Abstract
In this review, we present an overview on metal amidoboranes, which have recently been considered as hydrogen storage materials for fueling of the low temperature fuel cells. We focus on amidoborane salts containing only metal cations and amidoborate anions. During the last decades, 19 new compounds from this group were described in the literature. We provide a summary of various physical and chemical properties of amidoborane compounds reported up to date.
1. Introduction
Amidoboranes, also named as amidotrihydroborates (MNH2BH3, MAB), constitute a constantly growing family of hydrogen-rich ammonia borane [1,2,3,4] derivatives. Amidoboranes comprise metal cations and monovalent amidoborate anions [NH2BH3−, AB−]. Known for nearly 80 years, they were recently rediscovered and are now the subject of intense research [5] (Figure 1).
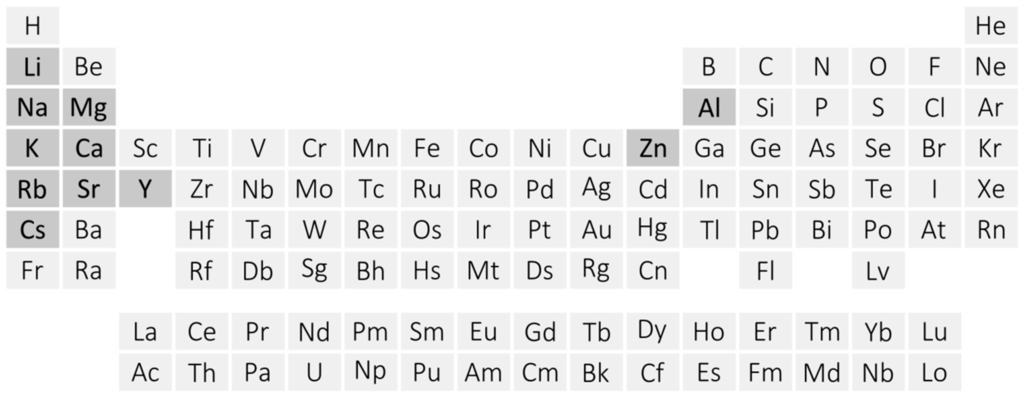
Figure 1.
Periodic table showing metals forming mono- or bimetallic amidoborane salts (dark grey).
Ammonia borane, the parent compound of amidoborane salts, was synthesized and characterized for the first time by Shore and Perry in 1955 [1]. Sixty years after its first synthesis, ammonia borane is still the subject of intensive experimental and theoretical studies [1,2,3,4,6,7,8,9], and has been the subject of several reviews [10,11]. Ammonia borane has one of the highest gravimetrical hydrogen capacities (19.6 wt%) among all known chemical compounds [1]. Thus, it is a very convenient starting material for preparation of new hydrogen storage materials [12].
The first amidoborane salt, sodium amidoborane (NaAB), was synthesized and described by Schlesinger and Burg in 1938 [13]. For almost half a century, there was no interest in the chemistry of amidoboranes. In the late 1980s and early 1990s, several amidoborane compounds were investigated in the group of Shore, namely: lithium amidoborane (LiAB), sodium amidoborane (NaAB), potassium amidoborane (KAB), magnesium amidoborane (Mg(AB)2) and zinc amidoborane (Zn(AB)2) [14,15,16]. In the mid-1990s, the possibility of using LiAB as reducing agent in organic chemistry was discussed by Myers et al. [17].
A huge acceleration of research focused on metal amidoboranes—and especially their hydrogen storage properties—has been observed during the last decade. Since 2008, a number of important papers and reports were released describing amidoboranes of: lithium, LiAB [18,19,20,21]; sodium, NaAB [18,21,22,23,24]; potassium, KAB [25]; rubidium, RbAB [26]; cesium, CsAB [26]; magnesium, Mg(AB)2 [27,28,29,30]; calcium, Ca(AB)2 [19,31,32]; strontium, Sr(AB)2 [33]; zinc, Zn(AB)2 [34]; aluminum, Al(AB)3 [35,36,37]; and yttrium, Y(AB)3 [38]. Several bimetallic amidoborane salts have been prepared, as well, i.e., lithium–sodium, LiNa(AB)2 [39]; lithium–aluminum, LiAl(AB)4 [35,40]; sodium–aluminum, NaAl(AB)4 [41]; sodium–magnesium, NaMg(AB)3 [42]; potassium–magnesium, KMg(AB)3 [43]; rubidium–magnesium, RbMg(AB)3 [43]; disodium–magnesium, Na2Mg(AB)4 [44]; and dipotassium–magnesium, K2Mg(AB)4 [44,45]. Amidoborane compounds containing metal centers coordinated with molecules of the solvent (which could not be later desorbed in post processing, e.g., THF complexes of europium amidoborane, Eu(AB)2 [15], ytterbium amidoborane, Yb(AB)2 [15], and calcium amidoborane, Ca(AB)2 [30]) have also been described. One can also find information about unsuccessful attempts of synthesis of other amidoboranes, e.g., those of titanium [19] and iron [46].
Apart from those, systems containing molecules of solvent trapped in the crystal structure of the compound are also known, e.g., amidoborane ammoniates of: lithium, LiAB∙NH3 [47]; magnesium, Mg(AB)2∙NH3 [48] and Mg(AB)2∙2NH3 [49]; calcium, Ca(AB)2∙NH3 [50] and Ca(AB)2∙2NH3 [51]; and aluminum, Al(AB)3∙NH3 [35,52]. The are also amidoborane hydrazinates of: lithium, LiAB∙N2H4 [53]; calcium, Ca(AB)2∙N2H4 [54]; and complex of lithium amidoborane with ammonia borane, LiAB∙AB [55]. Another group of amidoborane derivatives is that of compounds containing amidoborate anions with a hydrocarbon functional group, e.g., LiNMe2BH3 [56,57], LiNPr2BH3 [57], LiNH2B(C6F5)3 [58], NaNMe2BH3 [59], KNMe2BH3 [60], K(NMe2BH2NMe2BH3) [61], and many others, which have been described in the review by Stennett and Harder [62]. Complex compounds containing amidoborate anions and other hydrocarbon groups have also been synthesized [63,64,65,66,67].
Amidoboranes were subjects of several patent applications. Among them, the most important were probably the US patents of Torgersen et al. [68], Burrell et al. [69], and Balema et al. [70], which covered most of the known and probably synthesizable compositions.
There is also a number of theoretical papers on modeling of the properties and crystal structures of selected amidoboranes, e.g., electronic structure and dehydrogenation characteristics of LiAB [71,72,73]; crystal structure and dehydrogenation mechanism of LiAB [74,75], NaAB [74,75], KAB [75], LiNa(AB)2 [76] and Na2Mg(AB)4 [77]; crystal structure, and electronic and mechanical properties of Mg(AB)2, Ca(AB)2 and Sr(AB)2 [78]; spectroscopic properties of LiAB [79]; influence of homopolar dihydrogen bonding on hydrogen storage properties [80]; or supramolecular interactions [60].
In this review, we present a comprehensive summary of amidoborane salts containing only metal cations and amidoborate anions without any other constituents, substituents, solvent molecules, or additional functionalities. We compare their crystal structures, hydrogen storage properties, 11B NMR spectra, vibrational spectra, and other features. We analyze physicochemical properties of amidoborane salts as compared to those of ammonia borane to show an impact of introduction of different metal cations to the crystal structure in place of one acidic proton.
2. Crystal Structures of Metal Amidoboranes
Up till now, 19 amidoborane salts were reported (11 monometallic and eight bimetallic) [5,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,62]. Usually, amidoboranes are white or grayish solids. However, the color of the compound may strongly depend on the grain size of the material or even on the synthesis method. All amidoborane salts are highly sensitive against water and moisture, which cause hydrolysis to ammonia borane and respective metal hydroxide [23,81] or to boric acid [82]. Because of high reactivity against moisture and water, amidoborane compounds are typically stored and processed under inert atmosphere, mostly common argon, nitrogen or helium. Some of the compounds seem to be unstable at room temperature, thus they should be cooled down and stored at low temperature to preserve their properties [24].
All amidoborane salts exhibit properties typical of protonic–hydridic hydrogen storage materials because the amidoborate anions contain both the hydrogen atoms with partial positive charge (Hδ+ in NH2 groups) and those with partial negative charge (Hδ– in BH3 groups). Amidoborate anions differ from ammonia borane molecules by substitution of one protonic hydrogen atom in each molecule with a metal cation. The shortest M–N distances in amidoborane crystals (e.g., Li–N, 1.98 Å [18]; Na–N, 2.35 Å [18]; K–N, 3.10 Å [25]; Ca–N, 2.383 Å [19]; etc.) are obviously much longer than the H–N distances in ammonia borane molecule (N–H, 0.85 Å [3]); the M–N bonds in the former are usually quite ionic [18].
The parent ammonia borane crystallizes in the I4mm space group (HT form [3]) or in the Pmn21 (the LT form below 224 K [4]). Amidoborane salts are not isostructural with ammonia borane and crystallize in various lower symmetry space groups. In Table 1 and Table 2, we have listed crystallographic data available in the literature for a handful comparison of ammonia borane with mono- and bimetallic amidoborane salts, for which crystal structures have been solved or their diffraction patterns have been successfully indexed in a given unit cell.

Table 1.
Crystal structure information of various metal amidoboranes and ammonia borane. Crystallographic data obtained at room temperature apart from CsAB (263 K). Presented data are rounded and shown without standard deviation. For numbers with uncertainties, please refer to Section 5.
Table 2.
Interatomic distances in crystal structures of various amidoboranes salts (# mildly constrained). Note that the positions of H atoms cannot be determined with large accuracy and thus the BH and NH bond lengths usually have much larger errors than those indicated in brackets. Presented data are rounded and shown without standard deviation. For numbers with uncertainties please refer to Section 5. (For NaAB, data are combined from references [18] and [21] due to mild inconsistencies in both references.).
