
Hydrogen-Bonded Ladder Motifs in Naphthalene Dicarboxamides: Influence of Linear vs. Angular Amide Orientation

 , , ,
, , , 
Abstract
:
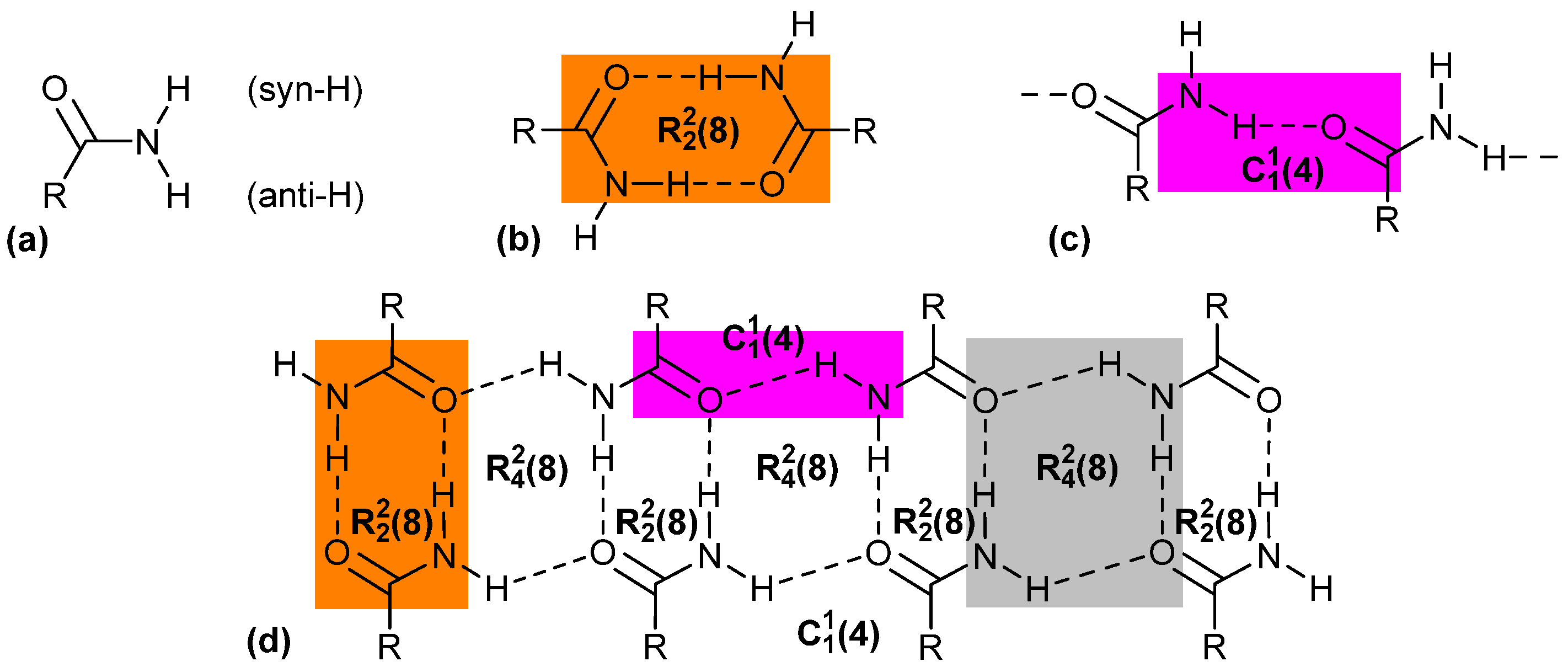
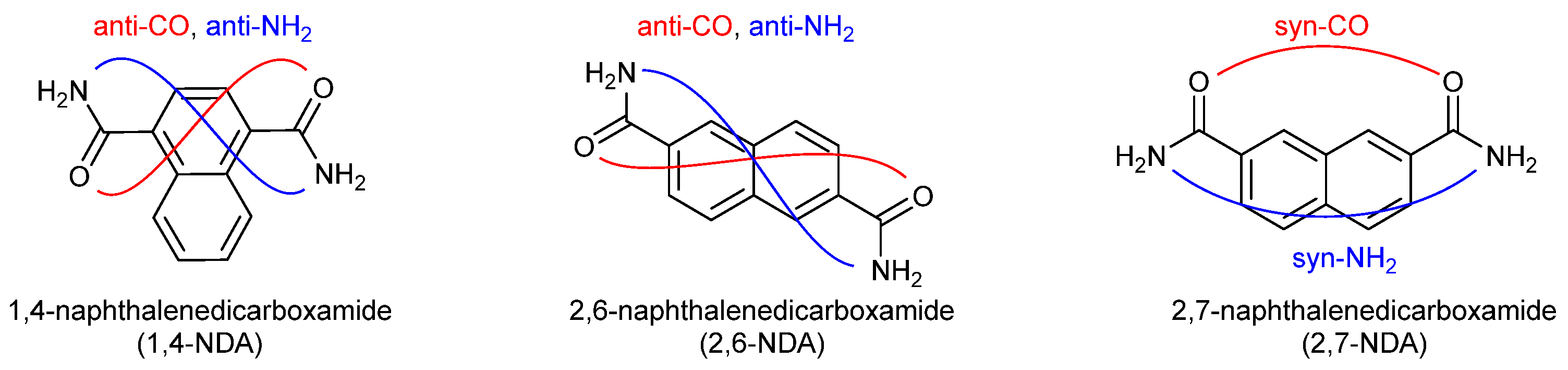
1. Introduction
2. Materials and Methods
2.1. Synthesis of Dicarboxamides
2.2. Crystallization of Dicarboxamides
3. Results and Discussion
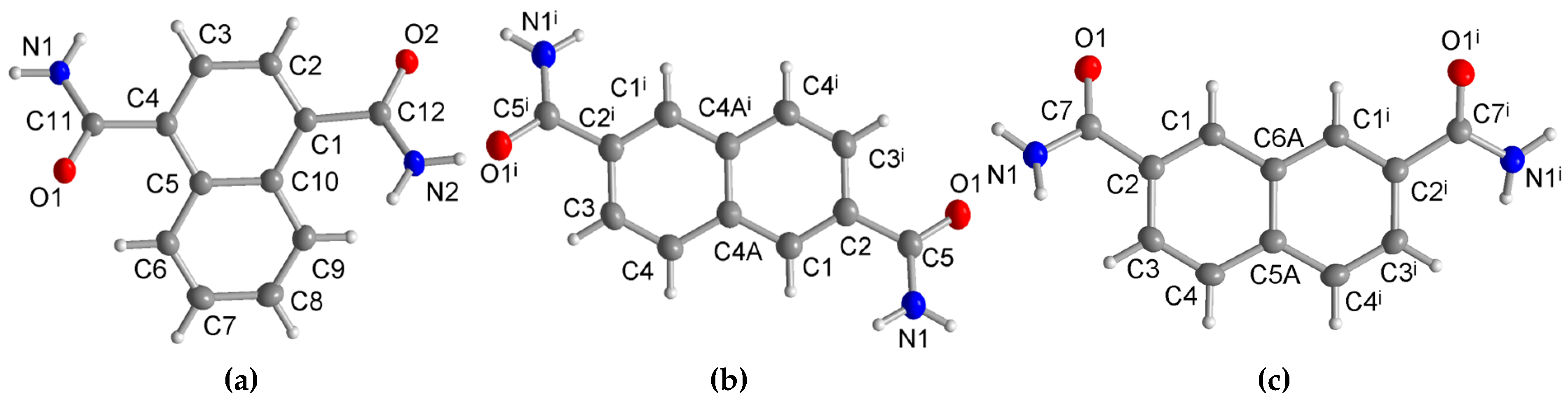
3.1. Crystal Structures of the Dicarboxamides 1,4-NDA, 2,6-NDA, and 2,7-NDA
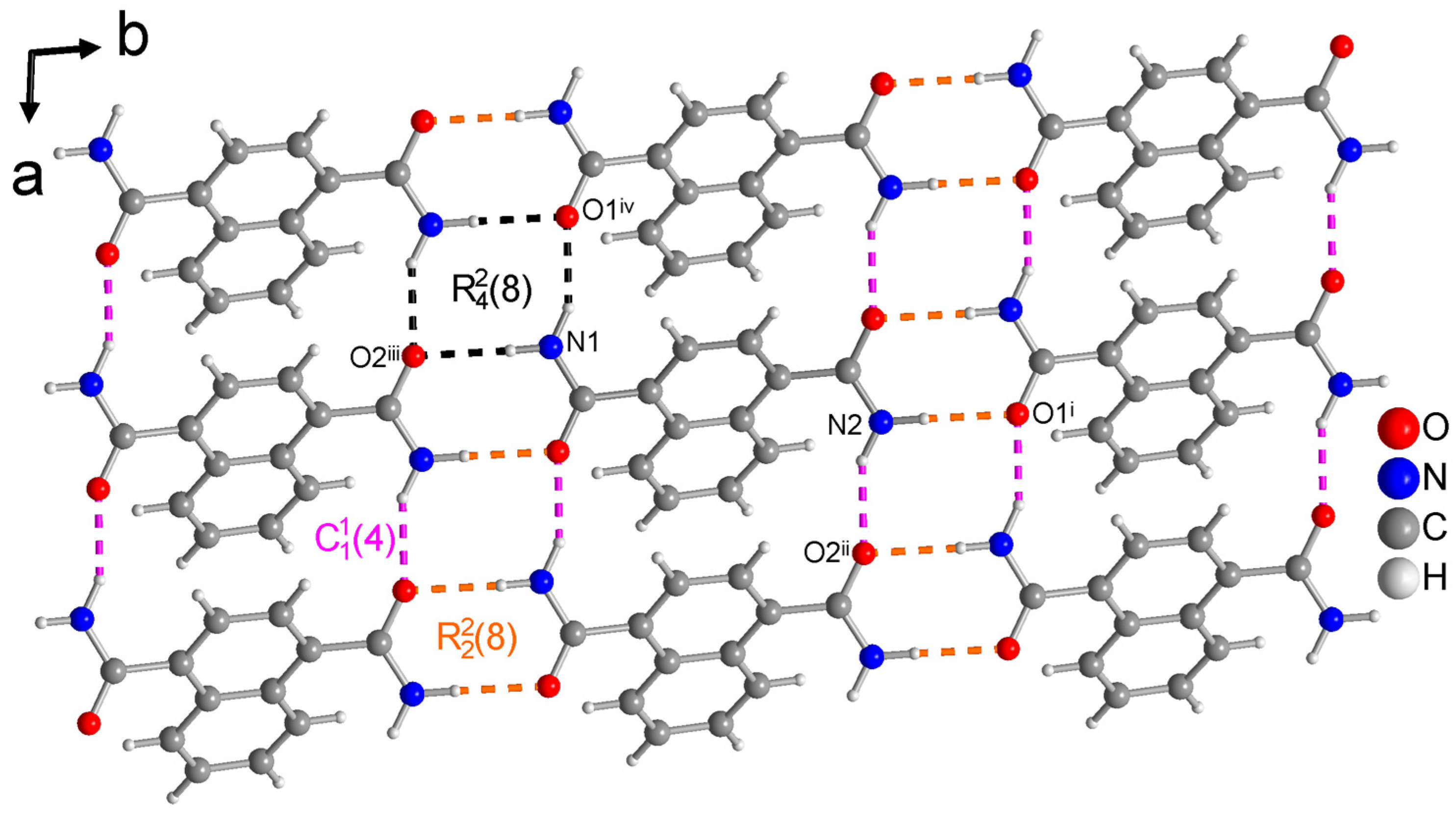
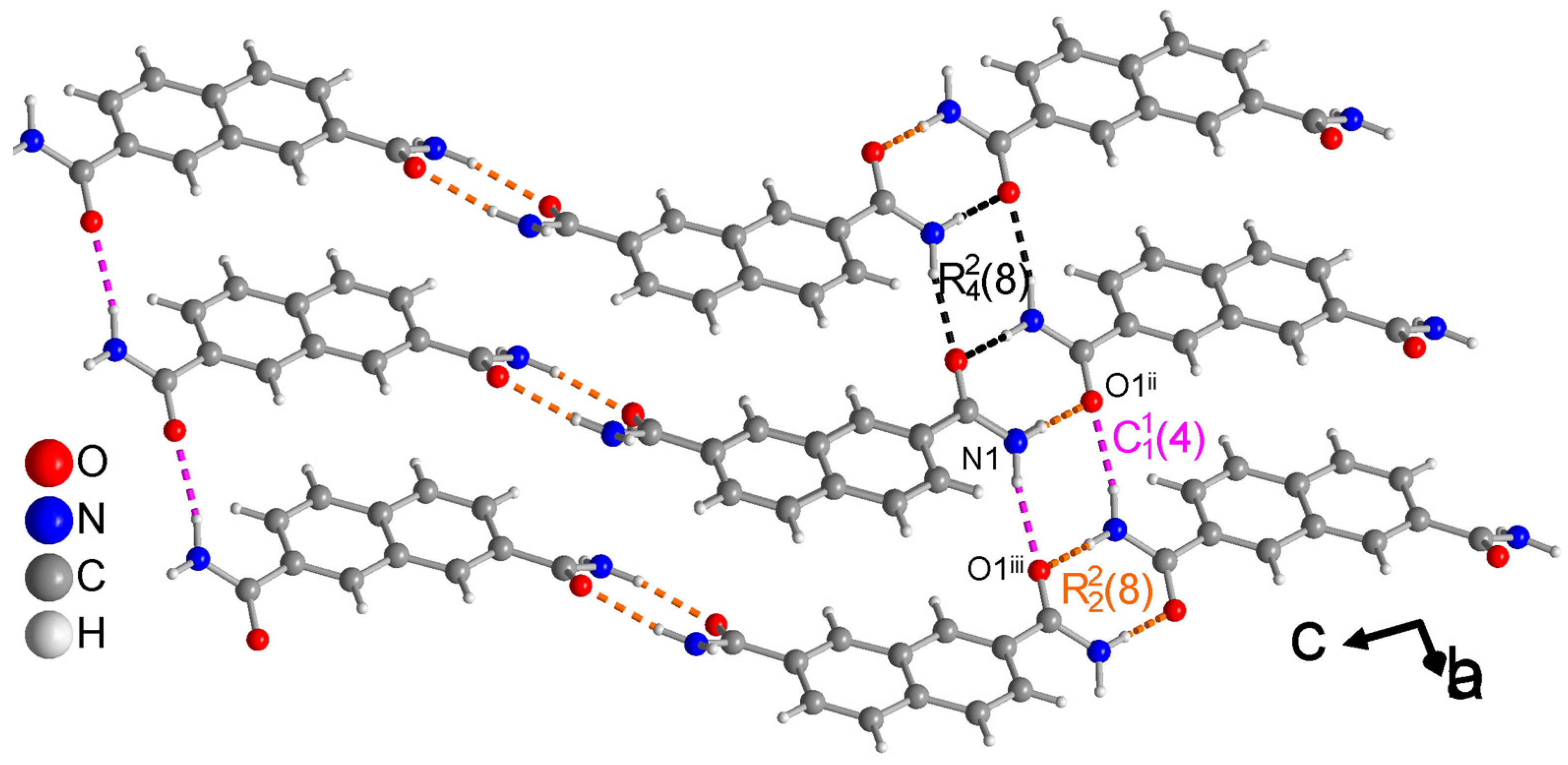
3.1.1. Hydrogen Bonding in 1,4-NDA
3.1.2. Hydrogen Bonding in 2,6-NDA
3.1.3. Hydrogen Bonding in 2,7-NDA
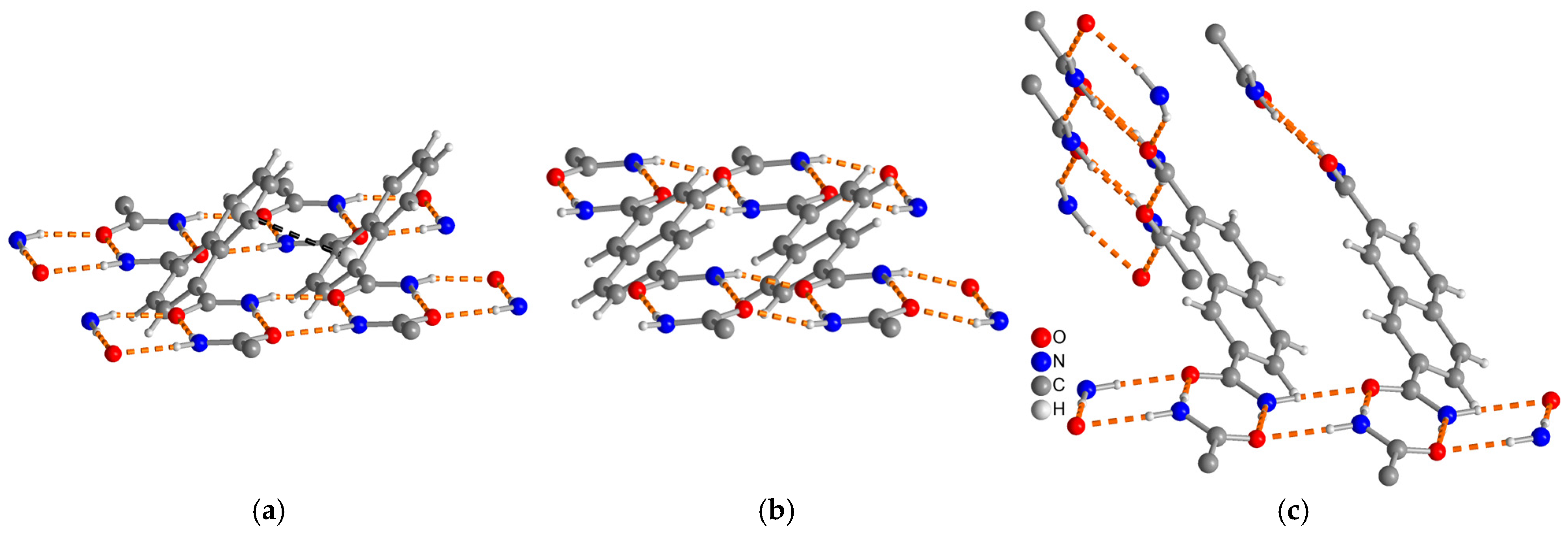
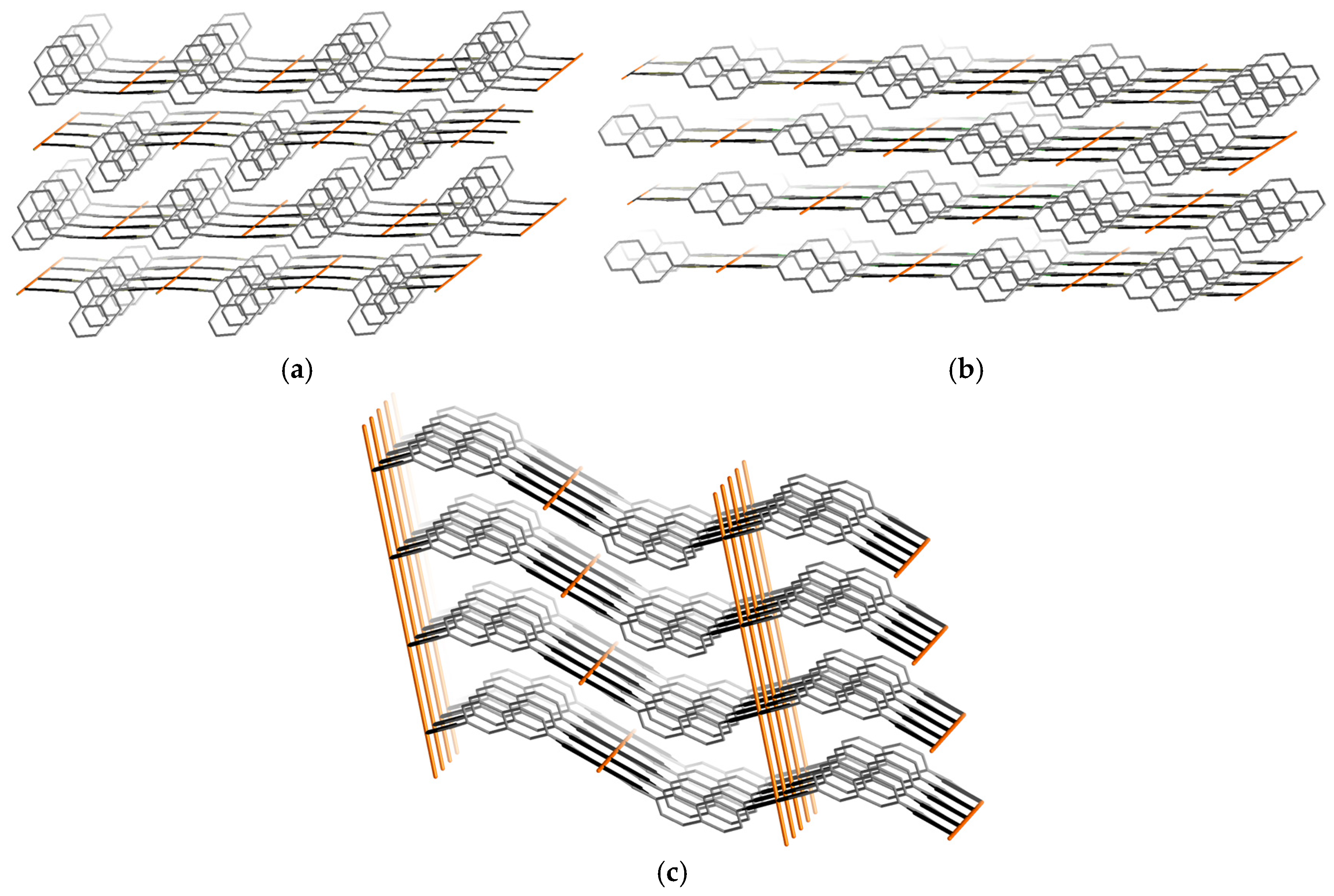
3.1.4. Comparative Topological Presentation
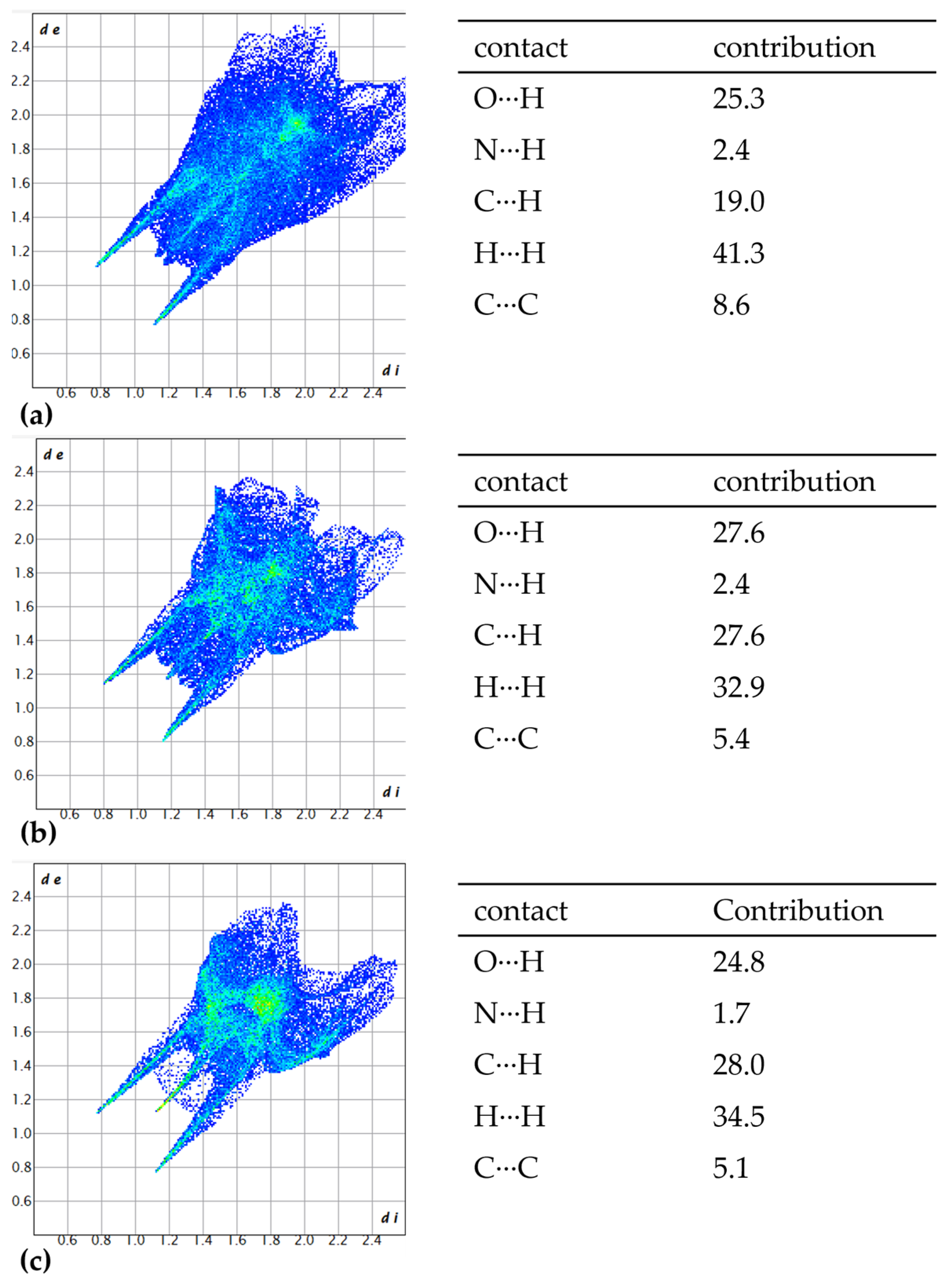
3.2. Hirshfeld Analysis
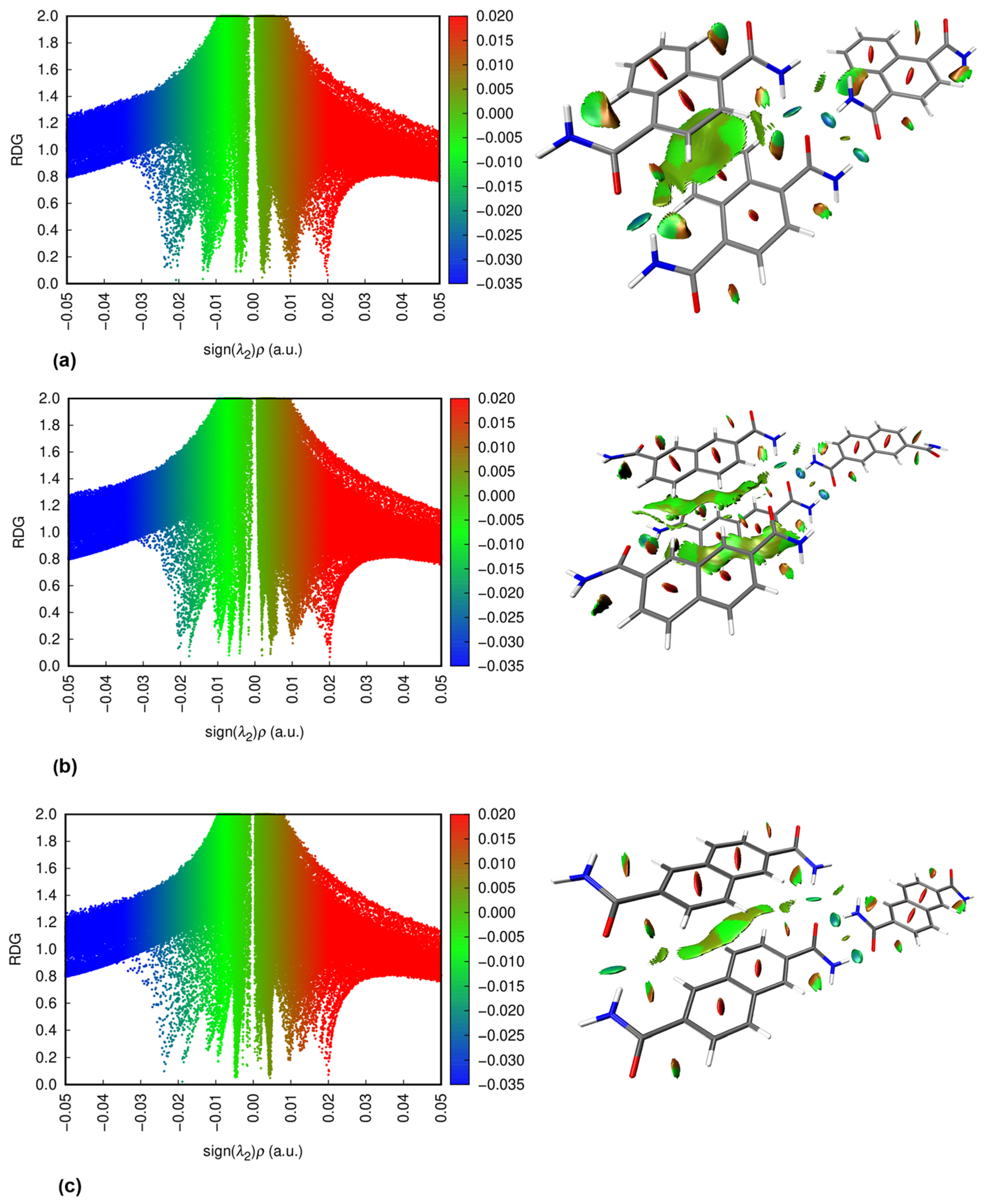
3.3. NCI Plots
3.4. Thermogravimetric Analysis and Melting Points
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme (Influence of configuration on the action of enzymes). Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. II. The one-electron bond and the three-electron bond. J. Am. Chem. Soc. 1931, 53, 3225–3237. [Google Scholar] [CrossRef]
- Lugger, S.J.D.; Houben, S.J.A.; Foelen, Y.; Debije, M.G.; Schenning, A.P.; Mulder, D.J. Hydrogen-bonded supramolecular liquid crystal polymers: Smart materials with stimuli-responsive, self-healing, and recyclable properties. Chem. Rev. 2021, 122, 4946–4975. [Google Scholar] [CrossRef] [PubMed]
- Stang, P.J.; Olenyuk, B. Self-assembly, symmetry, and molecular architecture: Coordination as the motif in the rational design of supramolecular metallacyclic polygons and polyhedra. Acc. Chem. Res. 1997, 30, 502–518. [Google Scholar] [CrossRef]
- Keller, E.F. Self-organization, self-assembly, and the origin of life. In Mapping the Future of Biology: Evolving Concepts and Theories; Barberousse, A., Morange, M., Pradeu, T., Eds.; Springer: Dordrecht, The Netherlands, 2009; Volume 266, pp. 131–140. [Google Scholar] [CrossRef]
- Palma, C.-A.; Cecchini, M.; Samorì, P. Predicting self-assembly: From empirism to determinism. Chem. Soc. Rev. 2012, 41, 3713–3730. [Google Scholar] [CrossRef]
- Dijkstra, M.; Luijten, E. From predictive modelling to machine learning and reverse engineering of colloidal self-assembly. Nat. Mater. 2021, 20, 762–773. [Google Scholar] [CrossRef]
