.4 Hz), phenyl: 7.21–7.33 (m, 10H). 13C NMR (DMSO-d6, δ): 160.1 (C9), 158.5 (C8), 147.5 (C2), 140.0 (C5), 130.0 (C4, C6), 115.8 (C1, C3), 61.6 (C19), 35.1 (C20), 31.6 (C21), phenethylamine: N10H-CH2 41.4, CH2-Ph 35.1, phenyl: Ci 139.6, Co 129.0, Cm 129.3, Cp 126.8. Anal. calcd. % (found %) for C31H36N4O5: C 68.36 (67.61), H 6.66 (6.89), N 10.29 (9.91).
2.3. Molecular Docking Protocol
Molecular docking was performed using the AutoDock Vina tool implemented in UCSF Chimera version 1.16 [8]. The three-dimensional structures of the compounds 3a–c were constructed using Maestro version 13.3 Schrodinger, LLC [9]. The protein structure of GPR35 was retrieved from the protein data bank with the accession code 8H8J [10]. All water molecules and the co-crystallized ligands were removed from the crystallographic structure. The grid box was defined surrounding the co-crystallized ligand lodoxamide (x = 145.78, y = 139.23, z = 162.25) with a box size of 14.510, 10.479, and 13.487 (x, y, z) within the GPR35 active site. In all simulations, the ligands were flexible, and the protein remained static. Protein–ligand interaction diagrams in 2D and 3D were generated through Discovery Studio Visualizer 2021 [11].
Validation of the protocol was performed using AutoDock Vina tool in UCSF Chimera 1.16 by redocking the co-crystallized ligand lodoxamide. The 3D structure of lodoxamide was built through Maestro 13.3 [9] and docked within the active site of GPR35 (8H8J) [10]. The grid box was centered at the crystallographic coordinates of the co-crystallized ligand (x = 145.78, y = 139.23, z = 162.25) with a box size of 14.510, 10.479, and 13.487 (x, y, z). This validation was carried out based on important interactions.
2.4. Single-Crystal X-Ray Diffraction
Single-crystal X-ray diffraction data for molecules 3a–b were collected at 100(2) K and 173(2) K, respectively, on Bruker Apex II (Germany) [12], and 3c at 293 K on Nonius Kappa CCD (Bruker, Germany) diffractometers equipped with area detectors [13], both using Mo Kα radiation, λ = 0.71073 Å. Crystal data and collection parameters are listed in Table 1. The program SAINT [12] was used for integration of the diffraction profiles for 3a–b, while for 3c, the program hkl2000 software was used [13]. The structures were solved with direct methods using SHELXS97 [14]. The final refinement was performed using the full-matrix least-squares methods on F2 with SHELXL2018/3 [14]. H atoms on C, N, and O were positioned geometrically and treated as riding atoms, with C−H = 0.93–0.99 Å, and with Uiso (H) = 1.2 Ueq (C). Intrinsic pseudo-disorder al the C2-Me group in compound 3a was not treated. The H atoms bonded to N atoms were freely and isotropically refined. The program Mercury 3.8 [15] was used for visualization, molecular graphics, and analysis of crystal structures. Software used to prepare material for publication was PLATON [16] in the WinGX package version 2021.3 [17]. Suitable single crystals for diffraction of 3a, 3b, and 3c were obtained by dissolving 0.5 mg of each compound in DMSO and left to evaporate the solvent. CCDC deposition numbers are 818063 (3a), 818062 (3b) and 2406249 (3c).

Table 1.
Crystallographic parameters for compounds 3a, 3b, and 3c at 293 K.
2.5. Hirshfeld Surface Analysis
The crystal structure of compounds 3a–c was analyzed using the Hirshfeld surface [18] approach. The intermolecular interactions and their reciprocals fingerprints [19,20] were calculated in CrystalExplorer 21.5 software [21]. The individual energetic components were obtained by generating a wavefunction linking the software to the Gaussian 09 package [22] using the B3LYP/6-31G(d,p) theory. Scale factors [23] were included in the calculation, and the % contribution of individual components to the stabilization energy were also determined. The results were depicted as frameworks [23,24] in which the Eelect (electrostatic), Edisp (dispersion), and Etot (total energy) were included in a 23 [19,20] unit cell, and a 3.8 Å radius, with a tube size of 180 kJ mol−1 and 10 kJ mol−1 cut-off values.
3. Results and Discussion
3.1. Synthesis and Characterization of 3a–c
The synthesis of 1,3-phenylene bis-oxamides 3a–c was carried out by reacting the 1,3-phenylene bis-oxalamate 2a or 2b with the corresponding amine, Scheme 1.
For the NMR spectra, the signals were assigned according to Scheme 1, and the whole assignments are listed in Table 2. Half of the expected signals were observed in the spectra of 3a–c, in agreement with a C2 symmetry axis in solution. The following 1H NMR characteristic signals were observed: the phenyl ring of the 1,3-pheylene bis-oxalamate base at 7.00 ppm for 3a, 7.93 ppm for 3b, and 7.93 ppm for 3c; the N7-H7 as a single signal at 10.18 ppm for 3a, 9.87 ppm for 3b, and 9.87 ppm for 3c. The formation of compounds 3a–c was evidenced by the appearance of the N10—H10 signal in the 1H-NMR spectra as a triplet at 8.69 ppm, 8.94 ppm, and 9.20 ppm, respectively. The characteristic 13C NMR signals of 3a–c are two carbonyl signals found in the 160.1–159.6 ppm range; the phenyl carbons of the 1,3-phenylene bis-oxamate base in 3a are found in the 129.3–134.5 ppm range, meanwhile, for 3b–c, the phenyl carbons appeared in the 115.8–147.5 ppm range. The NMR chemical shifts are consistent with previous reports about the synthesis of similar 1,3-phenylene bis-oxamides [7,25]. IR spectroscopy showed characteristic N—H stretching bands at 3253 cm−1 for 3a, 3349 cm−1 for 3b, and 3390 cm−1 and 3346 cm−1 for 3c; meanwhile, the C=O stretching bands were 1654 cm−1 for 3a, 1670 cm−1 for 3b, and 1668 cm−1 for 3c.
Table 2.
1H and 13C NMR chemical shifts in DMSO-d6 of compounds 3a–c (δ = ppm).