Amidoboranes form crystals where amidoborate anions coordinate metal cations with either (NH2) or/and (BH3) groups. The bond lengths in amidoborate anions (N–H, ca. 0.9–1.1 Å; B–H, ca. 1.1–1.2 Å; and B–N, ca. 1.56–1.58 Å) are comparable with the corresponding bond lengths in ammonia borane molecules (HT form: N–H, 0.85(7) Å; and B–H, 1.11(4) Å [3]) (Table 2). It is worth mentioning that the B–N bond length is usually slightly shorter (by 0.012–0.067 Å) in amidoborate anions than in ammonia borane molecules (HT form: 1.597(3) Å [3]). Volume of one formula unit of all amidoborane salts may be estimated with reasonable accuracy by simply adding the calculated volumes of amidoborate anion (ca. 60 Å3) and the respective metal cations.
Among all known amidoborane salts, only two pairs of compounds are monometallic α-LiAB and NaAB crystallizing in Pbca space group [18] and bimetallic Na2Mg(AB)4 and K2Mg(AB)4 crystallizing in I41/a space group [44,45].
Light alkali metal (α-LiAB, β-LiAB, NaAB, and KAB) amidoboranes crystallize in Pbca space group [18,20,21,25]. β-LiAB [20,21] and KAB [25], with Z = 16, have twice as big unit cell than α-LiAB and NaAB, both with Z = 8 [18]. RbAB and CsAB crystallize with Z = 4 in monoclinic space group P21/c and orthorhombic space group Pnam, respectively [26]. Ca(AB)2 and Sr(AB)2 both crystallize in C2 space group but they are not isostructural and their atomic coordinates are quite different from each other [19,43]. If space group assignment is correct, these would be the only amidoboranes that lack the inversion center (polar space group) and their unit cells exhibit an uncompensated dipole moment. In fact, the heavy-atom sublattice of Ca(AB)2 may be symmetrized to centrosymmetric C2/m at crude threshold. LiNa(AB)2 and NaAl(AB)4 have the lowest (triclinic) symmetry among all amidoborane salts [39,41]. Na2Mg(AB)4 and K2Mg(AB)4 form isostructural crystals with the highest symmetry among amidoborane salts (I41/a) [44,45]. Crystal structures of amidoborane salts listed above are presented in Figure 2 and Figure 3.
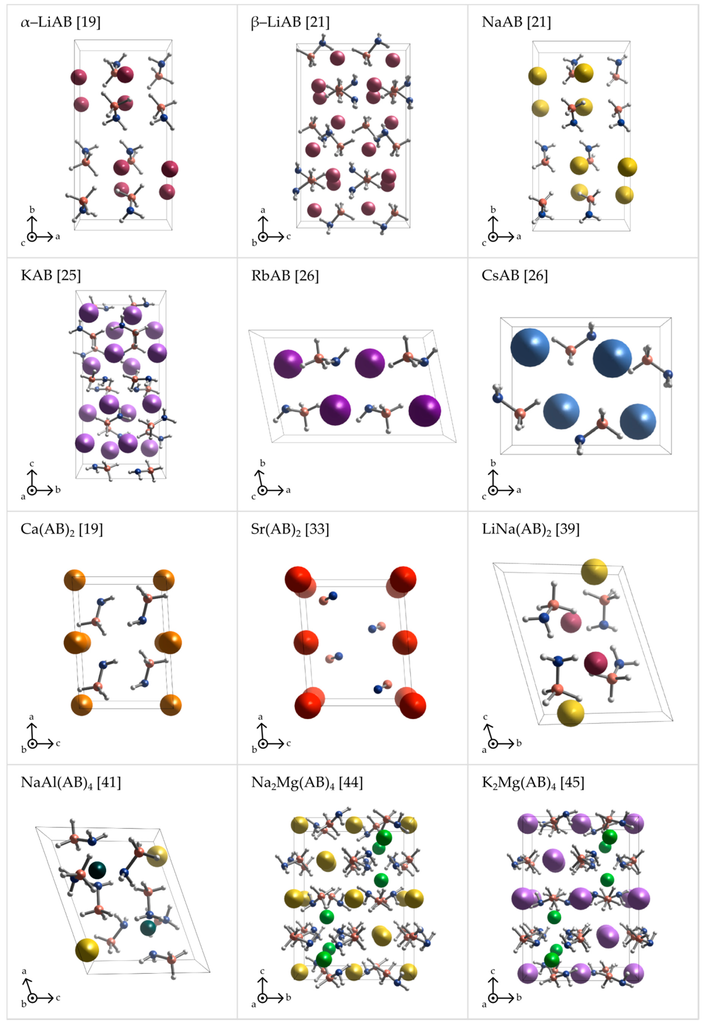
Figure 2.
Perspective projections of the crystal structures of various metal amidoboranes: α-LiAB [19], β-LiAB [21], NaAB [21], KAB [25], RbAB [26], CsAB [26], Ca(AB)2 [19], Sr(AB)2 [33], LiNa(AB)2 [39], NaAl(AB)4 [41], Na2Mg(AB)4 [44], and K2Mg(AB)4 [45].
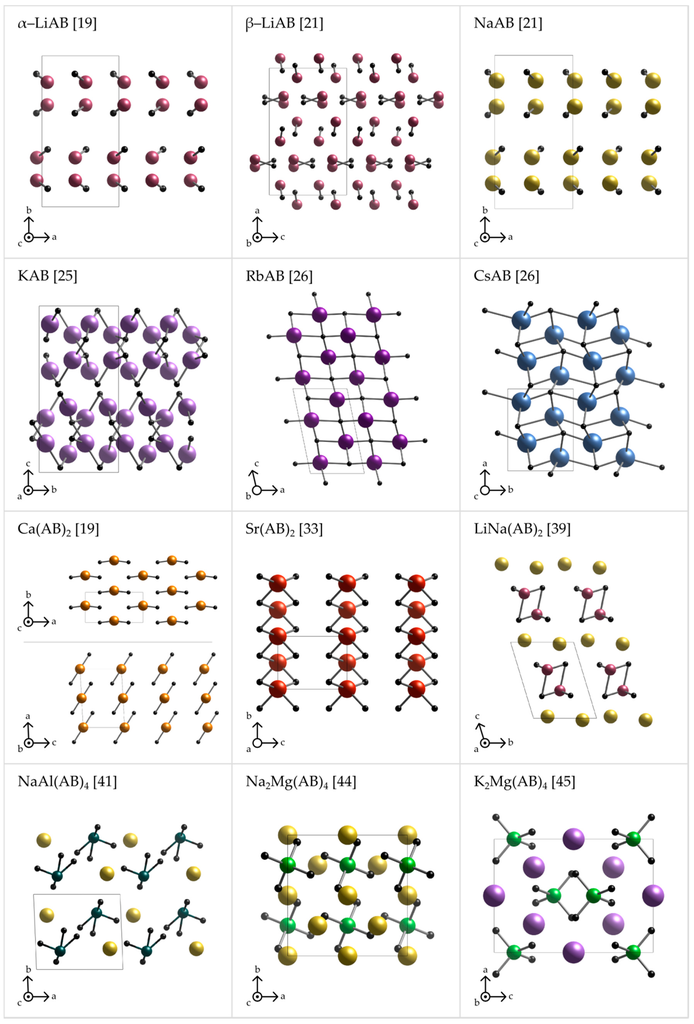
Figure 3.
Axonometric projections of the crystal structures of various metal amidoboranes (α-LiAB [19], β-LiAB [21], NaAB [21], KAB [25], RbAB [26], CsAB [26], Ca(AB)2 [19], Sr(AB)2 [33], LiNa(AB)2 [39], NaAl(AB)4 [41], Na2Mg(AB)4 [44], and K2Mg(AB)4 [45]) emphasizing the cation–anion connectivity. Anion is represented by a dummy atom (black ball) sitting at the middle of the BN bond.
Crystal structure of Y(AB)3 [38] and NaMg(AB)3 [41] were not solved but their powder X-ray patterns were successfully indexed and fitted using the LeBail method. Crystal structures of other synthesized amidoborane salts have not yet been determined.
The crystal structures of metal amidoboranes are often unprecedented and show great variety (Figure 2 and Figure 3); this is due to a low symmetry of the amidoborate anion, and its flexibility to bind to metal cations, involving both the lone pair on N atom, and the three hydride terminal at B atom. We presented general perspective views over each crystal structure showing unit cells along selected axis perpendicular to the plane of the picture (Figure 2). To analyze crystal packing and cation–anion connectivity, for simplicity, we have also represented the entire amidoborate anion by a single dummy atom, X, sitting in the middle of the B–N bond in an axonometric view over the unit cells (Figure 3). Such representation leads to facile analysis of anion–cation sublattice as well as inter-ionic interactions. Moreover, the nearest anionic neighbors of a given cation may now be identified. To show interactions between metal cations and amidoborate anions, we also present fragments of the unit cells of amidoborane salts showing coordination polyhedra of metal cations (Figure 4).
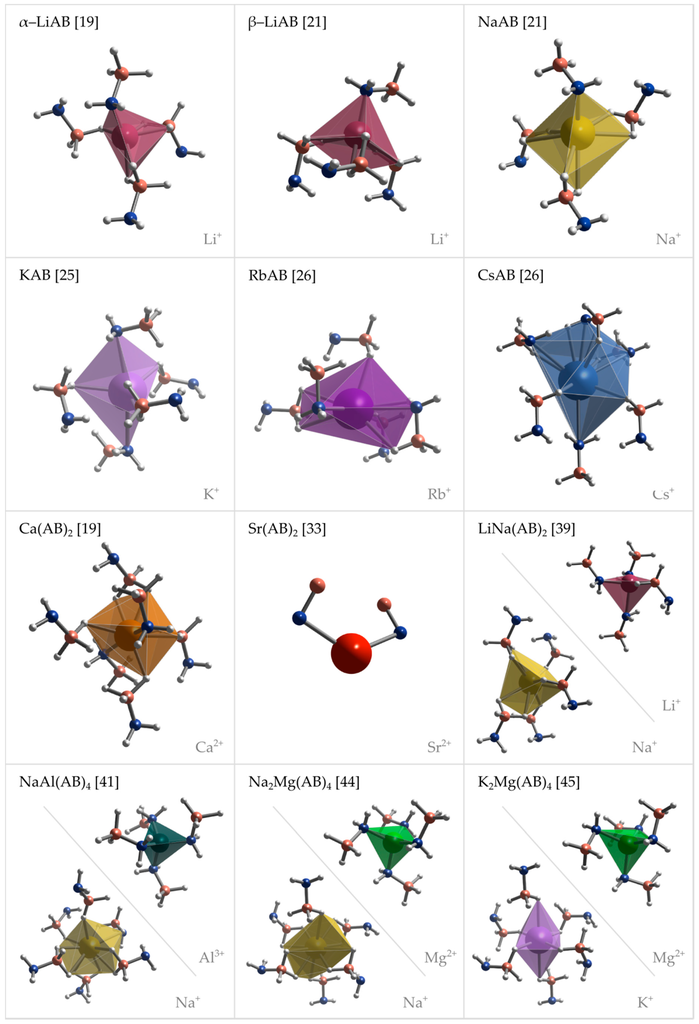
Figure 4.