- Li, F.; Han, J.; Cao, T.; Lam, W.; Fan, B.; Tang, W.; Chen, S.; Fok, K.L.; Li, L. Design of self-assembly dipeptide hydrogels and machine learning via their chemical features. Proc. Natl. Acad. Sci. USA 2019, 116, 11259–11264. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, R.; Yan, X.; Fan, K. Machine learning facilitating the rational design of nanozymes. J. Mater. Chem. B 2023, 11, 6466–6477. [Google Scholar] [CrossRef]
- Gardin, A.; Perego, C.; Doni, G.; Pavan, G.M. Classifying soft self-assembled materials via unsupervised machine learning of defects. Commun. Chem. 2022, 5, 82. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Shen, J.; Gao, C.; van der Bruggen, B. The potential of Kevlar aramid nanofiber composite membranes. J. Mater. Chem. A 2020, 8, 7548–7568. [Google Scholar] [CrossRef]
- Prasad, V.V.; Talupula, S. A review on reinforcement of basalt and aramid (Kevlar 129) fibers. Mater. Today Proc. 2018, 5, 5993–5998. [Google Scholar] [CrossRef]
- Majed, A.A.; Al-Duhaidahawi, D.; Omran, H.A.; Abbas, S.; Abid, D.S.; Hmood, A.Y. Synthesis, molecular docking of new amide thiazolidine derived from isoniazid and studying their biological activity against cancer cells. J. Biomol. Struct. Dyn. 2024, 42, 13485–13496. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tang, H.; Xu, Y.; Guo, J.; Zhao, X.; Meng, Q.; Xiao, J. Design, synthesis, bioactivity evaluation, crystal structures, and in silico studies of new α-amino amide derivatives as potential histone deacetylase 6 inhibitors. Molecules 2022, 27, 3335. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: Role and strength. Encycl. Life Sci. 2010, 1, 1–6. [Google Scholar] [CrossRef]
- Dong, Y.; Yao, C.; Zhu, Y.; Yang, L.; Luo, D.; Yang, D. DNA functional materials assembled from branched DNA: Design, synthesis, and applications. Chem. Rev. 2020, 120, 9420–9481. [Google Scholar] [CrossRef]
- Hutchins, K.M. Functional materials based on molecules with hydrogen-bonding ability: Applications to drug co-crystals and polymer complexes. R. Soc. Open Sci. 2018, 5, 180564. [Google Scholar] [CrossRef]