3.2. Crystal Structure of 3a–c
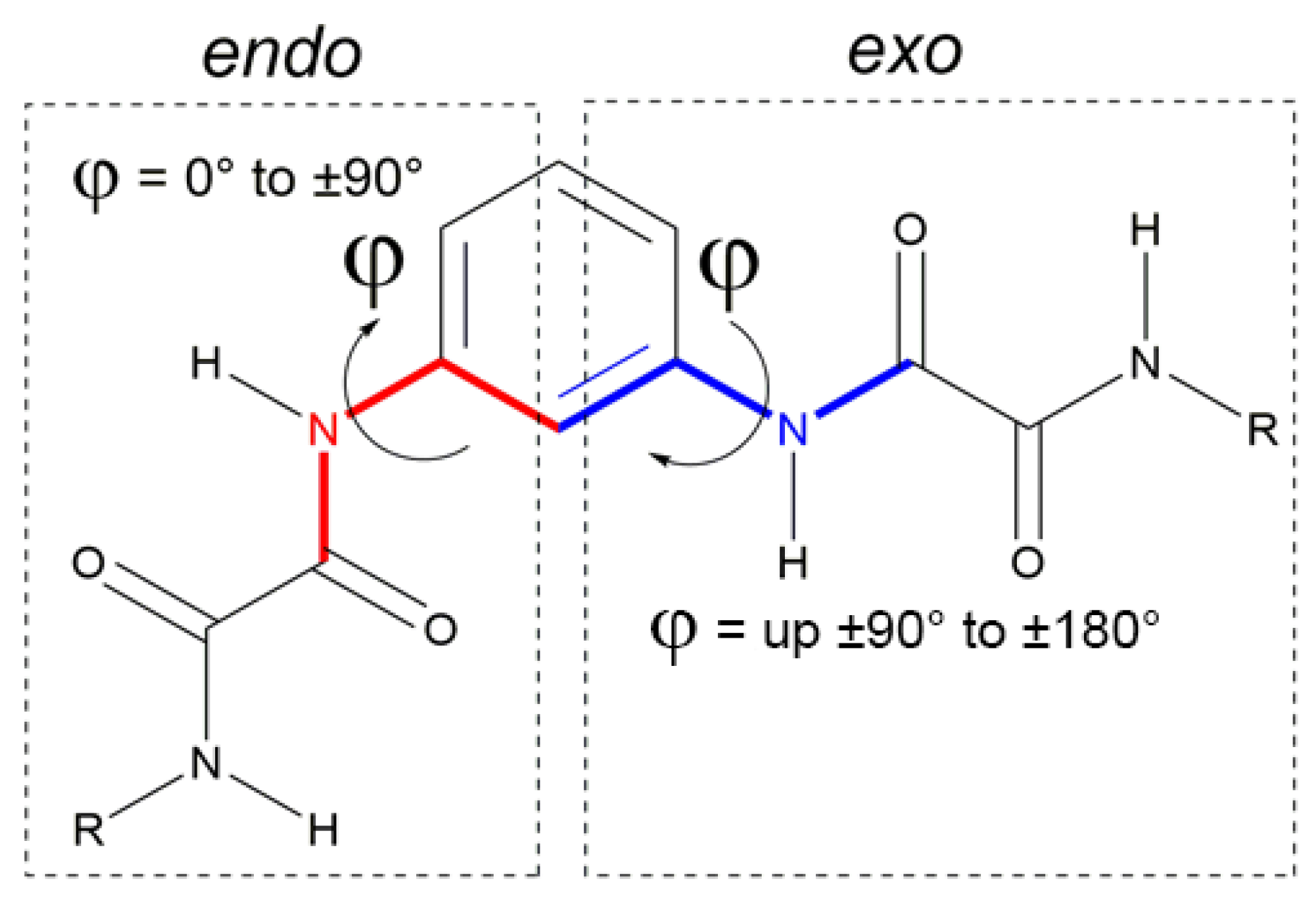
The 1,3-phenyl bis-oxalamidic compounds can adopt the endo or exo conformation according to the disposition of the oxalic groups with respect to the plane of the aromatic ring by measuring the torsion angle φ, Figure 2. In the crystal structures of compounds 3a–c, the oxalic carbonyl groups adopted the characteristic anti conformation (with O=C—C=O torsion angles ranging from 161.7(3)° to 175.0(3)°, with OC—CO distances (1.528(4) Å to 1.541(5) Å), similar to those reported [7].
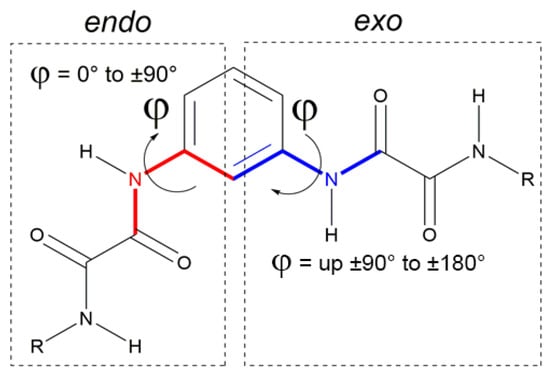
Figure 2.
Endo–exo conformations according to the torsion angle φ.
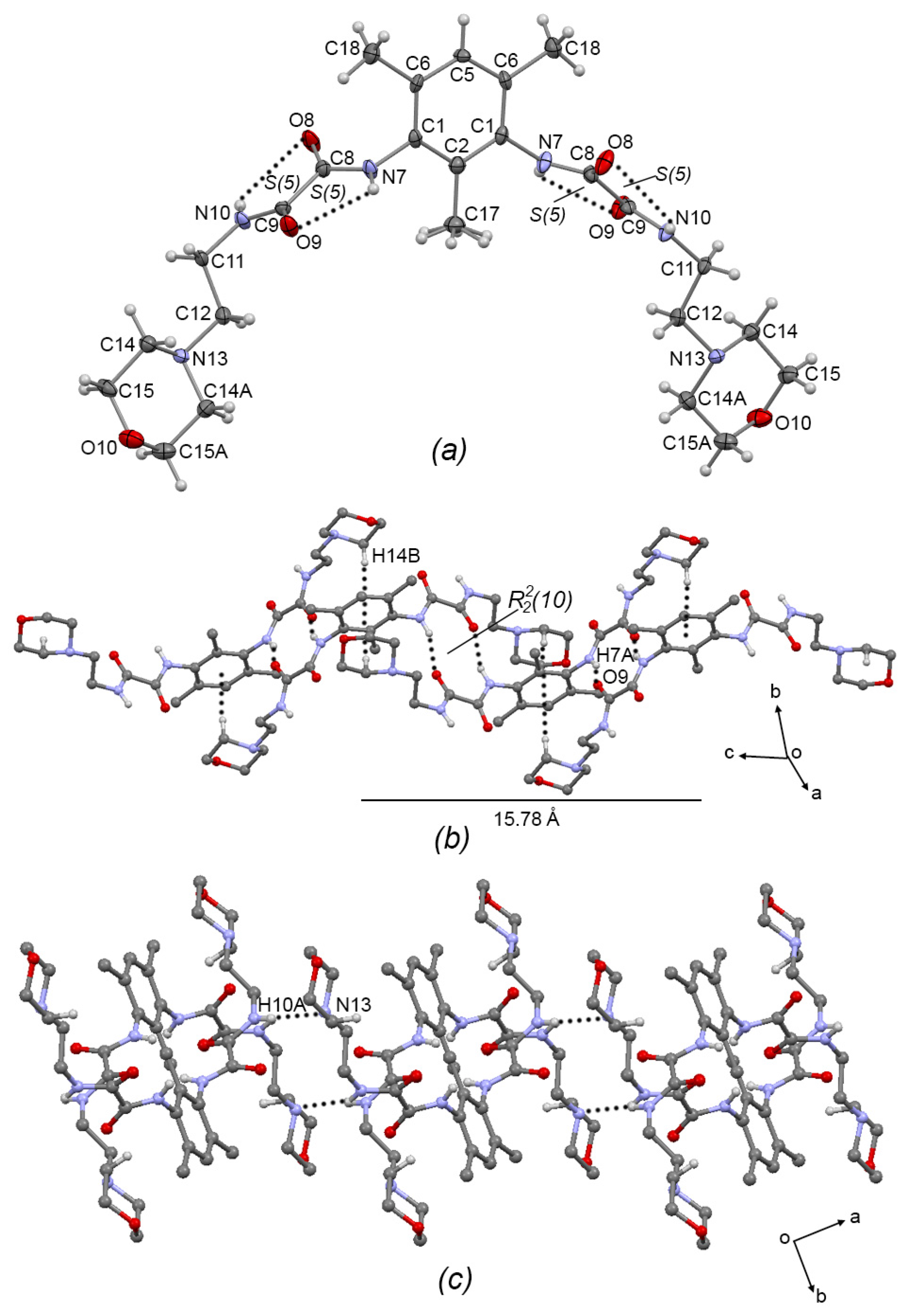
Compound 3a, Figure 3a, crystallized in the monoclinic system with space group C2/c; therefore, only one half of the molecule is observed. The oxalamidic groups adopt the exo–exo conformation with respect to the aromatic ring (torsion angle C2—C1—N7—C8 = −105.1(4)°). The N10—C11—C12—N13 torsion angle of the aminoethylmorpholine fragment is 170.3(3)° adopting an anti conformation. This molecule presents two adjacent sets of intramolecular hydrogen bonds [S(5)S(5)]; geometric intramolecular interactions parameters are listed in Table 3. Moreover, molecules of 3a are self-assembled in 1D via the N7-H7···O9 hydrogen bond (forming the R22(10) motif) and the C14—H14···Cg(2) interaction (Cg(2) = C1/C2/C6/C5/C6/C1), developing a 1D supramolecular meso-helix running along the c-axis, Figure 3b, similar to the related compound N′,N1′-(1,3-(2,4,6-Trimethyl)-phenyl)-bis-(N2-(2-(2-hydroxyethoxy)ethyl)oxalamide) [7]. Geometric intermolecular interaction parameters are listed in Table 4 for compounds 3a–c. The turn of the helix, measured as the space between phenyl rings, is 15.78 Å, which corresponds with the length of the c-axis of the unit cell. Two-dimensional supramolecular arrangement is given by interconnected helixes along the ab plane by the formation of the N10—H10A···N13 hydrogen bond, Figure 3c.