Perspective projections of coordination spheres of metal cations in the crystal structures of various metal amidoboranes: α-LiAB [19], β-LiAB [21], NaAB [21], KAB [25], RbAB [26], CsAB [26], Ca(AB)2 [19], Sr(AB)2 [43], LiNa(AB)2 [39], NaAl(AB)4 [41], Na2Mg(AB)4 [44], and K2Mg(AB)4 [45].
The crystal structures of α-LiAB [18,19] and NaAB [18,21] consist of double layers of cations and anions, with dihydrogen bonds linking the layers together. Interestingly, one of the anions is much closer to the cation than two others; thus, the crystal structures of α-LiAB [18,19] and NaAB [18,21] can be viewed as quasi-molecular as far as weaker stabilizing interactions are omitted. By quasi-molecular, we mean here that there is just one close cation–anion contact, the others being substantially longer. β-LiAB [20,21] has a similar structure but with somewhat more complex pattern of cation–anion bonding with the local electric dipoles alternating along the c axis and the ab diagonal. KAB [25] repeats the double-layer motif, but the layers are now more puckered and each potassium cation is linked to three close anions thus forming a polymeric network. Both heaviest alkali metal amidoboranes, those of rubidium and cesium, do not show the presence of any low-dimensional polymeric sublattices; they are ionic compounds where anions are shared by at least three and even up to five metal cations [26]. For RbAB and CsAB, the distances M–B and M–N (Rb-B: 3.189–5.591 Å, Rb-N: 3.076–3.143 Å [26]; Cs-B: 3.680–3.854 Å, and Cs-N: 3.340–3.535 Å [26]) are comparable with respective distances in borohydrides (Rb-B: 3.541 Å [83]; and Cs-B: 3.71 Å [83]) and amides (Rb-N: 3.140–3.355 Å [84]; and Cs-N: 3.515 Å [85]). The changes observed in the Li–Cs series are obviously due to an increasing ionic radius of alkali metal cations, and enhances ionicity of chemical bonding down the Group 1.
A similar trend may be observed when comparing the crystal structures of Ca(AB)2 [19] and Sr(AB)2 [43]. While the former is quasi-molecular, with two closest anions linked to Ca site in the quasi-linear fashion, the latter comprises bent Sr(AB)2 units that are in fact interconnected in 2D sheets, with each AB− anion shared between two neighboring Sr2+ cations. One may expect that crystal structure of not-yet-synthesized Ba(AB)2 would form extended polymeric network, just like RbAB or CsAB [26].
Crystal structure of LiNa(AB)2 [39] contains isolated Li2(AB)42− dianions (linked only via weak dihydrogeen bonds into 1D chains) as well as Na+ cations. This form clearly originates from transfer of AB− anions from a stronger Lewis base, NaAB, to a Lewis acid, LiAB. The same situation is observed for Na2Mg(AB)4 [44], which consists of isolated Mg(AB)42− dianions and Na+ cations. K2Mg(AB)4 [45] and NaAl(AB)4 [41] also comprise the isolated Mg(AB)42− and Al(AB)4− anions, respectively, together with alkali metal cations.
Coordination spheres of metal cations in the lattice of amidoborane salts are dependent on the size of the cation and type of crystal structure. Li+ cations are coordinated by four amidoborate anions in α-LiAB [19], β-LiAB [21] and LiNa(AB)2 [39]. In LiNa(AB)2 [39], Li+ cations are coordinated by three N atoms and one hydridic H atom, each in a different top of a distorted tetrahedron, while in α-LiAB [19] and β-LiAB [21] Li+ cations are surrounded by one N atom and hydridic H atoms, five and six, respectively, where one or two H atoms sit in one top of a distorted tetrahedron. Na+ is coordinated by four AB− anions in NaAB [21], by five AB− anions in LiNa(AB)2 [39], and by six AB− anions in NaAl(AB)4 [41] and Na2Mg(AB)4 [44], but only in NaAB [21] Na+ are coordinated by N atoms. Metal cations in KAB [25], RbAB [26], CsAB [26] and Ca(AB)2 [19] are coordinated by five or six AB− anions, where two of them are facing metal cations with N atmos. Sr2+ cations are coordinated at least by two N atoms form neighbor AB− anions [33].
Coordination number of Sr2+ cations cannot be derived due to lack of hydrogen positions in the solution of Sr(AB)2 crystal structure [33]. In bimetallic LiNa(AB)2 [39], NaAl(AB)4 [41], Na2Mg(AB)4 [44] and Na2Mg(AB)4 [45], tetrahedral complex anions are formed by Li+, Al3+ and Mg2+ cations that are coordinated by four AB anions through their N atoms [39,41,44,45]. Respective counter ions, Na+ [39,41,44] and K+ [45], are coordinated only by hydridic H atoms form amidoborate groups.
3. NMR, IR and Raman Spectra of Metal Amidoboranes
Metal amidoboranes exhibit certain similarities in physicochemical properties and spectroscopic characterization (NMR, FTIR, and Raman). Several diagnostic features can be seen for all of them, which help in identification of these compounds (Figure 5, for more detailed view of selected amidoborane salts see S1, S6, S8, S10, S12, S13, S16, S19, S22, S23, S25, S27, S31 Supplementary Materials).
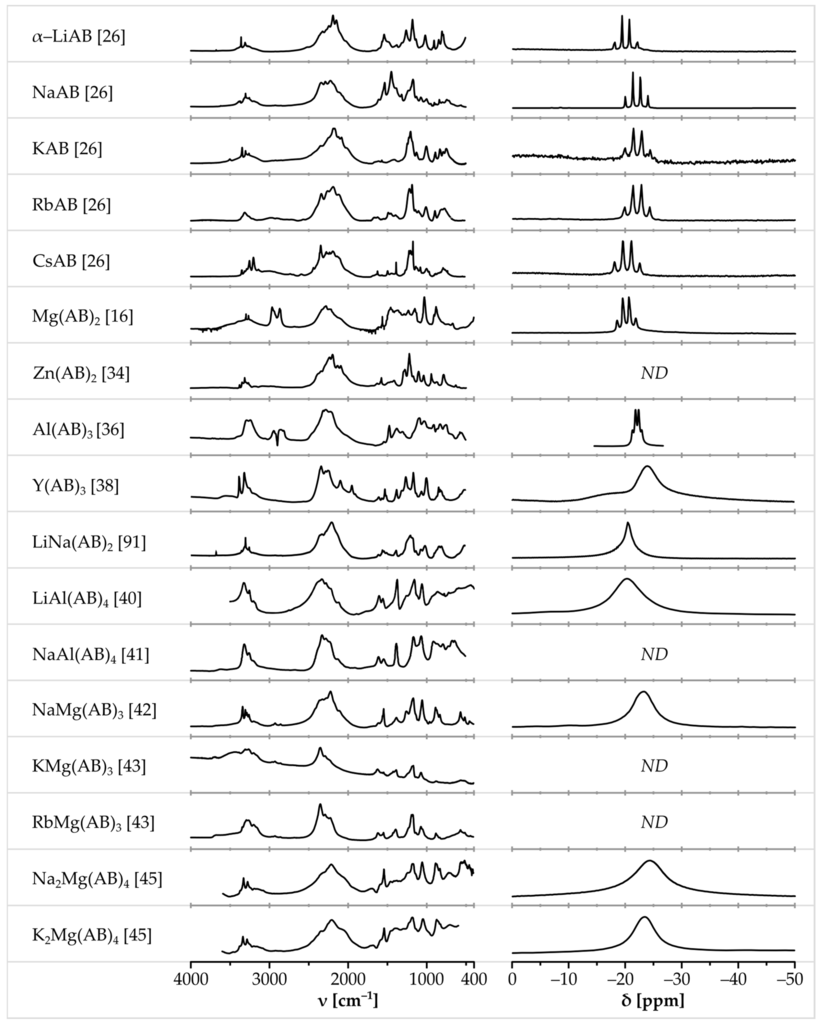
Figure 5.
Comparison of FTIR spectra (left column) and 11B NMR spectra (right column) of various amidoborane salts. 11B NMR spectra in solution were selected if available. Spectra of Mg(AB)2 adapted from [16] with permission from Allison L. DeGraffenreid. Spectra of: α-LiAB, NaAB, KAB, RbAB and CsAB adapted from [26]; LiAl(AB)4 adapted from [40]; and NaMg(AB)3 adapted from [42], all with permission from The Royal Society of Chemistry. Spectrum of Zn(AB)2 adapted from [34] with permission from Rafal Owarzany. Spectra of Al(AB)3 adapted from [36]. Spectra of Y(AB)3 adapted from [38] with permission from the Journal of Alloys and Compounds. Spectra of NaAl(AB)4 adapted from [41] with permission from John Wiley and Sons. Spectra of KMg(AB)3 and RbMg(AB)3 adapted from [43] with permission from Elsevier. Spectra of Na2Mg(AB)4 and K2Mg(AB)4 adapted with permission from Chua Y.S. et al. Chem. Mater., 2012, 24, 3574. Copyright 2012 American Chemical Society. Spectra of LiNa(AB)2 adapted from [91] with permission from Karol J. Fijalkowski.
Boron-11 nuclear magnetic resonance spectroscopy (either in solution or in the solid state) is one of the most common and accurate techniques for characterization of amidoboranes. 11B NMR spectra of these salts contain a single characteristic signal at ca. −21 ppm (similar to that seen for the parent ammonia borane [18,23,35]), which comes from [BH3] groups. It can easily be distinguished from the signals originating from [BH4] groups (ca. −45 ppm), [BH2] groups (ca. −10 ppm), or other chemical moieties containing boron atoms in a form different to [BH3] groups.