- Deng, L.; Deng, S.-S.; Pan, S.-Y.; Wu, Z.-Y.; Hu, Y.-Y.; Li, K.; Zhou, Y.; Li, J.-T.; Huang, L.; Sun, S.-G. Multivalent amide-hydrogen-bond supramolecular binder enhances the cyclic stability of silicon-based anodes for lithium-ion batteries. ACS Appl. Mater. Interfaces 2021, 13, 22567–22576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Huang, J.; Wang, K.; Huang, W. Recent structural engineering of polymer semiconductors incorporating hydrogen bonds. Adv. Mater. 2022, 34, 2110639. [Google Scholar] [CrossRef]
- Galuska, L.A.; Ocheje, M.U.; Ahmad, Z.C.; Rondeau-Gagné, S.; Gu, X. Elucidating the role of hydrogen bonds for improved mechanical properties in a high-performance semiconducting polymer. Chem. Mat. 2022, 34, 2259–2267. [Google Scholar] [CrossRef]
- Yu, X.; Li, C.; Gao, C.; Zhang, X.; Zhang, G.; Zhang, D. Incorporation of hydrogen-bonding units into polymeric semiconductors toward boosting charge mobility, intrinsic stretchability, and self-healing ability. SmartMat 2021, 2, 347–366. [Google Scholar] [CrossRef]
- Bojarska, J.; Łyczko, K.; Breza, M.; Mieczkowski, A. Recurrent Supramolecular Patterns in a Series of Salts of Heterocyclic Polyamines and Heterocyclic Dicarboxylic Acids: Synthesis, Single-Crystal X-ray Structure, Hirshfeld Surface Analysis, Energy Framework, and Quantum Chemical Calculations. Crystals 2024, 14, 733. [Google Scholar] [CrossRef]
- Boeré, R.T. Hydrogen Bonds Stabilize Chloroselenite Anions: Crystal Structure of a New Salt and Donor-Acceptor Bonding to SeO2. Molecules 2023, 28, 7489. [Google Scholar] [CrossRef] [PubMed]
- Haro-Saltos, L.A.; Bonilla-Valladares, P.M.; Alcívar-León, C.D. Theoretical Study of Intermolecular Interactions in Benzopyrans Substituted with Polyhaloalkyl Groups. Chem. Proc. 2024, 16, 32. [Google Scholar] [CrossRef]
- Leiserowitz, L.; Schmidt, G.M. Molecular packing modes. Part III. Primary amides. J. Chem. Soc. A 1969, 2372–2382. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C. Aggregate structures of carboxylic acids and amides. Isr. J. Chem. 1985, 25, 312–319. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Leiserowitz, L. Molecular packing modes. Carboxylic acids. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1976, 32, 775–802. [Google Scholar] [CrossRef]
- Lima, P.S.V.; Weimer, G.H.; Oliveira, L.P.; Souza, H.D.S.; Fiss, G.F.; Bonacorso, H.G.; Martins, M.A.P. Mesoionic compounds: The role of intermolecular interactions in their crystalline design. CrystEngComm 2023, 25, 4976–4991. [Google Scholar] [CrossRef]