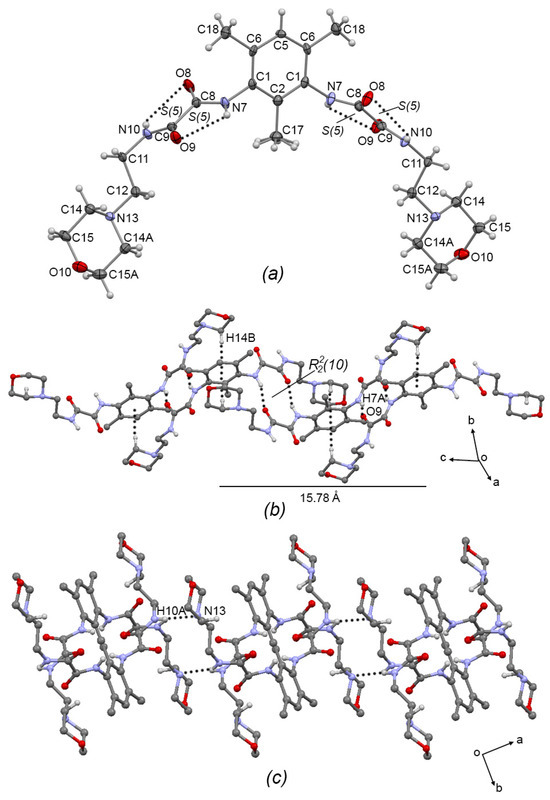
Figure 3.
(a) ORTEP diagram of 3a with the displacement ellipsoid drawn at 70% probability, showing intramolecular interactions. (b) 1D supramolecular meso-helix of 3a formed by N—H···O=C hydrogen bonds and C—H···π intermolecular interactions. (c) Interlinking of supramolecular helixes of 3a by N-H···N intermolecular hydrogen bonds. Some H atoms have been omitted for clarity. Dashed lines represent hydrogen bonds or non-covalent interactions.
Table 3.
Intramolecular interactions in 3a–c.
Table 4.
Intermolecular interactions in 3a–c.
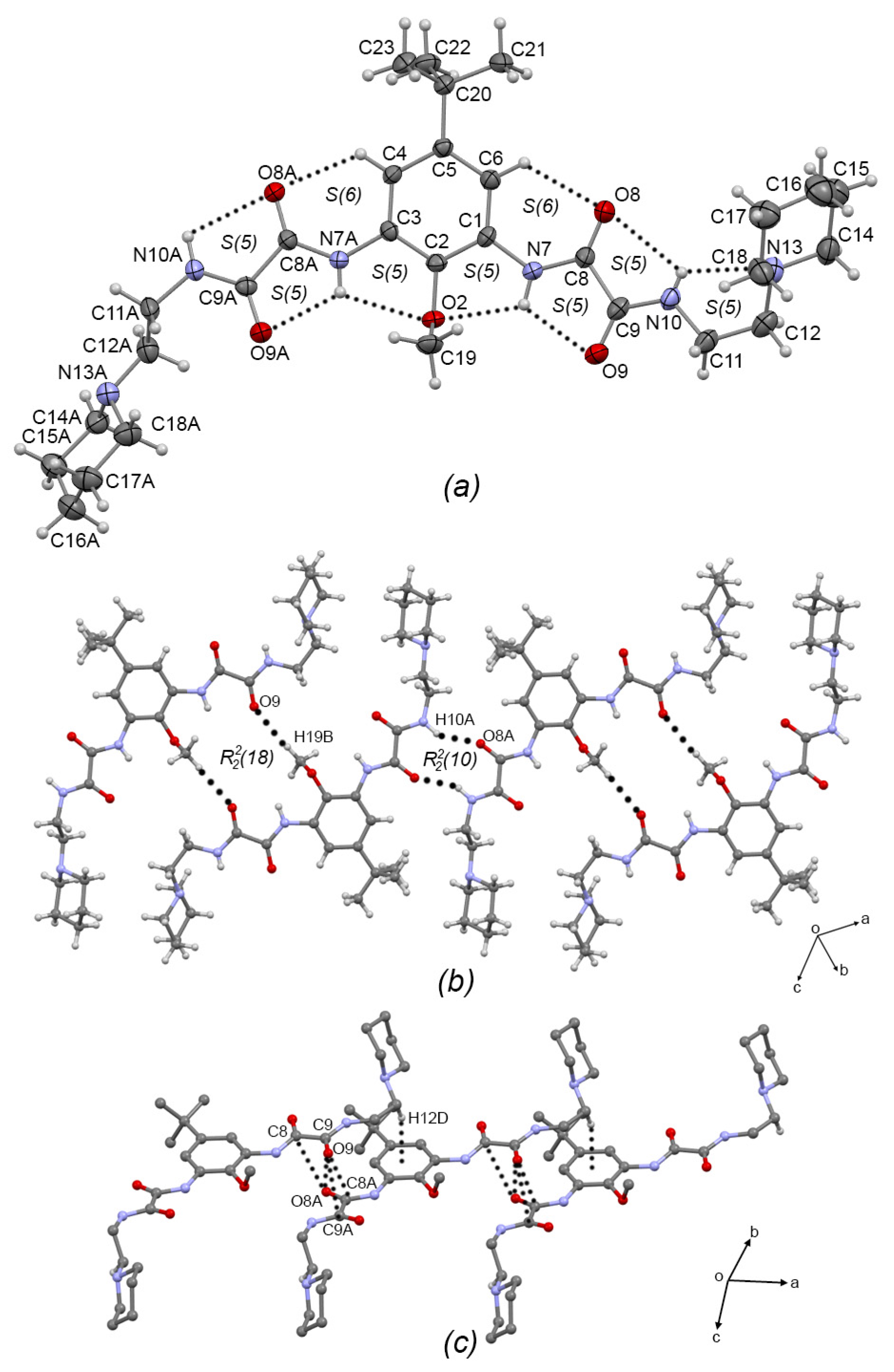
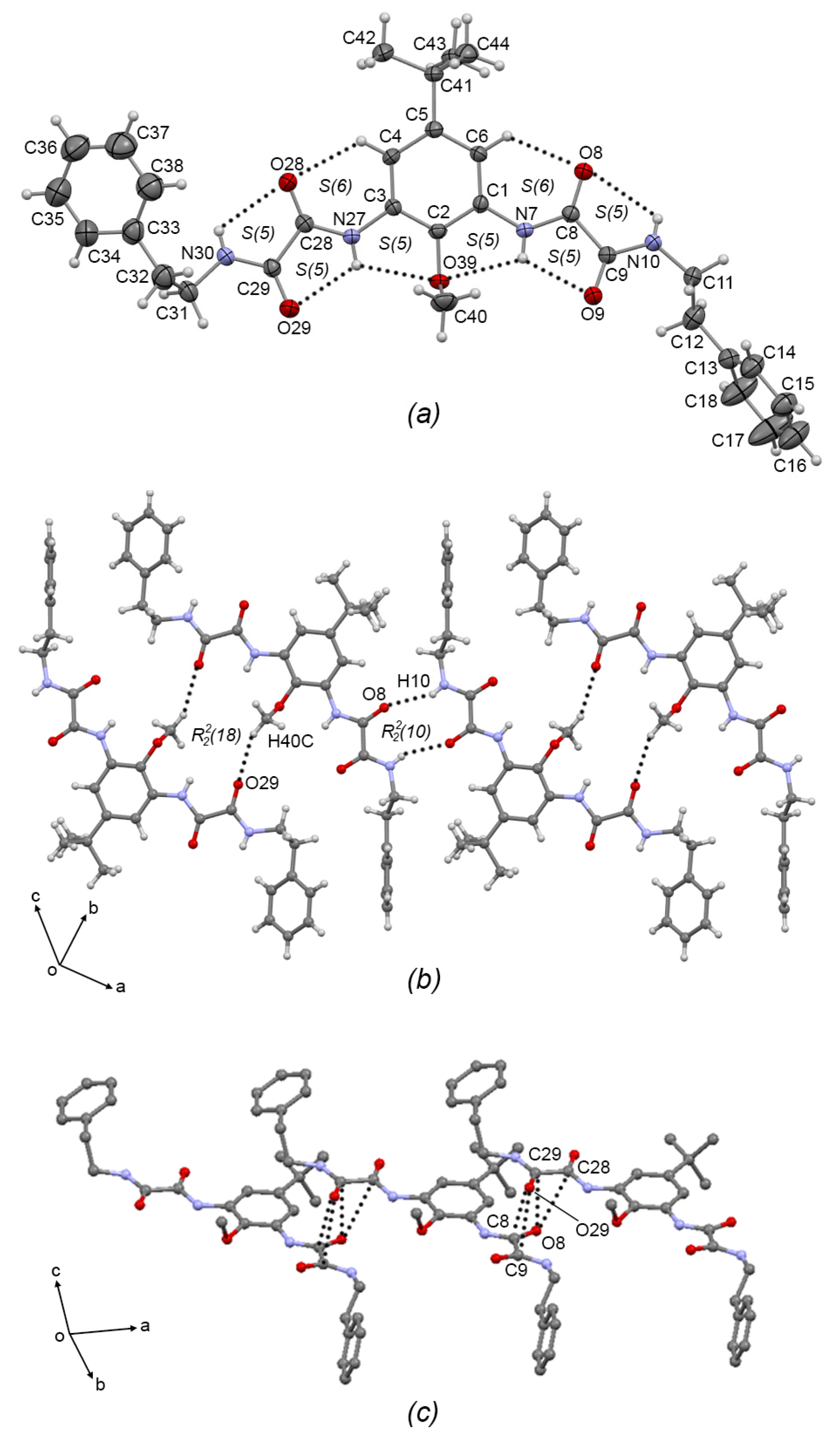
Compounds 3b and 3c crystallized in the triclinic system with P-1 space group. The ORTEP plots are depicted in Figure 4a and Figure 5a, respectively. In both compounds, the oxalic arms adopt the exo–exo conformation, showing torsion angles C2—C1—N7—C8 = −171.2(4)° and C2—C3—N7A—C8A = −179.9(4)° for 3b; and C2—C1—N7—C8 = −178.5(3)° and C2—C3—N27—C28 = 173.1(3)° for 3c, depicting the [S(5)S(5)S(6)S(5)S(5)S(6)S(5)S(5)] set of intermolecular hydrogen bonds (similar to the starting compound 2b [7]. In 3b, one of the aminoethylpiperidine substituent groups adopts an anti conformation, with torsion angle N10A—C11A—C12A—N13A = 173.2(4)°, and the other adopts the syn conformation