11B NMR measurements are usually carried out in THF-d8 solution despite relatively low solubility of metal amidoboranes in THF. 11B NMR spectra of amidoboranes consist of a quartet with an intensity ratio of 1:3:3:1. The signal is split due to coupling of boron-11 with three hydrogen-1 nuclei. The quartet is centered at ca. −21 ppm, with J-coupling value in the range of 80–100 Hz [16,23,25,26,31,35,86,87]. On the other hand, the 11B MAS NMR measurements for the samples in the solid state give one broad peak at about −21 ppm [18,25,29,38,39,40,43,45]. In Figure 4, we have shown a collection of the 11B NMR spectra, while, in Table 3, we have listed 11B NMR chemical shifts and J-coupling constants for various amidoboranes present studied so far.
Table 3.
11B NMR experimental data for ammonia borane and various metal amidoboranes. σ—chemical shift; J—coupling constant; ND—Not Determined.
Two papers have reported “non-standard” NMR spectra of amidoborane salts; Shimoda et al. studied NaAB with 23Na NMR [88] and KAB with temperature resolved 39K NMR spectroscopy [89].
Infrared absorption and Raman scattering spectroscopy are two other common—and complementary—methods for characterization of metal amidoboranes. The IR and Raman spectra of metal amidoboranes, similar to neat ammonia borane, consist of the two main distinct groups of bands related to stretching vibrations of NH and BH bonds, which fall in the 3200–3400 cm−1 and 2000–2400 cm−1 range, respectively (Table 4 and Table 5). Other bands originate from deformations of the NH2 group (1400–1650 cm−1), deformations of BH3 group (1000–1350 cm−1), stretching vibrations of the BN bond (below 1000 cm−1), and others. It is hard to determine precise range of wavenumbers for these bands because of their partial overlap. Unfortunately, there is no single well defined band with a fixed wavenumber that would be distinctive and common for all metal amidoboranes as the chemical vicinity of amidoborate anion is different for each of them; thus, the band positions in the vibrational spectra turn out to be sensitive markers of interatomic interactions in the solid state. The only generalization that one may make is that the upper bound of the NH stretching region in the spectra of amidoboranes (3200–3400 cm−1) is usually blueshifted with respect to the respective band for ammonia borane (3200–3320 cm−1) [2,23]. This indicates some stiffening of the N–H vibrons.
Table 4.
FTIR experimental data for ammonia borane and various metal amidoboranes. In the table, only the strongest bands for each spectrum region are presented: ν-NH, ν-BH, δ-NH and δ-BH. Data shown in italics were estimated from graphs shown in the cited papers. ND = Not Determined.
Table 5.
Raman experimental data for ammonia borane and various metal amidoboranes. In the table, only the strongest bands for each spectrum region are presented: ν-NH, ν-BH, δ-NH and δ-BH.
4. Thermal Decomposition of Metal Amidoboranes
Thermal decomposition of amidoborane salts is an exothermic multistep process leading to amorphic polymeric (BNH)n–type products at a relatively low temperature (100–250 °C) or to BN at higher temperature (>350 °C). Hydrogen is the main gaseous product of thermal decomposition of amidoboranes. The first step of the thermal decomposition of amidoborane salts occurs at ca. 60–110 °C resulting in desorption of a single molecule of hydrogen from each formula unit (Table 5). Further decomposition steps occur at temperatures higher than 100 °C. The second molecule of hydrogen per formula unit is usually desorbed below 250 °C. From the point of view of hydrogen storage for support of low temperature fuel cell systems for automotive use, only hydrogen available below 100 °C can be useful [93]. In this context, most amidoborane salts can offer at most only 2/5 of their hydrogen content.
For comparison, neat ammonia borane undergoes three exothermic decomposition steps (70–112 °C, 135–180 °C, and >450 °C) while releasing one hydrogen molecule per formula unit in each step, which is equivalent to ca. 6.5 wt% mass loss [2,7,74,76,94]. Neat ammonia borane decomposes via a rapid uncontrollable process while evolving hydrogen contaminated with borazine and monomeric aminoborane [94]. The decoposition can be accelerated using an addition of specific catalyst [77] or changing of the process conditions [95,96,97].
Most amidoborane salts decompose with melting via exothermic reactions (ca. −5 kJ/mol∙H2) which are less exothermic than decomposition of ammonia borane (−20 kJ/mol∙H2). Thermal decomposition of amidoboranes is thermodynamically favorable because of both the enthalpic and the entropic contributions to Gibbs free energy of this reaction being negative. Surprisingly, several bimetallic amidoborane salts decompose via slightly endothermic processes, namely: Ca(AB)2 (+3.5 kJ/mol) [31], NaMg(AB)3 (+3.4 kJ/mol) [42], Na2Mg(AB)4 [45] and K2Mg(AB)4 [45]. This result proves that thermodynamics of the decomposition process may to some extend be tuned by designing a composition of bimetallic amidoborane salts. A rule of thumb here is that the acid–base reactions stabilize the complex (bimetallic) system [12], e.g.,
where KAB serves as a Lewis base, and Mg(AB)2 as a Lewis acid. This, in turn, leads to less negative (or even positive) enthalpy of thermal decomposition of the product.
2 KAB + Mg(AB)2 → K2Mg(AB)4
Possibility of onboard regeneration of a decomposed material is very important when considering amidoborane salts as hydrogen source for automotive use. According to DOE targets, reloading of an “empty” hydrogen store should be possible under mild pressure of hydrogen (ca. 5 bars) within 5 min [93]. The condition of thermodynamic reversibility of absorption–desorption reactions at close-to-ambient (p,T) conditions is an equivalent (except for so-called high-entropy storage systems) to the enthalpy of thermal decomposition falling around +40 kJ/mol. Clearly, since the thermal decomposition of amidoboranes is exothermic or only slightly endothermic, the regeneration process is thermodynamically unfavorable. Indeed, regeneration of decomposed amidoborane salts with gaseous hydrogen under high pressure has not yet been successfully completed, even in the case of the salts decomposing in endothermic reactions [18,42]. Thus, chemical pathways of regeneration of the discharged material are now under development.
There has been some discussion in the literature whether hydrogen desorbed upon heating of light alkali metal amidoboranes is pure or it contains contaminants which might be harmful for fuel cells catalysts and membranes (Figure 6). A number of papers were published reporting alkali metal amidoboranes as sources of pure hydrogen, e.g., LiAB [18,19,86], NaAB [18,86] and KAB [25]. On the other hand, several other groups reported for these materials the desorption of hydrogen contaminated with ammonia: α-LiAB [20,25,39,89,90], β-LiAB [20] and NaAB [23,24,87,88,98]. Some amidoborane salts (KAB [25,89], Mg(AB)2 [29], Al(AB)3 [35], and LiAl(AB)4 [35,40]) were reported to desorb pure hydrogen and, currently, there are no contradictory reports. The remaining known amidoborane compounds (RbAB [26], CsAB [26], Sr(AB)2 [43], Y(AB)3 [38], LiNa(AB)2 [39], NaAl(AB)4 [41], NaMg(AB)3 [42], Na2Mg(AB)4 [44]) decompose releasing hydrogen contaminated with ammonia, diborane or borazine, all of which are harmful for PEM and alkaline fuel cells.
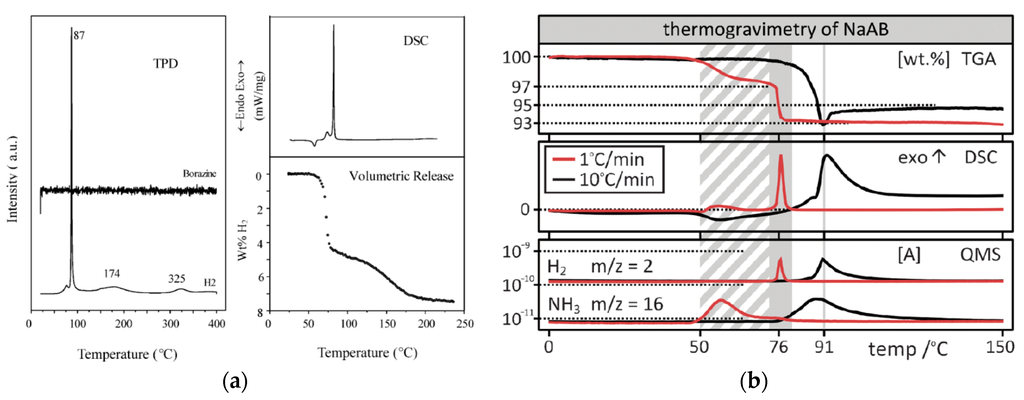
Figure 6.
Comparison of thermal decomposition of NaAB presented by: (a) Xiong et al. [22] reporting NaAB as source of borazine-free hydrogen (Left. Reproduced from reference [22] with permission from The Royal Society of Chemistry.); and (b) Fijalkowski et al. [24] reporting contamination of hydrogen with ammonia (Right. Reproduced from reference [24] with permission from the PCCP Owner Societies.). Xiong et al. [22] have not presented any data related to ammonia evolution.
Mechanism of thermal decomposition of amidoboranes is not well determined because of differences in reported observation, however, a few groups proposed possible scenarios of this process. The first step of the most often cited mechanism of hydrogen evolution is identical to that determined for ammonia borane [99], and it relies on intermolecular recombination of protonic and hydridic hydrogen atoms to form dihydrogen molecule [18,23,24].