- Pagliari, A.B.; Meyer, A.R.; Solner, V.B.; Rosa, J.M.L.; Hörner, M.; Bonacorso, H.G.; Zanatta, N.; Martins, M.A.P. Effect of hydrogen bonds and π⋯π interactions on the crystallization of phenyl-perfluorophenyl amides: Understanding the self-organization of a cocrystal. CrystEngComm 2022, 24, 5348–5363. [Google Scholar] [CrossRef]
- Mohabbat, A.; Salama, J.; Seiffert, P.; Boldog, I.; Janiak, C. Single-Crystal Structure Analysis of Dicarboxamides: Impact of Heteroatoms on Hydrogen Bonding of Carboxamide Groups. Crystals 2024, 14, 811. [Google Scholar] [CrossRef]
- Shishkina, S.V.; Konovalova, I.S.; Kovalenko, S.M.; Trostianko, P.V.; Geleverya, A.O.; Bunyatyan, N.D. Hydrogen bonding vs. stacking interaction in the crystals of the simplest coumarin derivatives: A study from the energetic viewpoint. CrystEngComm 2019, 21, 6945–6957. [Google Scholar] [CrossRef]
- Niu, D.; Jiang, Y.; Ji, L.; Ouyang, G.; Liu, M. Self-assembly through coordination and π-stacking: Controlled switching of circularly polarized luminescence. Angew. Chem. 2019, 131, 6007–6011. [Google Scholar] [CrossRef]
- Li, G.-L.; Zhuo, Z.; Wang, B.; Cao, X.-L.; Su, H.-F.; Wang, W.; Huang, Y.-G.; Hong, M. Constructing π-stacked supramolecular cage based hierarchical self-assemblies via π⋯π stacking and hydrogen bonding. J. Am. Chem. Soc. 2021, 143, 10920–10929. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.-R.; Wang, Y.; Cui, P.-F.; Xing, L.; Lee, J.; Kim, D.; Jiang, H.-L.; Oh, Y.-K. Applications of π-π stacking interactions in the design of drug-delivery systems. J. Control. Release 2019, 294, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-W.; Mohamed, M.G.; Zhu, C.-Y.; Kuo, S.-W. Functional supramolecular polypeptides involving π–π stacking and strong hydrogen-bonding interactions: A conformation study toward carbon nanotubes (CNTs) dispersion. Macromolecules 2016, 49, 5374–5385. [Google Scholar] [CrossRef]
- Yagai, S. Supramolecularly engineered functional π-assemblies based on complementary hydrogen-bonding interactions. Bull. Chem. Soc. Jpn. 2015, 88, 28–58. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Merino, D.H.; Weng, W.; Seppala, J.; Colquhoun, H.M.; Hayes, W.; Mackay, M.E.; Hamley, I.W.; Rowan, S.J. A healable supramolecular polymer blend based on aromatic π−π stacking and hydrogen-bonding interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Calhorda, M.J. Weak hydrogen bonds: Theoretical studies. Chem. Commun. 2000, 801–809. [Google Scholar] [CrossRef]