with torsion angle N10—C11—C12—N13 = 49.5(6)°), allowing the formation of the N10—H10···N13 S(5) intramolecular hydrogen bond. In the same way, in 3c, one of the phenethylamine substituent groups adopts the anti conformation, torsion angle N10—C11—C12—N13 = −172.2(3)°), and the other adopts the syn conformation torsion angle N30—C31—C32—N33 = 67.3(4)°.
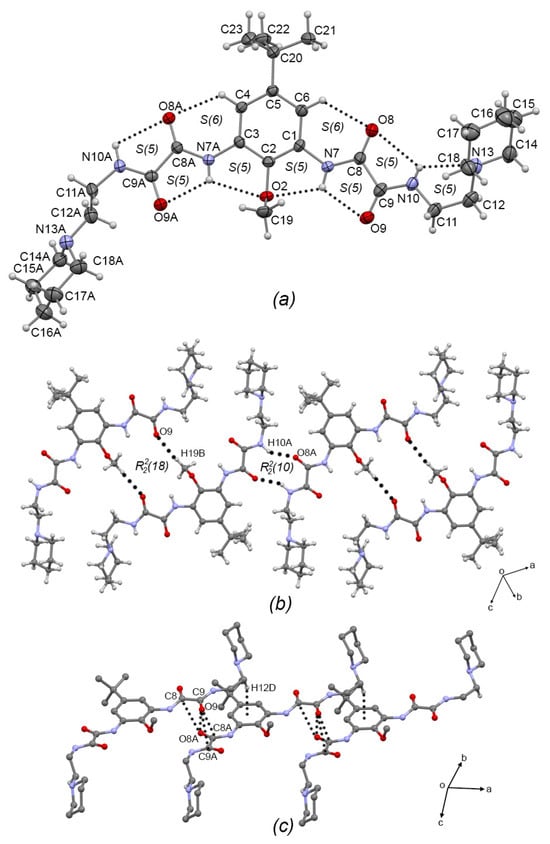
Figure 4.
(a) ORTEP diagram of 3b with the displacement ellipsoid drawn at 50% probability, showing intramolecular interactions. (b) 1D supramolecular tape of 3b formed by C—H···O intermolecular interactions showing the R22(18) motif. (c) Interlinking of supramolecular tapes of 3b by dipolar C=O···O=C and C—H···π intermolecular interactions. Some H atoms have been omitted for clarity. Dashed lines represent hydrogen bonds or non-covalent interactions.
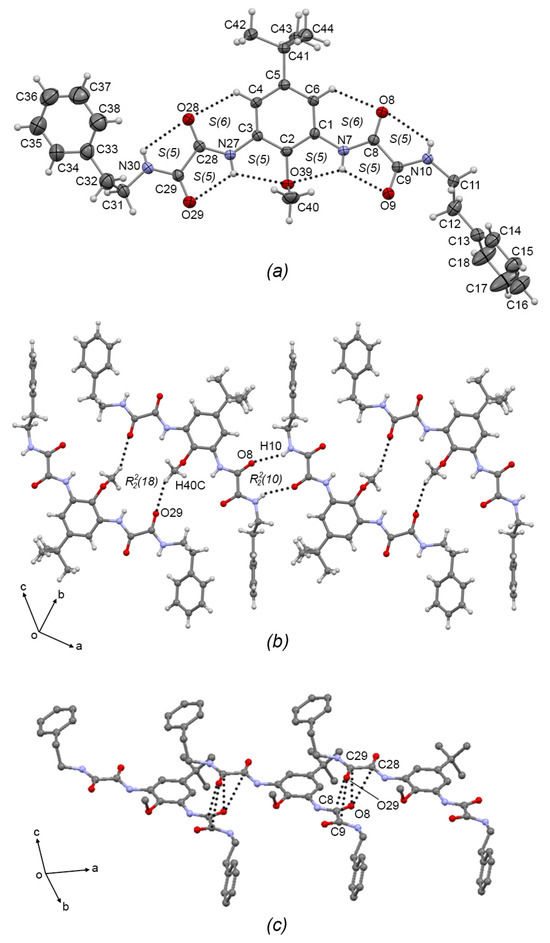
Figure 5.
(a) ORTEP diagram of 3c with the displacement ellipsoid drawn at 50% probability, showing intramolecular interactions. (b) 1D supramolecular tape of 3c formed by C-H···O intermolecular interactions showing the R22(18) motif. (c) Interlinking of supramolecular tapes of 3c by dipolar C=O···O=C intermolecular interactions. Some H atoms have been omitted for clarity. Dashed lines represent hydrogen bonds or non-covalent interactions.