Spielmann et al. performed a detailed study on the mechanism of dehydrogenation of Ca(AB)2 suggesting a head-to-tail dimerization of amidoborate anions yielding [HN-BH-NH-BH3]2− ions [32], which is in good agreement with mechanism presented earlier by Stowe et al. [99]. Xiong et al., describing thermolysis of LiAB and NaAB, pointed out that the key feature enabling formation of dihydrogen is presence of both positively and negatively charged hydrogen atoms in amidoborate anions [18]. Local recombination of Hδ+ and Hδ− atoms does not involve mass transport through different phases [18]. In the next paper, Xiong et al. reported formation of amorphous BN along with reformation of NaH upon heating NaAB to 200 °C (see S4 in Supplementary Materials) [22]. Luedke et al. proposed metal ion assisted hydride transfer involving the scission of M–N and B–H bonds resulting in the formation of MH and dimerization of two amidoborate anions upon thermolysis of amidoborane salts [86]. MH further reacts with an imide group of the dimer to release hydrogen molecule. Next, hydrogen molecule is formed in recombination of Hδ+ and Hδ− atoms [86]. Fijalkowski et al. [23] further proposed an intermolecular transformation of two amidoborate anions forming hypothetical product containing BH3NHNaBH3− anions and Na[NH3]+ cations, which could easily evolve weakly coordinated ammonia [23,24]. Later, this hypothesis was substantiated by Fijalkowski et al. [87]. The intermolecular step responsible for both ammonia evolution and initiation of the polymerization of the solid residue was finally suggested to correspond to trimerisation of amidoborate anions with the concomitant formation of BH3NH2BH2NH2BH3− anions and ammonia molecules [87]. The latter hypothesis finds strong support from the X-ray, Raman and FTIR studies [87]. One can find signals from LiBH3NH2BH2NH2BH3 and NaBH3NH2BH2NH2BH3 in the X-ray powder patterns of, e.g., α-LiAB [18], β-LiAB [20], NaAB [21], LiNa(AB)2 [39] and LiAB∙AB [55], synthesized in recent years. LiBH3NH2BH2NH2BH3 and NaBH3NH2BH2NH2BH3 were earlier observed by Evans [100] and Ryan [21] but at that time their role as intermediates of the thermal decomposition of LiAB and NaAB had not yet been understood. Selected mechanism discussed above are presented in Figure 7 [19,23,32,86,87].
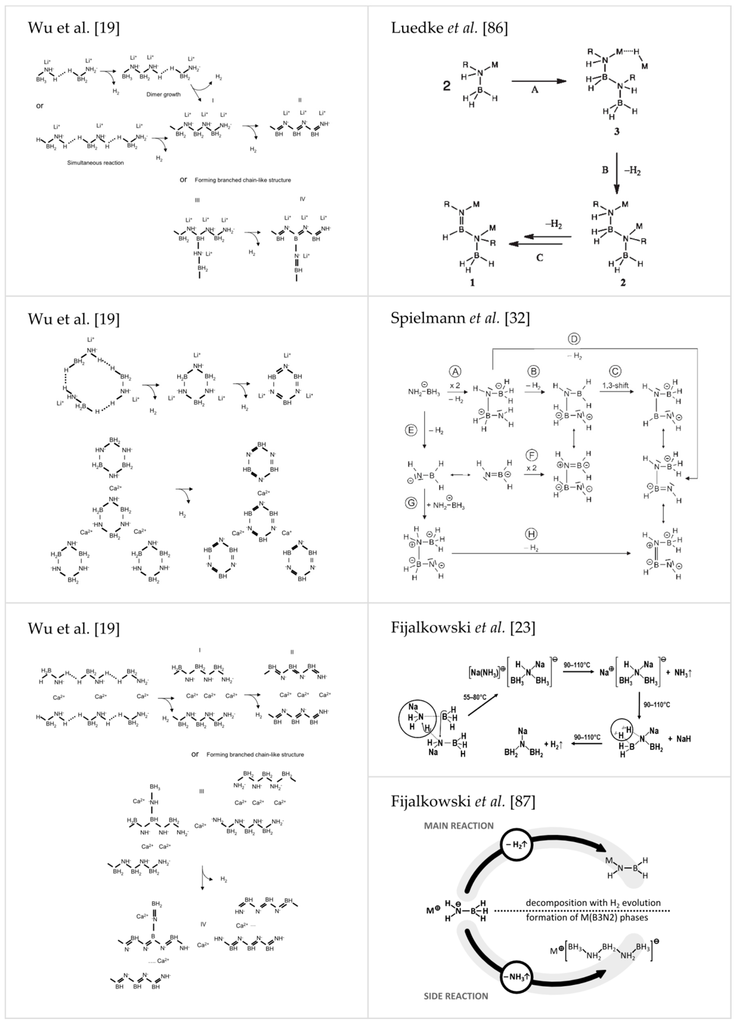
Figure 7.
Reproduction of several discussed mechanisms of thermal decomposition of amidoborane salts, proposed by: Wu et al. [19] (Reprinted with permission from Wu et al. J. Am. Chem. Soc. 2008, 130, 14834. Copyright 2008 American Chemical Society.), Spielmann et al. [32] (Reproduced from [32] with permission from John Wiley and Sons.), Luedke et al. [86] (Reprinted with permission from Luedke et al. Inorg. Chem. 2010, 49, 3905. Copyright 2010 American Chemical Society.) and Fijalkowski et al. [23] (Reproduced from reference [23] with permission from The Royal Society of Chemistry.) [87] (Reproduced from reference [87] with permission from the PCCP Owner Societies.).
In Table 6, we summarize experimentally obtained data on thermal decomposition of amidoborane salts. We would like to point out that, for most of the presented compounds, mass loss is not connected exclusively with evolved hydrogen but also other compounds (e.g., ammonia, borazine, and diborane) that are found to be undesired hydrogen contaminants.
Table 6.
Dehydrogenation data of various metal amidoborane salts. Experimental data of the first step of thermal decomposition: temperature range, mass loss, contamination of hydrogen evolved. Nominal hydrogen content [wt%] is indicated. ND—Not Determined.
5. Synthesis, Reactivity and Selected Properties of Metal Amidoboranes
In this section, we describe synthesis, structure and properties of each of the currently known nineteen mono- and bimetallic amidoboranes.
5.1. Alkali Metal Amidoboranes
5.1.1. Alpha-Lithium Amidoborane, α-LiNH2BH3 [α-LiAB]
The most common method of synthesis of α-LiAB is a direct reaction of ammonia borane and LiH in molar ratio 1:1, where solid α-LiAB is formed together with gaseous dihydrogen according to reaction Equation (2). Degrafenreid conducted this process in THF solution for ca. 4 h [16]. Alternatively, the synthesis can be driven in a 3 h solid–solid reaction in a ball mill, as proposed by Xiong et al. [18] and Wu et al. [19].
LiH + NH3BH3 → LiNH2BH3 + H2↑
Alternatively, α-LiAB can be obtained from ammonia borane and LiNH2 as proposed by Genova et al. [38]. This reaction considers formation of an intermediate product (lithium amidoborane amide) in a 6 min high energy disc milling process. The intermediate further decomposes over ca. 1.5 h to α-LiAB and gaseous ammonia according to Equation (3):
NH3BH3 + LiNH2 → LiNH2BH3(NH3) → LiNH2BH3 + NH3↑
The last known method of synthesis of α-LiAB considers reaction of ammonia borane with metaloorganic lithium compounds: n-butyllithium [16,17] or methyllithium [16] (Equation (4)):
RLi + NH3BH3 → LiNH2BH3 + RH
α-LiAB undergoes thermal decomposition with the first step occurring at ca. 90 °C. According to Xiong et al., α-LiAB evolve 10.9 wt% of borazine-free hydrogen during a 19 h isothermal heating at 92 °C (see S2 in Supplementary Materials) [18]. α-LiAB thermally decomposes in exothermic reaction releasing 3–5 kJ/mol∙H2, which is significantly less than the heat evolved during decomposition of ammonia borane (−20 kJ/mol∙H2) [18]. According to Wu et al., α-LiAB evolves 8.8 wt% of hydrogen at 91 °C and 10.8 wt% upon 180 °C [20]. Wu et al. [20] and Diyabalanage et al. [25] detected ammonia contamination of hydrogen desorbed upon thermolysis of α-LiAB, while Luedke et al. [86] did not observed any evolution of ammonia sampling the gas evolved through a Drägar tube detecting ammonia in the range of 10–1000 ppm. Fijalkowski et al. observed desorption of ca. 3 wt% of ammonia preceding evolution of ca. 5 wt% of hydrogen when heating α-LiAB in the temperature range of 65–95 °C [24]. This observation was correlated with the growth of ionic conductivity of the material determined from impedance spectroscopy [23] and the formation of Li(BH3NH2BH2NH2BH3) salt [24,87]. According to Fijalkowski et al., α-LiAB is unstable at room temperature and it slowly decomposes over several months giving among others Li(BH3NH2BH2NH2BH3) salt [24,87]. Regeneration of decomposed α-LiAB with gaseous hydrogen at high pressure was not achieved [18]. Tang et al. reported chemical regeneration of α-LiAB with 98% yield using combination of CH3OH, LiAlH4 and NH4Cl [101].
α-LiAB was studied at high pressure by Najiba et al. [102]. Surprisingly, no dihydrogen bonds were observed at the pressures up to ~20 GPa [102].
α-LiAB crystallizes in the orthorhombic space group Pbca [18]. Crystal structure of α-LiAB was solved from powder data by Xiong et al. [18], Wu et al. [19] and Lee et al. [69]. The lattice parameters are: a = 7.1051(8) Å, b = 13.930(1) Å, c = 5.1477(7) Å, V = 509.489 Å3 with Z = 8 [19]. Volume of a formula unit is equal to 63.6875 Å3. Lithium ion is in tetrahedral coordination of a N atom of [NH2] group (Li–N distance 2.063 Å) and three [BH3] groups (Li–B distance 2.510–2.969 Å). B–N distance in α-LiAB (1.56 Å [18]; 1.5467 Å [19]) is shorter than in ammonia borane molecules at RT (1.597 Å) [3]. Other bonds distances: N–H: 1.0255–1.0264 Å; B–H: 1.2367–1.2476 Å; Li–N: 2.0634 Å [19].
5.1.2. Beta-Lithium Amidoborane, β-LiNH2BH3 [β-LiAB]
Synthesis of β-LiAB considers a special 5 h regime of ball milling of a mixture containing ammonia borane and LiH in molar ratio 1:1 [20]. Firstly, as an intermediate product LiNH2BH3∙NH3BH3 is formed, which later reacts with LiH while yielding β-LiAB. The process proceeds according to Equation (5). During further milling β-LiAB transforms to α-LiAB. Full phase conversion was observed after 16 h of milling. Such behavior suggests β-LiAB is less thermodynamically stable than α-LiAB. Indeed, volume per one formula unit for β-LiAB (67.7 Å3) is by ca. 6% larger than that for α-LiAB, which suggests that β- LiAB is the high-temperature form.
2LiH + 2NH3BH3 → LiH + LiNH2BH3∙NH3BH3 + H2↑ → 2LiNH2BH3 + 2H2↑
According to Wu et al., β-LiAB exhibits essentially identical features in dehydrogenation as α-LiAB [20]. Evolution of 8.8 wt% of hydrogen contaminated with 300 ppm of ammonia was observed upon isothermal thermolysis of β-LiAB at 91 °C [20].