- Kaur, S.; Grover, P.; Wetmore, S.D.; Sharma, P. Role of stacking interactions in the stability of primitive genetics: A quantum chemical view. J. Chem. Inf. Model. 2021, 61, 4321–4330. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, A.M.; Acharya, A.; Titelmayer, A.V.; Mamontova, A.V.; Bravaya, K.B.; Kolomeisky, A.B.; Lukyanov, K.A.; Krylov, A.I. Turning on and off photoinduced electron transfer in fluorescent proteins by π-stacking, halide binding, and Tyr145 mutations. J. Am. Chem. Soc. 2016, 138, 4807–4817. [Google Scholar] [CrossRef] [PubMed]
- Ardejani, M.S.; Powers, E.T.; Kelly, J.W. Using cooperatively folded peptides to measure interaction energies and conformational propensities. Acc. Chem. Res. 2017, 50, 1875–1882. [Google Scholar] [CrossRef]
- Beall, E.; Ulku, S.; Liu, C.; Wierzbinski, E.; Zhang, Y.; Bae, Y.; Zhang, P.; Achim, C.; Beratan, D.N.; Waldeck, D.H. Effects of the backbone and chemical linker on the molecular conductance of nucleic acid duplexes. J. Am. Chem. Soc. 2017, 139, 6726–6735. [Google Scholar] [CrossRef]
- Heller, B.A.; Gindt, Y.M. A biochemical study of noncovalent forces in proteins using phycocyanin from Spirulina. J. Chem. Educ. 2000, 77, 1458. [Google Scholar] [CrossRef]
- Wang, S.; Guo, G.; Lu, X.; Ji, S.; Tan, G.; Gao, L. Facile soaking strategy toward simultaneously enhanced conductivity and toughness of self-healing composite hydrogels through constructing multiple noncovalent interactions. ACS Appl. Mater. Interfaces. 2018, 10, 19133–19142. [Google Scholar] [CrossRef]
- Meli, M.; Engel, H.; Laor, D.; Gazit, E.; Colombo, G. Mechanisms of metabolite amyloid formation: Computational studies for drug design against metabolic disorders. ACS Med. Chem. Lett. 2019, 10, 666–670. [Google Scholar] [CrossRef]
- Yang, D.; Gao, S.; Fang, Y.; Lin, X.; Jin, X.; Wang, X.; Ke, L.; Shi, K. The π–π stacking-guided supramolecular self-assembly of nanomedicine for effective delivery of antineoplastic therapies. Nanomed. 2018, 13, 3159–3177. [Google Scholar] [CrossRef]