In the 1D supramolecular architecture of 3b, a dimer is formed by the C19—H19B···O8A R22(18) interaction, and the propagation of this dimer by the N10—H10A···O8A R22(10) hydrogen bond form a supramolecular tape of molecules of 3b running along the (2–4 7) plane, Figure 4b. Interlinking the supramolecular tapes along the a-axis by dipolar C=O···C=O interactions (C8···O8A = 3.205(5) Å; C9···O8A = 3.183(6) Å; C8A···O9 = 3.219(5) Å; C9A···O9 = 3.056(5) Å) and C—H···π interactions (C12—H12D···Cg(3) = 2.68 Å; Cg(3) = C1-C6) gives rise the 2D supramolecular array, Figure 4c. Compound 3c showed the same supramolecular architecture as 3b: 1D supramolecular tape formed by C40—H40C···O29 dimers extended by N10—H10···O8 hydrogen bonds depicting R22(10) and R22(18) motifs, respectively, Figure 5b; and 2D interlinking of tapes, Figure 5c by C=O···C=O interactions (C28···O8 = 3.224(4) Å; C29···O8 = 3.109(4) Å; C8···O29 = 3.166(4) Å; C9···O29 = 3.096(4) Å). Comparing the supramolecular architectures of 3b and 3c with the crystal structure of the precursor 2b and with the related oxalamidic derivative N′,N1′-(1,3-(5-tert-butyl-2-methoxy)-phenyl)-bis-(N2-(2-hydroxyethyl)oxalamide) [7] (TEOA), it was found that both compounds also are paired by C—H···O=C interactions depicting the R22(18) motif, which is extended by C—H···O=C interactions in 2b and O—H···O=C hydrogen bonds in TEOA to develop 1D supramolecular tapes (the R22(10) motif is absent); and the 2D array in both compounds showed stacked tapes by π···π interactions and by C=O···C=O interactions, unlike 3b and 3c, where the steric effect of the aminoethylpiperidine and phenethylamine fragments avoids the formation of π···π interactions, but the C=O···C=O interactions remain.
3.3. Hirshfeld Surface (HS) Analysis
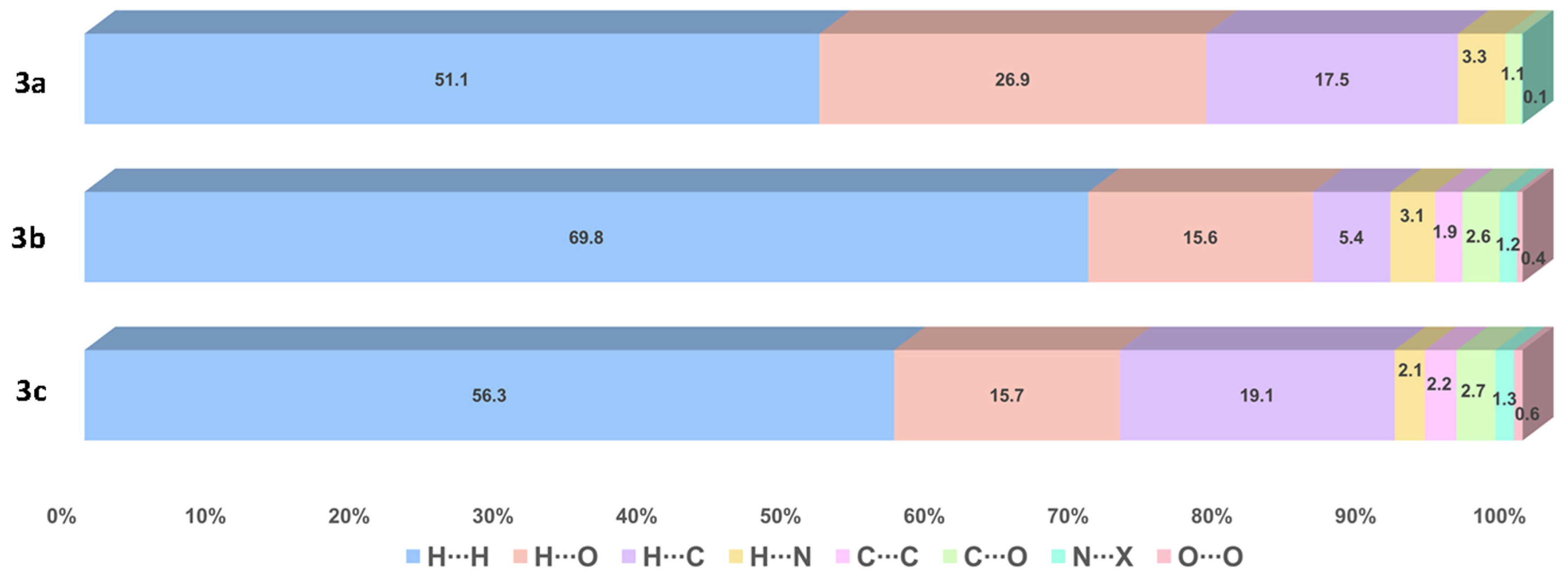
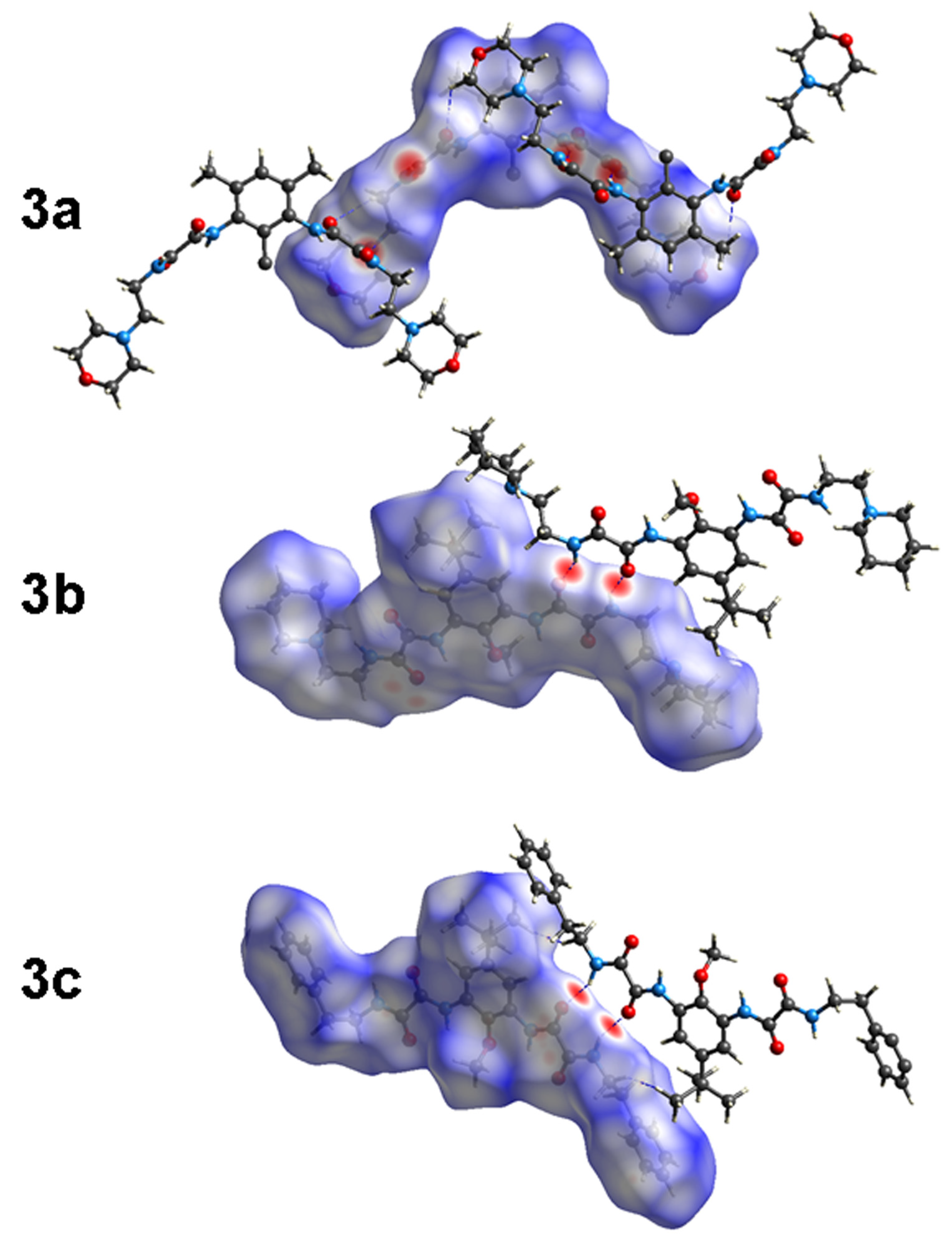
The contributions of several interactions to the HSs are differentiated between the compounds. The percentage of contribution of the most significant interactions and their reciprocals are shown in Figure 6. Among all the close interactions, H⋯H contacts represent the largest set of interactions contributing to the surface with 51.1–69.8%. The strongest interactions (A⋯H/H⋯A, A = O, N) are given by N―H∙∙∙O and N―H∙∙∙N HBs, contributing with 30.2% to the surface of 3a and 17.8–18.8% to the surface of 3b–c. These interactions are shown in Figure 7 as red bright spots. Finally, the H⋯C/C⋯H interactions, represent the 5.4–19.1% of the surface.