β-LiAB crystallizes in the orthorhombic space group Pbca but it is not isostructural with α-LiAB [20,21]. It was solved from powder data [20,21]. The lattice parameters are a = 15.146(6) Å, b = 7.721(3) Å, c = 9.268(4) Å, V = 1083.7(8) Å3 with Z = 16. β-LiAB unit cell is ca. two times bigger than the corresponding unit cell of α-LiAB [21]. Crystal structure of β-LiAB is an intergrowth of two different LiAB substructures. An asymmetric unit has a volume of 135.5 Å3 [20,21]. Interatomic distances: N–H: 0.990–0.991 Å; B–H: 1.180–1.219 Å; B–N: 1.580–1.581 Å; Li–B: 2.501–2.927 Å; Li–N: 1.930–2.042 Å [21].
5.1.3. Sodium Amidoborane, NaNH2BH3 [NaAB]
NaAB can be synthesized in a reaction of ammonia borane and NaH in molar ratio 1:1 according to reaction Equation (6). The reaction can be carried out in THF solution in −78 °C as proposed by Degrafenreid [16]. Xiong et al. [18] conducted the same reaction in solid state in a 3 h ball milling, while Fijalkowski et al. [23] carried the milling in a high energy tungsten carbide disc mill for 3 min.
NaH + NH3BH3 → NaNH2BH3 + H2↑
Another synthetic route described by Xiong et al. considers 20 h reaction between ammonia borane and NaNH2 in THF according to Equation (7) [22]. Formation of NaAB ammoniate was not observed during this process.
NaNH2 + NH3BH3 → NaNH2BH3 + NH3↑
NaAB is an off-white crystalline material that thermally decomposes via a two-step process. According to Xiong et al. [18], NaAB evolve 7.4 wt% of pure hydrogen during isothermal heating at 89 °C (see S2 in Supplementary Materials). Further investigation of Xiong et al. [22] showed two step thermal decomposition with evolution of 4.4 wt% of borazine-free hydrogen below 87 °C when heating with a high heating rate. The second decomposition step occurs at temperatures exceeding 100 °C and results in evolution of 3.1 wt% of hydrogen together with reformation of crystalline NaH (see S4 in Supplementary Materials) [22]. Fijalkowski et al. [23] reported desorption of hydrogen contaminated with ammonia (see S5 in Supplementary Materials), which was later confirmed by Sandra et al. [98]. Luedke et al. [86] did not confirmed evolution of ammonia upon thermolysis of NaAB sampling the gas evolved through a Drägar tube detecting ammonia in the range of 10–1000 ppm. Later Fijalkowski et al. [24] observed—upon slow heating (1 °C/min) of NaAB—the evolution of 3 wt% ammonia at 50–60 °C separated from evolution of 4 wt% of hydrogen at 70–80 °C (see S7 in Supplementary Materials). Evolution of ammonia was correlated with the growth of ionic conductivity of the material determined from impedance spectroscopy and formation of the Na(BH3NH2BH2NH2BH3) salt (Figure 5) [24,87]. According to Fijalkowski et al. [24], NaAB undergoes slow spontaneous decomposition at a room temperature. Na(BH3NH2BH2NH2BH3) salt can be detected in this solid residue after this slow room-temperature decomposition [87]. Regeneration of decomposed NaAB with gaseous hydrogen at high pressure was not achieved [18].
A comprehensive high pressure study of NaAB using in situ Raman spectroscopy and DFT calculations was performed by Magoś-Palasyuk et al. [103]. An isosymmetric phase transition to α/-NaAB was observed at pressure about 3 GPa [103].
NaAB crystallizes in the orthorhombic space group Pbca and it is isostructural with α-LiAB [18,21]. Crystal structure of NaAB was solved from powder data by Xiong et al. [18] and Ryan [21]. Both works contain comparable data, but there are some differences between them. Xiong et al. [18] described lattice parameters and B–N bond length (1.56 Å); while Ryan [21] gave lattice parameters, atomic positions and some interatomic distances. B–N bond length (1.670 Å) derived from atomic positions given by Ryan [21] seems to be little unreasonable since it is more than 0.1 Å longer than B–N bond length observed in other alkali metal amidoboranes (Table 3). The crystal structure of NaAB has not yet been deposited in any crystallographic database. The lattice vectors presented by Xiong et al. (a = 7.46931(7) Å, b = 14.65483(16) Å, c = 5.65280(8) Å, V = 618.764(20) Å3 [18] are a bit different from the parameters given by Ryan (a = 7.4680(16) Å, b = 14.6474(32) Å, c = 5.6548(09) Å, V = 618.56 Å3) [21]. In NaAB Z = 8, thus the volume of a formula unit is equal to 77.3 Å3. Sodium ions are in tetrahedral coordination of N atom of [NH2] group and three H atoms of [BH3] group. B–N bond length in amidoborate anon is equal to 1.56 Å [18], which is shorter than B–N distance in ammonia borane molecules at RT (1.597 Å) [3].
5.1.4. Potassium Amidoborane, KNH2BH3 [KAB]
KAB was synthesized in a reaction of ammonia borane and KH in molar ratio 1:1 in THF by Diyabalanage et al. [25] according to reaction Equation (8). The synthesis is carried out in THF solution at room temperature for 4 h [25], or in THF solution at −78 °C for 48 h as described by DeGraffenreid [16].
KH + NH3BH3 → KNH2BH3 + H2↑
An alternative synthetic approach described by DeGraffenreid [16] applies a 72 h reaction of metallic potassium with ammonia borane in molar ratio 1:1 in NH3 at −78 °C according to Equation (9). Further purification by filtration of KAB solution in NH3 can be done [16].
K + NH3BH3 → KNH2BH3 + ½H2↑
According to Diyabalanage et al. [25], KAB undergoes a two-step decomposition steps releasing pure hydrogen: 4.0 wt% (1.5 mol equivalent H2) is released at ca. 65–110 °C with a hydrogen release peak at 98 °C; and additional 2.0 wt% (0.75 mol equivalent H2) evolves at ca. 110–225 °C with a peak at 179 °C (see S9 in Supplementary Materials). Unlike other alkali metal amidoboranes KAB evolves pure hydrogen [25]. KAB is stable at room temperature, in contrast to its lighter congeners. Tang et al. reported chemical regeneration of KAB with up to 98% yield using combination of CH3OH, LiAlH4 and NH4Cl [87].
KAB was a subject of in-situ Raman and X-ray investigation under high pressure conducted by Magoś-Palasyuk et al. [104]. From 5 GPa one of the NH bands in Raman spectrum of KAB exhibited a soft mode behavior resulting in decrease of frequency value by 150 cm−1 which is ca. three time more than that observed for ammonia borane at a comparable pressure of 25 GPa [104]. This observation can be assigned to formation of conventional hydrogen bonds between amidoborate anions in the crystal structure of KAB [104].
KAB crystallizes in the orthorhombic space group Pbca [25] but it is not isostructural with lighter alkali metal amidoboranes [18]. Crystal structure of KAB was solved from the crystals grown from diglyme/hexane by Diyabalanage et al. [25]. The lattice parameters are a = 9.4304(1) Å, b = 8.26112(1) Å, c = 17.3403(2) Å, V = 1319.42 Å3 with Z = 16. Volume of a formula unit is equal to 82.5 Å3. In the lattice there are two distinct sites for every K, N and B atom. Potassium ions are in octahedral coordination of three N atoms from [NH2] groups and five H atoms from three [BH3] groups. The Hδ+–Hδ− distance in KAB (2.2650 Å) is much shorter than those in LiAB (2.372 Å) [18] and NaAB (2.717 Å) [18], which may explain different dehydrogenation properties of these compounds, and evolution of pure H2 from KAB. The B–N bond length in amidoborate anion is equal to 1.532 Å [25], which is shorter than in ammonia borane molecules at RT (1.597 Å [3]) and in LiAB or NaAB [18]. Other intramolecular distances: N–H: 0.785–1.047 Å; B–H: 1.078–1.259 Å; B–N: 1.531–1.532 Å; K–N: 2.907–3.364 Å; K–B: 3.279–3.587 Å [25].
5.1.5. Rubidium Amidoborane, RbNH2BH3 [RbAB]
RbAB was synthesized by Owarzany et al. [26] from metallic rubidium and ammonia borane in molar ratio 1:1 in THF solution at room temperature in 24 h reaction according to Equation (10):
Rb + NH3BH3 → RbNH2BH3 + ½H2↑
According to Owarzany et al. [26], RbAB thermally decomposes while evolving hydrogen and significant amount of ammonia. Decomposition was observed in the temperatures of ca. 65–90 °C with 9 wt% mass loss and a hydrogen release peak at ca. 88 °C during slow heating (1 °C/min) [26]. Heating of RbAB in the range of 90–200 °C does not result in further decomposition. RbAB undergoes slow spontaneous decomposition at room temperature with formation of Rb(BH3NH2BH2NH2BH3) salt [26,92].
RbAB crystallizes in the monoclinic space group P21/c unlike other alkali metal amidoboranes which crystallize in orthorhombic system. Crystal structure was solved by Owarzany et al. from powder X-ray diffraction data [26]. The lattice parameters are: a = 6.9305(7) Å, b = 5.0145(5) Å, c = 11.0729(11) Å, β = 101.688°, V = 376.837 Å3 with Z = 4. Volume of a formula unit is equal to 94.2 Å3 [26]. Interatomic distances in rubidium amidoborane: N–H: 1.036–1.043 Å; B–H: 1.178–1.189 Å; B–N: 1.542 Å; Rb–N: 3.076–3.143 Å; Rb–B: 3.189–3.591 Å [26].
5.1.6. Cesium Amidoborane, CsNH2BH3 [CsAB]
CsAB was obtained by Owarzany et al. [26] from metallic cesium and ammonia borane in molar ratio 1:1 in THF solution in a 24 h reaction according to Equation (11).
Cs + NH3BH3 → CsNH2BH3 + ½H2↑
CsAB thermally decomposes in the temperature range of ca. 55–85 °C (1 °C/min) while evolving hydrogen and ammonia [26]. The process can be divided into two steps which differ in the rate of decomposition: slow mass loss of 4 wt% at ca. 55–80 °C and quick mass loss of ca. 3 wt% at 80–85 °C [26]. Further decomposition does not occur under 200 °C. CsAB slowly decomposes at a room temperature resulting in formation of the Cs(BH3NH2BH2NH2BH3) salt [19,92].