- Wei, X.; Wang, Y.; Xiong, X.; Guo, X.; Zhang, L.; Zhang, X.; Zhou, S. Codelivery of a π–π stacked dual anticancer drug combination with nanocarriers for overcoming multidrug resistance and tumor metastasis. Adv. Funct. Mater. 2016, 26, 8266–8280. [Google Scholar] [CrossRef]
- Li, P.; Ryder, M.R.; Stoddart, J.F. Hydrogen-bonded organic frameworks: A rising class of porous molecular materials. Acc. Mater. 2020, 1, 77–87. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, Y.; Xiang, S.; Chen, B. Exploring multifunctional hydrogen-bonded organic framework materials. Acc. Chem. Res. 2022, 55, 3752–3766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, C.; Li, C.; Zhang, T.; Jiang, Y.; Cheng, X.; Wang, K.; Ma, C.; Li, Y. Topological hydrogen-bonded organic frameworks (HOFs) and their electronic applications in sensor, memristor, and neuromorphic computing. Appl. Phys. Rev. 2024, 11, 031303. [Google Scholar] [CrossRef]
- Xiong, Z.; Xiang, S.; Lv, Y.; Chen, B.; Zhang, Z. Hydrogen-Bonded Organic Frameworks as an Appealing Platform for Luminescent Sensing. Adv. Funct. Mater. 2024, 34, 2403635. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, B.; Chen, L.; Liu, H.; Hu, Y.; Qiao, S. Hydrogen-bonded organic frameworks: Design, applications, and prospects. Mater. Adv. 2022, 3, 3680–3708. [Google Scholar] [CrossRef]
- Ueda, A.; Hatakeyama, A.; Enomoto, M.; Kumai, R.; Murakami, Y.; Mori, H. Modulation of a molecular π-electron system in a purely organic conductor that shows hydrogen-bond-dynamics-based switching of conductivity and magnetism. Chem. Eur. J. 2015, 21, 15020–15028. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.M.; Nishihara, S.; Akutagawa, T.; Noro, S.; Nakamura, T. Design of a Magnetic Bistability Molecular System Constructed by H-Bonding and π···π-Stacking Interactions. Inorg. Chem. 2006, 45, 2229–2234. [Google Scholar] [CrossRef]
- Ueda, A.; Yamada, S.; Isono, T.; Kamo, H.; Nakao, A.; Kumai, R.; Nakao, H.; Murakami, Y.; Yamamoto, K.; Nishio, Y. Hydrogen-bond-dynamics-based switching of conductivity and magnetism: A phase transition caused by deuterium and electron transfer in a hydrogen-bonded purely organic conductor crystal. J. Am. Chem. Soc. 2014, 136, 12184–12192. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond, Version 5.0.0. Crystal and Molecular Structure Visualization, Crystal Impact. K. Brandenburg & H. Putz Gbr: Bonn, Germany, 1997–2023.