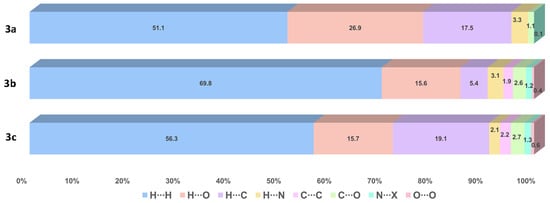
Figure 6.
Percentage of contribution of the most significant interactions and their reciprocals in compounds 3a–c. N∙∙∙X (X = C, N, O).
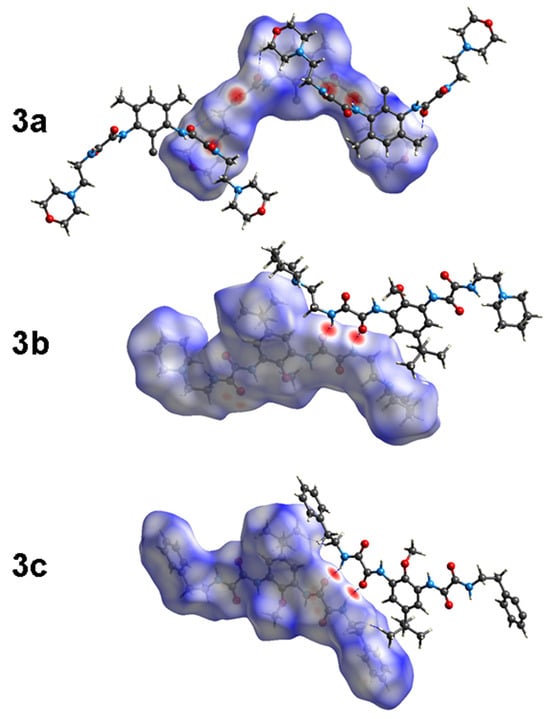
Figure 7.
Hirshfeld surfaces (HS) of 3a–c.
3.4. Quantitative Interaction Energy Analysis and Energy Framework Diagrams
Quantitative interaction energies of 3a–c were calculated at the B3LYP/6-31G(d,p) level of theory; selected results are listed in Table 5, and those values (Etot) smaller than −20.0 kJ mol−1 were neglected. In all crystals, the most energetic interactions involve the amide N—H∙∙∙O hydrogen bonding, assembling as R22(10) intermolecular ring motif, whose geometric parameters agree with a strong interaction. This motif contributes with −113.9 (3a), −97.0 (3b), and −110.5 (3c), kJ mol−1 to the energy in the corresponding crystal. These N―H∙∙∙O hydrogen bonding (HB) interactions are more dispersive than electrostatic in nature, contributing to the stabilization energy with 46.6–53.9% and 39.0–46.4% of Edis and Eele percent ranges, respectively. No further crystal energy results were obtained for 3a, probably due to disorder located at the C2-methyl group, even when the bright red spots marked by N10―H10∙∙∙N13 HB are observed in the HS.
Table 5.
Calculated interaction energies (kJ mol−1), %Ecomp contributions to stabilization energy for selected HB and close contacts in compounds 3a–c.
In the case of 3b–c, the second, third, and subsequent interaction contributors to the crystal energy are mainly composed of the interplay of C―H∙∙∙A (A = O, Cg), C―O∙∙∙CO and Cg∙∙∙Cg interactions, whose nature is mainly dispersive. Each assembly contributes 58.9–80.9% (%Edis) to stabilize the crystal network. Among Cg∙∙∙Cg interactions, the partial double bond character of the amide N30―C29 bond, Cg(1), is worth highlighting. The N30―C29 bond length is particularly short in both 3b (1.325 Å) and 3c (1.322 Å), compared with the Nn7―Cn8 (n = 0, 2) mean bond length in the anilide counterparts (1.346 ± 4). Thus, the C17―H17A∙∙∙ Cg(1) and C15―H15∙∙∙Cg(1) interactions arise in 3b and 3c, respectively, after the analysis of interaction energies. The energy values associated with C―H∙∙∙ Cg(1) interaction are −79.7 kJ mol−1 (%Edis = 74.1) and −59.2 kJ mol−1 (%Edis = 75.0), in 3b and 3c, respectively, mainly being dispersive in nature. The geometric parameters associated with this interaction are given in the footnote of Table 5.
The visual representation of the energy-framework diagrams for Eele (red), Edis (green), and Etot (blue) for a cluster of nearest-neighbor molecules is shown in Figure 8. The crystal networks of 3a–c are mainly dominated by the Edis component, showing zig-zagging energy frameworks that overcome the Eelec component to give the final shape to the Etot energy framework in all cases.

Figure 8.
Energy frameworks of 3a–c. (a) Electrostatic; (b) Dispersive; (c) Total.
3.5. Molecular Docking Analysis
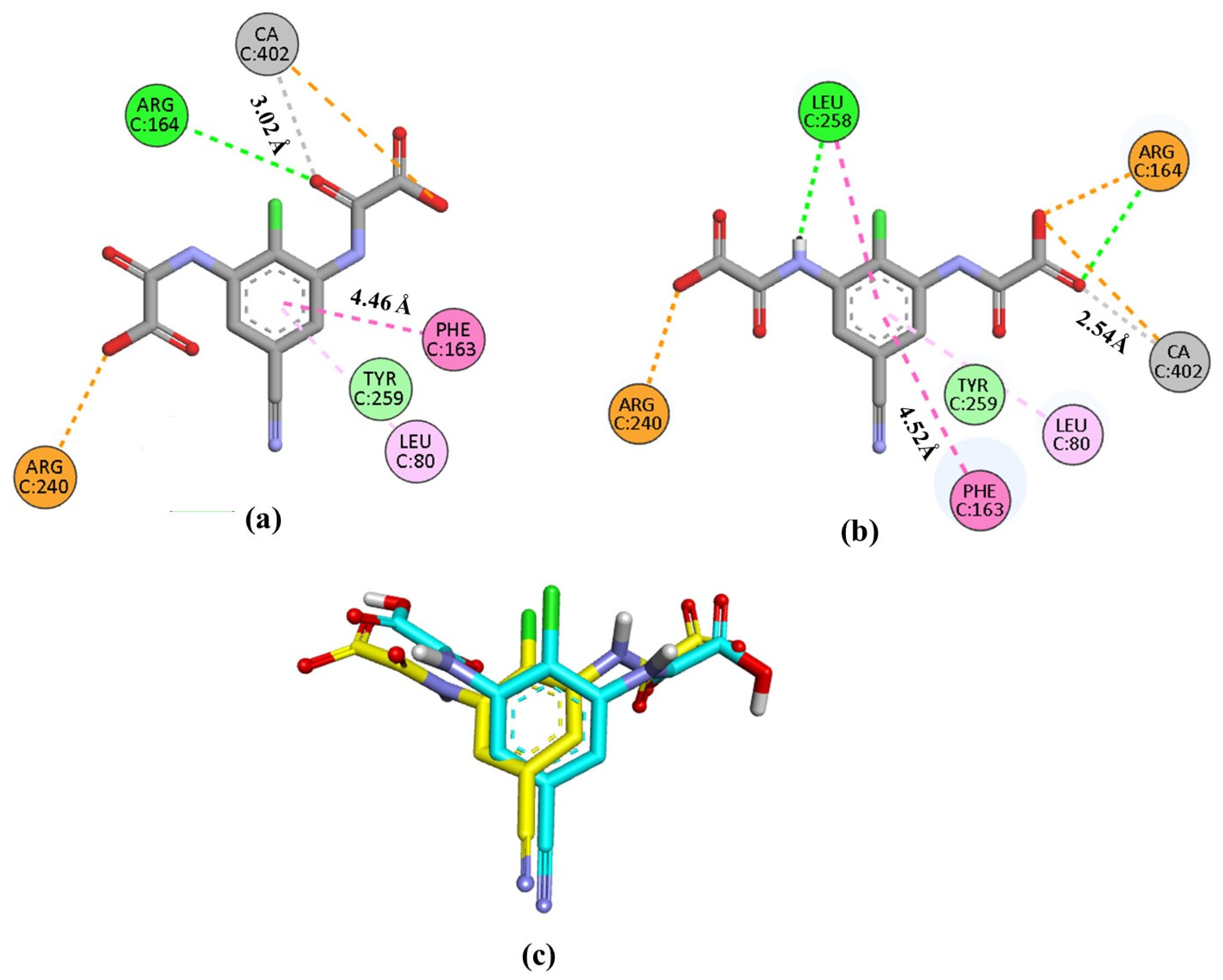
Validation of the method was performed by reproducing the experimental binding of the co-crystallized ligand lodoxamide to the GPR35 receptor. Our protocol replicated the experimental binding mode of lodoxamide with an RMSD of 2.05 Å, showing hydrogen bonding interaction with Arg164, electrostatic interaction with Arg 240, π-π stacking and π-alkyl interactions with Phe163 and Leu80, respectively, and coordination with the catalytic calcium ion, Figure 9. Additionally, the calculated binding mode of lodoxamide exhibited hydrogen bonding and π-π stacking interactions with Leu258.