CsAB crystallizes in orthorhombic space group Pnam. Crystal structure was solved by Owarzany et al. from X-ray powder data [26]. The lattice parameters are a = 9.1182(8) Å, b = 7.3437(7) Å, c = 5.9678(5) Å, V = 399.612 Å3 with Z = 4. Volume of CsAB formula unit is equal to 99.9 Å3 [26]. Interatomic distances in cesium amidoborane: N–H: 1.053 Å; B–H: 1.195–1.198 Å; B–N: 1.526 Å; Cs–N: 3.340–3.535 Å; Cs–B: 3.680–3.854 Å [26].
5.2. Alkaline Earth Metal Amidoboranes
5.2.1. Magnesium Amidoborane Mg(NH2BH3)2 [Mg(AB)2]
A number of research groups tried to synthesize Mg(AB)2 for several years but without success [19,27,28] (see S11 in Supplementary Materials). All of these approaches were unsuccessful because of the low reactivity of MgH2 against ammonia borane. Mg(AB)2 was finally synthesized by Luo et al. [29] via a two-step process. The first step involves a 2 h mechanochemical reaction of MgH2 or metallic magnesium with ammonia borane in molar ratio 1:2, while the second key step consists of aging of the post mill mixture [29]. Synthesis of Mg(AB)2 can be described overall by Equations (12) and (13).
MgH2 + 2NH3BH3 → Mg(NH2BH3)2 + 2H2↑
Mg + 2NH3BH3 → Mg(NH2BH3)2 + H2↑
Luo et al. [29] proposed also an alternative metathetic synthesis of Mg(AB)2 considering mechanochemical reaction of LiAB and Mg(BH4)2 according to Equation (14). The product of this reaction is a sticky mixture comprising only Mg(AB)2 and LiBH4.
Mg(BH4)2 + 2LiNH2BH3 → Mg(NH2BH3)2 + 2LiBH4
DeGraffenreid [16] described a metathetic reaction between LiAB and MgI2 carried out in THF solution for 3–5 days with continuous stirring (Equation (15)).
MgI2 + 2LiNH2BH3 → Mg(NH2BH3)2 + 2LiI
Mg(AB)2 thermally decomposes via a multi-step process while evolving pure hydrogen upon heating to 300 °C [29]. In the first step at ca. 75–110 °C Mg(AB)2 evolves ca. 2% of hydrogen with a peak of evolution at 104 °C (see S14 in Supplementary Materials) [29]. At higher temperatures a continuous mass loss and slow evolution of hydrogen with a maximum rate at ca. 225 °C can be observed [29]. Overall mass loss is equal to 9 wt% below 300 °C [29]. Luo et al. observed no ammonia contamination of hydrogen evolved, what may result from a very strong NH3 binding capability of Mg2+ cations [29].
Crystal structure of Mg(AB)2 has not yet been determined. A computational modeling of a crystal structure of Mg(AB)2 was performed [30] but without comparative study with experimental results of Luo et al. [29].
5.2.2. Calcium Amidoborane Ca(NH2BH3)2 [Ca(AB)2]
Ca(AB)2 was obtained by Wu et al. [19] via a 60 min ball milling of CaH2 together with ammonia borane in molar ratio 1:2 according to Equation (16). Wu et al. reported no difference of the quality of the product when milling with graphite additive or without [19].
CaH2 + 2NH3BH3 → Ca(NH2BH3)2 + 2H2↑
Diyabalanage et al. performed synthesis of Ca(AB)2 from CaH2 and ammonia borane in THF solution at room temperature in molar ratio 1:2 [31]. In this reaction a THF complex of Ca(AB)2 is formed according to Equation (17). The complex loses most of its THF content when heated to 70 °C in vacuum (Equation (18)). THF can be completely desorbed only during thermal decomposition of Ca(AB)2 [31].
CaH2 + 2NH3BH3 + 2THF → Ca(THF)2(NH2BH3)2 + 2H2↑
Ca(THF)2(NH2BH3)2 → Ca(NH2BH3)2 + 2THF↑
Thermolysis of Ca(AB)2 was described in several papers. According to Wu et al. [19], decomposition of Ca(AB)2 starts at ca. 80 °C, and a vigorous dehydrogenation can be observed at ca. 100 °C and ca. 140 °C. Below 250 °C, Ca(AB)2 desorbs four equivalents of borazine-free hydrogen which corresponds to 8 wt% mass loss (see S3 in Supplementary Materials) [19]. According to Diyabalanage et al. [31], 0.3 equivalents of hydrogen is being evolved at 90 °C, 1.1 equivalent at 120 °C, 2.4 at 150 °C and 3.6 at 170 °C (see S15 in Supplementary Materials). In summary Ca(AB)2 desorbs 7.4 wt% of hydrogen slightly contaminated with ammonia and borazine below 170 °C which gives 90% of theoretical dehydrogenation yield [31]. According to Spielmann et al. [32], Ca(AB)2 undergoes a head-to-tail dimerization of amidoborate anions leading towards [HN-BH-NH-BH3]2− ions upon dehydrogenation.
Ca(AB)2 crystallizes in the monoclinic space group C2 [19]. The crystal structure was solved by Wu et al. from X-ray powder data [19]. The lattice parameters are: a = 9.100(2) Å, b = 4.371(1) Å, c = 6.441(2) Å, V = 276.869 Å3 with Z = 2. Volume of a formula unit is equal to 138.5 Å3. Calcium cations are surrounded by six amidoborate anions. Two of them face Ca2+ cations with [NH2] groups with Ca–N distance 2.472 Å. The other four anions coordinate Ca2+ cation with [BH3] groups with Ca–B distance 2.995–3.175 Å Å. B–N bond length in amidoborate anions (1.552 Å [19]) is shorter than in ammonia borane molecules (1.597 Å) [3], and comparable with α-LiAB and β-LiAB (1.547) Å [18,19,20]. Other interatomic distances in calcium amidoborane: N–H: 1.044–1.105 Å; B–H: 1.251–1.321 Å [19].
5.2.3. Strontium Amidoborane Sr(NH2BH3)2 [Sr(AB)2]
Sr(AB)2 was obtained by Zhang et al. [33] in two step process considering gentle milling of SrH2 and ammonia borane in molar ratio 1:2 at 0 °C, followed by heating of the post milled sample at moderate conditions. The milling temperature needs to be low to avoid thermally activated decomposition of the forming product. Following heating is performed at 45 °C for 2 h to desorb two equivalents of hydrogen. Temperature of the thermal treatment cannot be too high to avoid thermal decomposition of Sr(AB)2. Synthesis of Sr(AB)2 can be illustrated by the overall Equation (19).
SrH2 + 2NH3BH3 → Sr(NH2BH3)2 + 2H2↑
Sr(AB)2 thermally decomposes at temperatures higher than 40 °C (see S17 and S18 in Supplementary Materials) [33]. Sr(AB)2 undergoes two main decomposition steps which overlap: 40–80 °C and 80–200 °C. In the first step pure hydrogen is being released at temperature range of 40–65 °C, while at higher temperatures ammonia and diborane are clearly present in the desorbed hydrogen [33]. The second step occurs at 80–200 °C. Total mass loss of 11.2 wt% up to 200 °C was reported [33].
Sr(AB)2 crystallizes in monoclinic space group C2 [33]. Crystal structure of Sr(AB)2 was solved from powder data by Zhang et al. [33]. The lattice parameters are a = 8.1660(4) Å, b = 5.0969(3) Å, c = 6.7258(4) Å, β = 94.392(4)°, V = 279.114 Å3 with Z = 2. Volume of a formula unit is equal to 139.6 Å3. Sr(AB)2 crystallizes in the same space group as Ca(AB)2 [19] but their atomic coordinates are quite different [33]. Positions of hydrogen atoms in the unit cell of Sr(AB)2 were not determined [33]. Interatomic distances in strontium amidoborane: B–N: 1.528 Å; Sr–B: 3.109–3.189 Å; Sr–N: 2.681 Å [33].
5.3. Other Metal and Mixed Metal Amidoboranes
5.3.1. Zinc amidoborane Zn(NH2BH3)2 [Zn(AB)2]
Zn(AB)2 was synthesized in a metathetic reaction of NaAB and ZnCl2 in molar ratio 2:1 by DeGrafenreid [16]. The reaction was performed in anhydrous THF at −78 °C for several hours. The process can be described by Equation (20). As a product of this reaction, a mixture of Zn(AB)2 and NaCl is obtained. NaCl forms a “dead mass” that cannot be separated from Zn(AB)2 [16].
ZnX2 + 2NaNH2BH3 → Zn(NH2BH3)2 + 2NaX (T < 0 °C, X = Cl, I)
Zn(AB)2 can be also obtained in a metathetic mechanochemical reaction of NaAB and zinc halide in molar ratio 2:1, namely ZnCl2 or ZnI2, as presented by Owarzany [34]. The milling was performed using cryogenic high energy disc mill at the temperature ca. −30 °C. Overall milling time did not exceed 6 min [34]. The reaction performed by Owarzany can be described by Equation (19) [34]. The product contains a mixture of Zn(AB)2 and alkali metal halide, similarly to the product of DeGraffenreid [16].
Zn(AB)2 is a greyish solid which thermally decomposes below 0 °C yielding black powder which contains metallic zinc [16,34]. Zn(AB)2 was not investigated with the powder X-ray diffraction due to its high sensitivity to manual grinding (as required for loading of a capillary for an X-ray measurement).
5.3.2. Aluminum Amidoborane Al(NH2BH3)3 [Al(AB)3]
Hawthorne et al. investigated several routes of synthesis of Al(AB)3 and found a methatetic approach to be the most efficient [35,36]. Al(AB)3 can be obtained from light alkali metal amidoboranes (LiAB, NaAB, KAB) and AlCl3 at low temperature according to Equation (21) [35,36]. The product contain mixture of Al(AB)3 and MCl “dead mass” which cannot be removed from the post-reaction mixture.