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. Available online: https://gaussian.com/citation/ (accessed on 17 March 2025).
- Lu, T.; Chen, Q. Interaction region indicator: A simple real space function clearly revealing both chemical bonds and weak interactions. Chem.-Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Meng, M.; Wang, G.Y.; Feng, P.; Liu, C.Y. Optically probing the localized to delocalized transition in Mo2–Mo2 mixed-valence systems. Chem. Commun. 2017, 53, 3030–3033. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π Hydrogen Bond in Chemistry. Conformation, Supramolecules, Optical Resolution and Interactions Involving Carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873. [Google Scholar] [CrossRef]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π Hydrogen Bonds in Organic and Organometallic Chemistry. CrystEngComm 2009, 11, 1757. [Google Scholar] [CrossRef]
- Popa, M.M.; Răducă, M.; Man, I.C.; Dumitrascu, F. The Role of Hydrogen Bond Interactions in Crystal Formation of Pyrrolo-Azines Alcohols. Crystals 2025, 15, 78. [Google Scholar] [CrossRef]
- Castro, J.; Ferraro, V.; Bortoluzzi, M. Single-crystal X-ray structure determination of tris (pyrazol-1-yl) methane triphenylphosphine copper (I) tetrafluoroborate, Hirshfeld surface analysis and DFT calculations. Crystals 2024, 14, 162. [Google Scholar] [CrossRef]
- Gomes, L.R.; Low, J.N.; Pinheiro, A.C.; Wardell, J.L. Structural Study of N-(1, 3-Benzothiazol-2-yl)-4-Halobenzenesulfonylhydrazides: Hirshfeld Surface Analysis and PIXEL Calculations. Crystals 2024, 14, 330. [Google Scholar] [CrossRef]
- El Hafi, M.; Anouar, E.H.; Lahmidi, S.; Boulhaoua, M.; Loubidi, M.; Alanazi, A.S.; Filali, I.; Hefnawy, M.; El Ghayati, L.; Mague, J.T. Synthesis of New Pyrazolo [3, 4-d] pyrimidine Derivatives: NMR Spectroscopic Characterization, X-Ray, Hirshfeld Surface Analysis, DFT, Molecular Docking, and Antiproliferative Activity Investigations. Molecules 2024, 29, 5020. [Google Scholar] [CrossRef] [PubMed]
- Al-Resayes, S.I.; Azam, M.; Trzesowska-Kruszynska, A.; Kruszynski, R.; Soliman, S.M.; Mohapatra, R.K.; Khan, Z. Structural and theoretical investigations, Hirshfeld surface analyses, and cytotoxicity of a naphthalene-based chiral compound. ACS Omega 2020, 5, 27227–27234. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, A.; Oliver, C.L.; Platero-Prats, A.E.; Laurila, E.; Öhrström, L. Crystal structures and hydrogen bond analysis of five amino acid conjugates of terephthalic and benzene-1, 2, 3-tricarboxylic acids. CrystEngComm 2014, 16, 8243–8251. [Google Scholar] [CrossRef]
- Hachuła, B.; Polasz, A.; Książek, M.; Kusz, J.; Kozik, V.; Matussek, M.; Pisarski, W. Insight into hydrogen bonding of terephthalamides with amino acids: Synthesis, structural and spectroscopic investigations. Tetrahedron 2017, 73, 2901–2912. [Google Scholar] [CrossRef]
- Martin, A.D.; Hartlieb, K.J.; Sobolev, A.N.; Raston, C.L. Hirshfeld surface analysis of substituted phenols. Cryst. Growth Des. 2010, 10, 5302–5306. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Boto, R.A.; Izquierdo-Ruiz, F.; Reva, I.; Woller, T.; Alonso, M. A benchmark for the non-covalent interaction (NCI) index or… is it really all in the geometry? Theor. Chem. Acc. 2016, 135, 242. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A. Nature of bonding and cooperativity in linear DMSO clusters: A DFT, AIM and NCI analysis. J. Mol. Graph. 2018, 81, 50–59. [Google Scholar] [CrossRef]
- Heller, E.; Schmidt, G.M. Topochemistry. Part XXXIII. The Solid-State Photochemistry of Some Anthracene Derivatives. Isr. J. Chem. 1971, 9, 449–462. [Google Scholar] [CrossRef]
- Ossowski, J.; Wächter, T.; Silies, L.; Kind, M.; Noworolska, A.; Blobner, F.; Gnatek, D.; Rysz, J.; Bolte, M.; Feulner, P. Thiolate versus selenolate: Structure, stability, and charge transfer properties. ACS Nano 2015, 9, 4508–4526. [Google Scholar] [CrossRef]