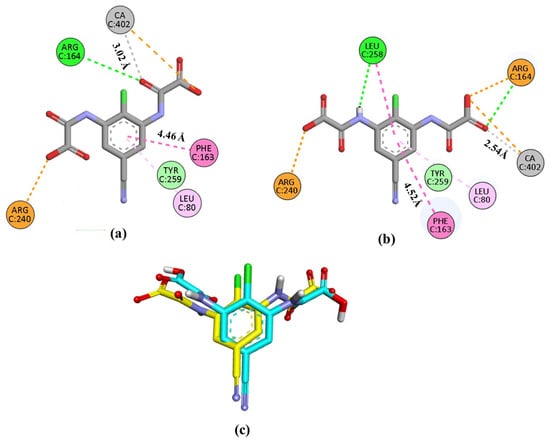
Figure 9.
Representation of the interactions in 2D of lodoxamide co-crystallized (a), lodoxamide docked (b), and (c) the superimposition of both (co-crystallized in yellow and docked in blue) within the GPR35 active site for validation purposes. Interactions are color-coded: green, hydrogen bonds; dark pink, π-π interactions; pale pink, alkyl-π interactions; orange, electrostatic and cation-π interactions; gray, metal coordination. CA; calcium ion.
The molecular docking studies of 3a–c showed that all ligands reached the catalytic binding site of GPR35, displaying favorable bindings. Free-binding energies ΔG (kcal/mol) are listed in Table 6. ΔG values range from −7.0 to −8.3 kcal/mol, close to the value obtained for the reference compound, lodoxamide (−8.5 kcal/mol). Our results also demonstrated that the binding modes of compounds 3a–c and lodoxamide are very similar, leading to comparable accommodation within the binding site, Figure 10.
Table 6.
Free-binding energies ΔG (kcal/mol) of compounds 3a–c docked into the active site of GPR35.
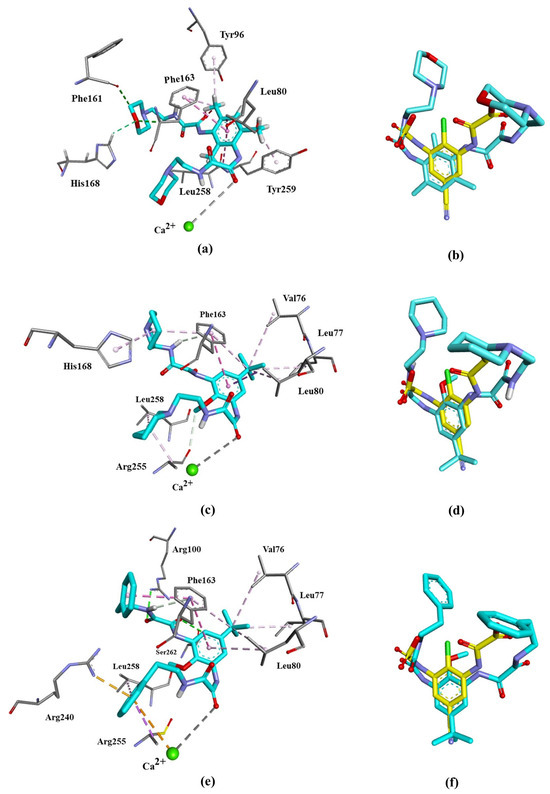
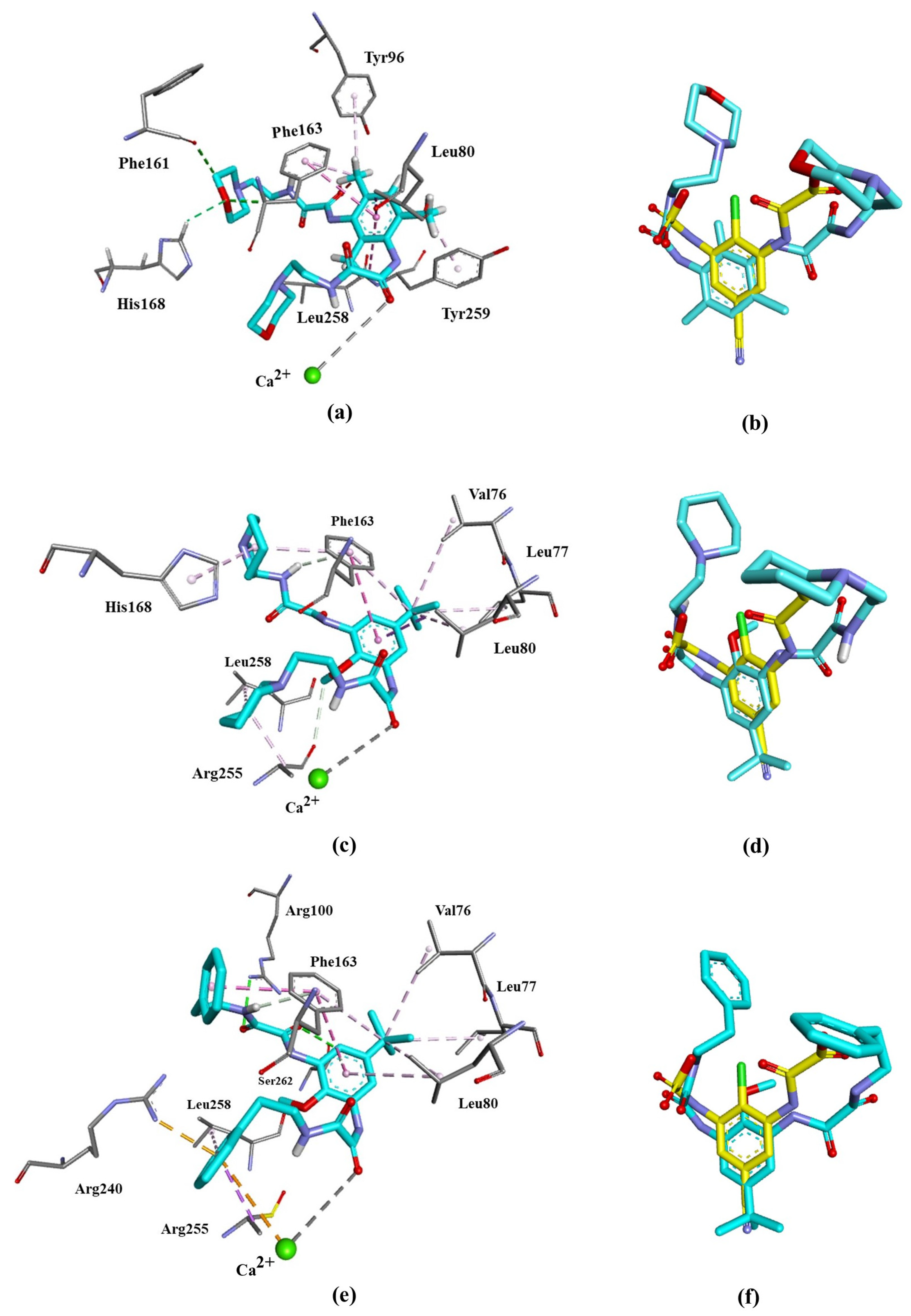
Figure 10.
Binding poses at GPR35 active site of the target compounds. Left panel: 3D ligand interaction diagram of (a) 3a, (c) 3b, and (e) 3d. Right panel: superimposition of (b) 3a, (d) 3b, and (f) 3d with lodoxamide (yellow) within the GPR35 active site.
All three ligands interact with Phe163 and Leu80 amino acid residues of GPR35 through π-π stacked and π-alkyl interactions and coordinate with the catalytic calcium ion through a carbonyl group, the same as lodoxamide. However, different substituents favor unique interactions. The binding of 3a, bearing a morpholine moiety, favors the formation of a hydrogen bond between its heterocyclic oxygen and Phe161 and His168. The pyridine moiety of 3b forms alkyl–alkyl interaction with His168, Arg255 and Leu258, while the tert–butyl in 3b and 3c shows interactions of the same type with Val76, Leu77 and Leu80. Compound 3c, bearing a phenyl moiety, exhibits π–alkyl interactions with Arg255 and Leu258, and π–cation interactions with Arg240 and the catalytic calcium ion. Most of these interactions have been reported for other GPR agonists [4,10,26]. Table 7 summarizes all the interactions of the compounds 3a–c with the residues of the binding site of GPR-35.
Table 7.
Interactions type of compounds 3a–c with the different residues of GPR-35.
It is worth mentioning that the docked structures of 3a–c adopt different conformations (bending of the N-H-R arms and O=C-C=O torsion angles different from the anti conformation, which is the most stable) compared to the X-ray structures, because they have to accommodate themselves in the GPR35 receptor binding site, which is a narrow place compared with the crystalline cell. This allows the formation of N—H···O=C, C—H···π and C—H···O intermolecular interactions with the amino acids of the binding site of the GPR35 receptor, which are responsible for the self-assembly of the 3a–c molecules in the crystal cell adopting the different supramolecular architectures (helix and tapes). Additionally, it should be noted that, during the docking process, the atoms of the protein are kept rigid, which prevents induced fit of the receptor residues. This limitation is particularly relevant when docking larger molecules, such as 3a–c, compared to smaller ligands like lodoxamide.
4. Conclusions
A series of 1,3 phenylene bis-oxalamides (3a–c) was prepared and characterized using 1H and 13C NMR, IR spectroscopy and single-crystal X-ray diffraction. The supramolecular architecture of 3a is driven by N—H···O=C and N—H···N hydrogen bonds depicting a meso-helix; meanwhile, 3b and 3c showed C—H···π, and O=C···O=C interactions depicting supramolecular tapes (3b–c). According to Hirshfeld surface analysis, N―H∙∙∙X (X = N, O) hydrogen bonding represents 30.2% to the surface of 3a and 17.8–18.8% to the surface of 3b–c. The most energetic interactions involve the amide N—H∙∙∙O hydrogen bonding, contributing in the −113.9 to −97.0 kJ mol−1 range to the crystal energy, being more dispersive than electrostatic in nature. The compounds showed favorable binding to the catalytic binding site of the GPR35 receptor, in a similar mode of binding and accommodation compared with lodoxamide.
Author Contributions
F.J.M.-M., I.I.P.-M. and J.S.G.-G. conceptualized the study. J.L.M.-A. and L.B.-M. developed Docking. J.S.G.-G., I.I.P.-M. and F.J.M.-M. performed research. J.M.S.-Q. and I.I.P.-M. provided Hirshfeld surface analysis. E.V.G.-B. and N.E.M.-V. collected X-ray diffraction. F.J.M.-M., J.S.G.-G. and I.I.P.-M. wrote, reviewed and edited the manuscript. F.J.M.-M. and I.I.P.-M. supervised the study. F.J.M.-M. and I.I.P.-M. confirm the authenticity of all the raw data. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
The present study was supported by SIP-IPN-20250238, Universidad de la Cañada and Universidad de Colima.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- MacKenzie, A.E.; Lappin, J.E.; Taylor, D.L.; Nicklin, S.A.; Milligan, G. GPR35 as a Novel Therapeutic Target. Front. Endocrinol. 2011, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, P.; Fan, H.; Zhang, C.; Yu, P.; Liang, X.; Chen, Y. GPR35 Acts a Dual Role and Therapeutic Target in Inflammation. Front. Immunol. 2023, 14, 1254446. [Google Scholar] [CrossRef] [PubMed]
- Shore, D.M.; Reggio, P.H. The Therapeutic Potential of Orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef]
- MacKenzie, A.E.; Caltabiano, G.; Kent, T.C.; Jenkins, L.; McCallum, J.E.; Hudson, B.D.; Nicklin, S.A.; Fawcett, L.; Markwick, R.; Charlton, S.J.; et al. The Antiallergic Mast Cell Stabilizers Lodoxamide and Bufrolin as the First High and Equipotent Agonists of Human and Rat GPR35. Mol. Pharmacol. 2014, 85, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Lu, D.; Sun, J.; Ye, Y.; Fang, J.; Wang, K.; Guo, S.; Zhang, Q.; He, X.; Xie, X.; et al. Discovery of a Novel GPR35 Agonist with High and Equipotent Species Potency for Oral Treatment of IBD. Bioorg. Med. Chem. 2023, 96, 117511. [Google Scholar] [CrossRef]
- Cao, M.; Gao, Y. Mast Cell Stabilizers: From Pathogenic Roles to Targeting Therapies. Front. Immunol. 2024, 15, 1418897. [Google Scholar] [CrossRef]
- González-González, J.S.; Martínez-Martínez, F.J.; Peraza-Campos, A.L.; Rosales-Hoz, M.d.J.; García-Báez, E.V.; Padilla-Martínez, I.I. Supramolecular Architectures of Conformationally Controlled 1,3-Phenyl-Dioxalamic Molecular Clefts through Hydrogen Bonding and Steric Restraints. CrystEngComm 2011, 13, 4748–4761. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Schrödinger, L. Maestro, Version 13.3; Schödinger Release 2025-1; Schrödinger: New York, NY, USA, 2025. Available online: https://www.schrodinger.com (accessed on 25 February 2025).
- Duan, J.; Liu, Q.; Yuan, Q.; Ji, Y.; Zhu, S.; Tan, Y.; He, X.; Xu, Y.; Shi, J.; Cheng, X.; et al. Insights into Divalent Cation Regulation and G13-Coupling of Orphan Receptor GPR35. Cell Discov. 2022, 8, 135. [Google Scholar] [CrossRef]
- Dassault Systèmes. BIOVIA Discovery Studio Visualizer 2021; Dassault Systèmes: Paris, France, 2025; Available online: https://www.3ds.com/products/biovia (accessed on 25 February 2025).
- Bruker. APEX2, SAINT; Bruker AXS Inc.: Madison, WI, USA, 2006. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1997; Volume 276, pp. 307–326. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer Model Energies and Energy Frameworks: Extension to Metal Coordination Compounds, Organic Salts, Solvates and Open-Shell Systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy Frameworks: Insights into Interaction Anisotropy and the Mechanical Properties of Molecular Crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Padilla-Martínez, I.I.; Martínez-Martínez, F.J.; Guillén-Hernández, C.I.; Chaparro-Huerta, M.; Cabrera-Pérez, L.C.; Gómez-Castro, C.Z.; López-Romero, B.A.; García-Báez, E.V. Switching from Twisted to Planar Oxamide Molecular Cavities through Intramolecular Three Centered Hydrogen Bonding. Arkivoc 2005, 2005, 401–415. [Google Scholar] [CrossRef]
- Zhao, P.; Lane, T.R.; Gao, H.G.L.; Hurst, D.P.; Kotsikorou, E.; Le, L.; Brailoiu, E.; Reggio, P.H.; Abood, M.E. Crucial Positively Charged Residues for Ligand Activation of the GPR35 Receptor. J. Biol. Chem. 2014, 289, 3625–3638. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).