- Cozzi, F.; Bacchi, S.; Filippini, G.; Pilati, T.; Gavezzotti, A. Competition between hydrogen bonding and arene–perfluoroarene stacking. X-Ray diffraction and molecular simulation on 5,6,7,8-tetrafluoro-2-naphthoic acid and 5,6,7,8-tetrafluoro-2-naphthamide crystals. CrystEngComm 2009, 11, 1122–1127. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1,4-NDA | 2,6-NDA | 2,7-NDA | |
|---|---|---|---|
| empirical formula | C12H10N2O2 | C12H10N2O2 | C12H10N2O2 |
| mol wt (g mol–1) | 214.22 | 214.22 | 214.22 |
| temperature (K) | 150 | 150 | 150 |
| crystal system | triclinic | triclinic | monoclinic |
| space group | C2/c | ||
| a (Å) | 5.0306 (1) | 5.0058 (3) | 6.9974 (2) |
| b (Å) | 9.9799 (3) | 7.2347 (7) | 7.2089 (2) |
| c (Å) | 10.0706 (3) | 7.4349 (7) | 19.4417 (5) |
| α (deg) | 98.620 (2) | 68.216 (9) | 90.00 |
| β (deg) | 92.433 (2) | 82.306 (7) | 98.110 (2) |
| γ (deg) | 96.459 (2) | 80.076 (7) | 90.00 |
| Volume, V (Å3) | 495.80 (2) | 245.57 (4) | 970.90 (5) |
| Z, Z′ | Z = 2, Z′ = 1 | Z = 1, Z′ = 0.5 | Z = 4, Z′ = 0.5 |
| Dcalc (g/cm3) | 1.435 | 1.449 | 1.466 |
| μ (mm−1) | 0.823 | 0.830 | 0.840 |
| F(000) | 224 | 112 | 448 |
| crystal size [mm3] | 0.65 × 0.05 × 0.02 | 0.13 × 0.07 × 0.04 | 0.13 × 0.07 × 0.05 |
| wavelength (Å) | 1.54184 | 1.54184 | 1.54184 |
| No. of unique reflections | 2112 | 938 | 1042 |
| No. of total reflections | 19,080 | 2180 | 5554 |
| No. of parameters | 161 | 73 | 82 |
| Rint | 0.0509 | 0.0304 | 0.0245 |
| R1[F2 > 2σ(F2)] (a) | 0.0486 | 0.0539 | 0.0358 |
| wR1[F2 > 2σ(F2)] (a) | 0.1438 | 0.1547 | 0.1006 |
| R2, wR2(F2) [all data] (a) | 0.0535, 0.1492 | 0.0609, 0.1604 | 0.0414, 0.1059 |
| S [all data] (a) | 1.045 | 1.134 | 1.095 |
| Δρmax, Δρmin (e·Å−3) (b) | 0.320, −0.215 | 0.279, −0.207 | 0.158, −0.193 |
| CCDC no. | 2431507 | 2431506 | 2431508 |
| D—H···A(a) (a) | D—H [ Å] | H···A [ Å] | D···A [ Å] | D—H···A [deg] | Bond Energy [kcal/mol] |
|---|---|---|---|---|---|
| N1—H1A···O2 iii | 0.90 (2) | 2.07 (2) | 2.9601 (16) | 170.8 (17) | −16 |
| N1—H1B···O1 iv | 0.93 (2) | 1.96 (2) | 2.8105 (16) | 152.4 (18) | −17 |
| N2—H2A···O1 i | 0.92 (2) | 2.01 (2) | 2.9255 (16) | 177.5 (16) | −16 |
| N2—H2B···O2 ii | 0.92 (2) | 1.98 (2) | 2.8386 (16) | 153.2 (19) | −19 |
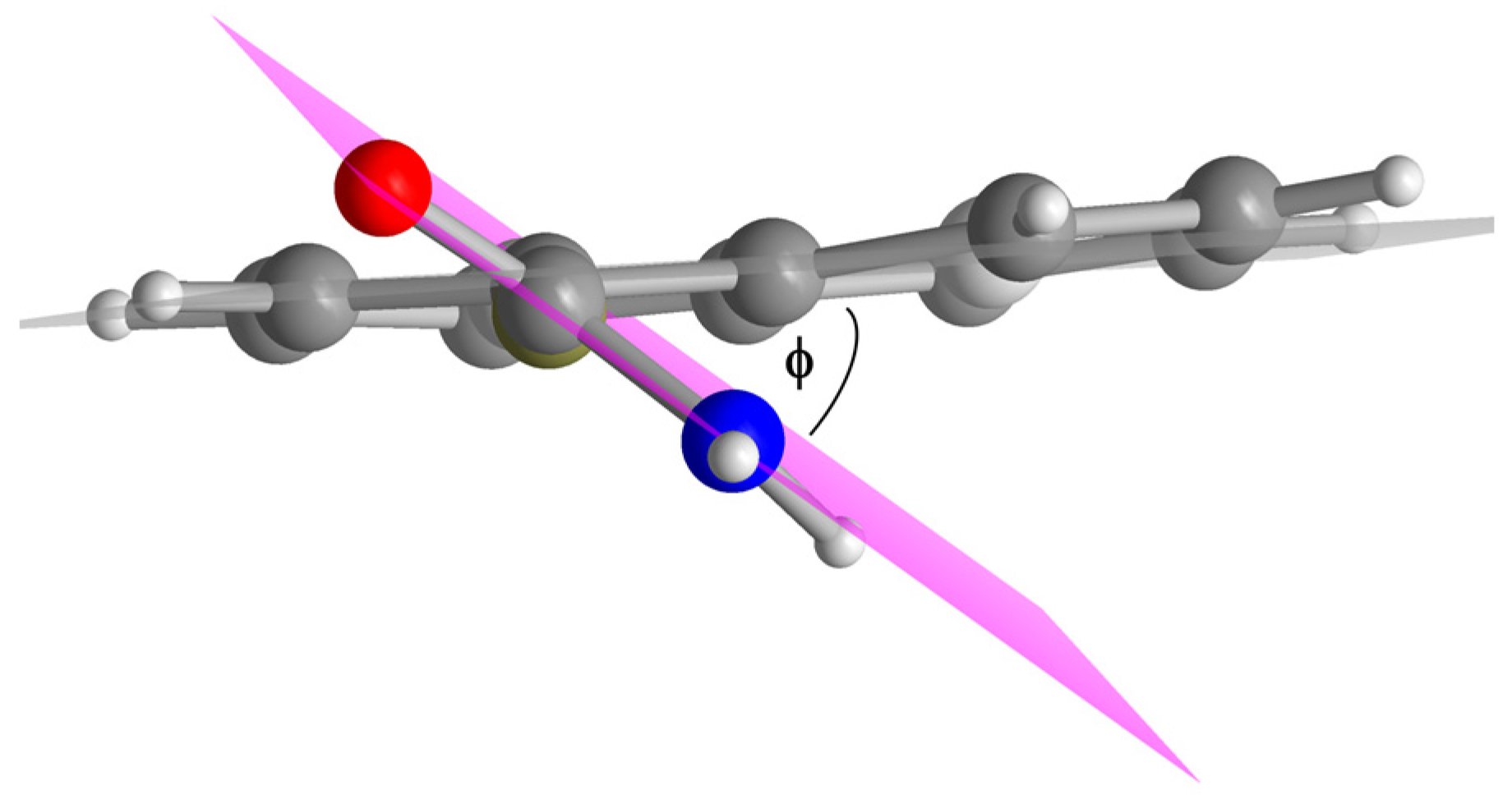
| Compound | Dihedral Angle ϕ [°] |
|---|---|
| 1,4-NDA | 40 |
| 2,6-NDA | 29 |
| 2,7-NDA | 22 |
| D—H···A(a) (a) | D—H [Å] | H···A [Å] | D···A [Å] | D—H···A [°] | Bond Energy [kcal/mol] |
|---|---|---|---|---|---|
| N1—H1A···O1 ii | 0.89 (3) | 2.04 (3) | 2.864 (2) | 154 (2) | −16 |
| N1—H1B···O1 iii | 0.88 (3) | 2.07 (3) | 2.942 (2) | 173 (2) | −16 |
| D—H···A (a) | D—H [ Å] | H···A [ Å] | D···A [ Å] | D—H···A [deg] | Bond Energy [kcal/mol] |
|---|---|---|---|---|---|
| N1—H1A···O1 ii | 0.906 (18) | 1.998 (19) | 2.8983 (14) | 171.9 (15) | −19 |
| N1—H1B···O1 iii | 0.924 (18) | 2.074 (18) | 2.9133 (13) | 150.4 (14) | −14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohabbat, A.; Boldog, I.; Sohi, T.H.H.; Reistel, N.; Seiffert, P.; Janiak, C. Hydrogen-Bonded Ladder Motifs in Naphthalene Dicarboxamides: Influence of Linear vs. Angular Amide Orientation. Crystals 2025, 15, 406. https://doi.org/10.3390/cryst15050406
Mohabbat A, Boldog I, Sohi THH, Reistel N, Seiffert P, Janiak C. Hydrogen-Bonded Ladder Motifs in Naphthalene Dicarboxamides: Influence of Linear vs. Angular Amide Orientation. Crystals. 2025; 15(5):406. https://doi.org/10.3390/cryst15050406
Chicago/Turabian StyleMohabbat, Abdulrahman, István Boldog, Takin Haj Hassani Sohi, Nils Reistel, Philipp Seiffert, and Christoph Janiak. 2025. "Hydrogen-Bonded Ladder Motifs in Naphthalene Dicarboxamides: Influence of Linear vs. Angular Amide Orientation" Crystals 15, no. 5: 406. https://doi.org/10.3390/cryst15050406
APA StyleMohabbat, A., Boldog, I., Sohi, T. H. H., Reistel, N., Seiffert, P., & Janiak, C. (2025). Hydrogen-Bonded Ladder Motifs in Naphthalene Dicarboxamides: Influence of Linear vs. Angular Amide Orientation. Crystals, 15(5), 406. https://doi.org/10.3390/cryst15050406