AlCl3 + 3MNH2BH3 → Al(NH2BH3)3 + 3MCl (M = Li, Na, K)
Thermal decomposition of Al(AB)3 can be observed in the temperature range 60–290 °C resulting in evolution of pure hydrogen without contamination of ammonia. Total sample mass loss after heating to 190 °C was 6 wt% which corresponds to 4.1 molecules per formula unit (see S20 in Supplementary Materials).
Al(AB)3 is a crystalline solid material which gives a characteristic X-ray powder pattern [35]. Crystal structure of Al(AB)3 has not yet been reported.
5.3.3. Yttrium Amidoborane Y(NH2BH3)3 [Y(AB)3]
Y(AB)3 was synthesized in a metathetic mechanochemical reaction of α-LiAB and yttrium chloride in molar ratio 3:1 by Genova et al. [38]. The milling was performed using high energy disc mill. The overall milling time of 9 min was divided into three 3 min sequences separated by 5 min intervals for cooling [38]. A composite of Y(AB)3 and LiCl was obtained as a product of this reaction (Equation (22)). LiCl forms a “dead mass” and cannot be separated from the product. Y(AB)3 constitutes only 58.4 wt% of the composite.
YCl3 + 3LiNH2BH3 → Y(NH2BH3)3 + 2LiCl
Y(AB)3 thermally decomposes via a multistep process in the temperature range of 80–300 °C [38]. It evolves hydrogen contaminated with ammonia (see S21 in Supplementary Materials) [38]. This process can be divided in two stages: 80–200 °C with 3.5 wt% mass loss and ammonia contamination (ca. 6 wt% for pure Y(AB)3); and 200–300 °C with 2.0 wt% mass loss and pure hydrogen (ca. 3.4 wt% for pure Y(AB)3). Total mass loss detected in the full decomposition process is equal to 5.5 wt% which is higher than theoretical hydrogen capacity of the composite (4.9 wt%) due to contamination of hydrogen desorbed [38].
Y(AB)3 was indexed by Genova et al. in triclinic space group C2/c using form X-ray powder data using Le Bail method [38]. The lattice parameters are: a = 13.18902(63) Å, b = 7.82233(38) Å, c = 14.874274(68) Å, β = 92.42620(40)°, V = 1533.19(13) Å3 with Z = 8. Volume of a formula unit is equal to 191.6 Å3. Estimated volume of Y(AB)3 formula unit (182.9–183.4 Å3) is less than 5% smaller than the experimental value proving correct identification of the product [38]. Crystal structure of Y(AB)3 has not yet been determined.
5.3.4. Lithium–Sodium Amidoborane, LiNa(NH2BH3)2 [LiNa(AB)2]
LiNa(AB)2 was synthesized by Fijalkowski et al. [39] in a metathetic mechanochemical reaction from LiH, NaH and ammonia borane in molar ratio 1:1:2. A high energy disc mill was used in the milling of total time of 9 min divided into three 3 min intervals separated by 5 min intervals for cooling [39]. The reaction runs according to Equation (23). Zhang et al. [105] conducted milling of LiH, NaH and ammonia borane in different molar ratios but no other bimetallic lithium–sodium amidoborane salt was form.
LiH + NaH + 2NH3BH3 → LiNa(NH2BH3)2 + 2H2↑
Li et al. [106] obtained LiNa(AB)2 by blending LiAB and NaAB in molar ratio 1:1 in THF, and then evaporating the THF solvent. This reaction can be described by an Equation (24) [106]. LiNa(AB)2 cannot be obtained in reaction 24 in a mechanochemical approach [39].
LiAB + NaAB → LiNa(NH2BH3)2 (in THF)
LiNa(AB)2 decomposes at elevated temperatures in two steps: 75–100 °C and 130–220 °C [39]. In the first decomposition step a 6% mass loss is observed which is attributed to evolution of hydrogen contaminated with ammonia and volatile NBH5 by-product (see S24 in Supplementary Materials). It is worth noticing that in the temperature range ca. 75–90 °C the level of impurities evolved is significantly lower than in the temperature range 90–100 °C [39]. In the second step 3 wt% of hydrogen is evolved. Evolution of ammonia was correlated with a growth of ionic conductivity of the material as determined from impedance spectroscopy [24] and formation of Na(BH3NH2BH2NH2BH3) salt [24,87]. Total observed mass loss is equal to 9 wt% below 220 °C [39]. LiNa(AB)2 slowly decomposes at room temperature [24] while forming Na(BH3NH2BH2NH2BH3) salt [87].
LiNa(AB)2 crystallizes in triclinic space group P–1 [39]. The synchrotron diffraction powder pattern was indexed by Fijalkowski et al. to unit cell with lattice parameters: a = 5.0197(4) Å, b = 7.1203(7) Å, c = 8.9198(9) Å, α = 103.003(6)°, β = 102.200(5)°, γ = 103.575(5)°, V = 289.98(5) Å3 with Z = 2. Volume of a formula unit is equal to 144.99(3) Å3 and is ca. 2% larger than a sum of the formula unit volumes of α-LiAB and NaAB (141.24 Å3) [39]. Lithium cations are coordinated by 3 nitrogen atoms from [NH2] groups and by one hydride atom from [BH3] group. A [Li(AB)2−]2 anionic dimers can be observed in the crystal structure. Sodium cations are in octahedral coordination surrounded by six hydride atoms from [BH3] groups. Li et al. [106] showed a theoretical crystal structure of LiNa(AB)2 calculated using a quasirandom structure method (SQS). Although the SQS approach failed to reproduce an actual crystal structure of LiNa(AB)2, it is a very promising method applicable to complex hydrides [106]. Mildly constrained interatomic distances in lithium-sodium amidoborane: N–H: 1.00–1.10 Å, B–H: Å, B–N: 1.22–1.32 Å; Na–B: 2.861–3.107 Å; Li–N: 2.157–2.261 Å [39].
5.3.5. Lithium–Aluminum Amidoborane, LiAl(NH2BH3)4 [LiAl(AB)4]
LiAl(AB)4 was synthesized by Xia et al. via a mechanochemical reaction of Li3AlH6 and ammonia borane [40]. Initially Xia et al. used several different molar ratios of the substrates but crystalline product was obtained only when substrates were milled in molar ratio 1:4. The milling was performed in a ball mill for 30 min with 30 min breaks. The overall milling time was 3 h [40]. A hypothetical nonbalanced reaction Equation (25) derived from the paper of Xia et al. [40] is given below:
Li3AlH6 + 4NH3BH3 → Li3AlH2(NH2BH3)4 + 4H2↑ + …
Hawthorne et al. [35] and Hoy [37] proposed an alternative LiAl(AB)4 synthesis from LiAlH4 and ammonia borane in molar ratio 1:4 according to Equation (26).
LiAlH4 + 4NH3BH3 → LiAl(NH2BH3)4 + 4H2↑
According to Xia et al. [40], 3.85 wt% of hydrogen is being released at 82–110 °C during the first step of decomposition of the “LiAl(AB)4” composite. Additional 6.25 wt% of hydrogen is being evolved at temperature range of 110–175 °C. Both steps of decomposition proceed with evolution of pure hydrogen (see S26 in Supplementary Materials) [40]. LiAl(AB)4 decomposed at 400 °C can be regenerated in a chemical way by reacting with liquid ammonia and hydrazine at 0 °C for 3 days and subsequent 12 h vacuum drying for removing of the residual ammonia [40]. According to Hawthorne et al. [35], thermolysis of LiAl(AB)4 occurs at 80–400 °C yielding pure hydrogen. Total observed mass loss after heating to 190 °C was 7 wt% which corresponds to 5.2 molecules per formula unit of LiAl(AB)4 [35].
LiAl(AB)4 is a crystalline solid. All attempts to index the X-ray diffraction pattern and to solve the crystal structure of LiAl(AB)4 failed, probably due to poor crystallinity of the sample [40].
5.3.6. Sodium–Aluminum Amidoborane Composite, NaAl(NH2BH3)4 [NaAl(AB)4]
NaAl(AB)4 was synthesized by Dovgaliuk et al. [41] via a mechanochemical reaction of NaAlH4 and ammonia borane in molar ratio 1:4. Reaction was performed in 240 cycles of 3 min milling and 5 min breaks with a 90% yield (Equation (27)). Yield of the synthesis increases with the milling time. Traces of substrates are present in the product.
NaAlH4 + 4NH3BH3 → Na[Al(NH2BH3)4] + 4H2↑
NaAl(AB)4 decomposes while releasing borazine- and diborane-free hydrogen, contaminated with traces of ammonia and yielding crystalline NaBH4 and an amorphous phase AlN4B3H(0-3.6) as solid residue [41]. Thermolysis of NaAl(AB)4 occurs via two overlapping steps at 115–130 °C and 130–250 °C with a mass loss of ca. 3 wt% and ca. 6 wt% (see S28 in Supplementary Materials) [41]. Both steps are exothermic. Total observed mass loss up to 250 °C is equal to 9 wt% which corresponds to eight equivalents of hydrogen per formula unit. Isothermal treatment at 120 °C causes evolution of approximately 5.3 equivalents of hydrogen. The once decomposed NaAl(AB)4 can be recharged (recovering about 27% of its hydrogen content) upon exposure to 150 bar H2 pressure, but without reformation of NaAl(AB)4 [41].
NaAl(AB)4 crystallizes in a triclinic space group P–1 [41]. It is a second amidoborane, after LiNa(AB)2, indexed in triclinic crystal system. Crystal structure was solved by Dovgaliuk et al. from synchrotron diffraction powder pattern [41]. Unit cell parameters were found to be: a = 9.4352 Å, b = 7.7198 Å, c = 7.6252 Å, α = 97.211°, β = 109.223°, γ = 89.728°, V = Å3 with Z = 2. Volume of a formula unit is equal to 259.946 Å3. The central Al3+ cation is coordinated by four nitrogen atoms from the [NH2BH3]− anions, whereas Na+ is octahedrally coordinated by six BH3 groups. Moderately strong dihydrogen bonds between oppositely charged hydrogen atoms in two different amidoborate anions are found at 1.96(1)–2.28(1) Å. DFT calculations of the unit cell vectors of NaAl(AB)4 were in good agreement with the experimentally obtained data. Interatomic distances in sodium-aluminum amidoborane: N–H: 1.027–1.031 Å; B–H: 1.123–1.398 Å; B–N: 1.581–1.629 Å; Na–B: 2.918–2.970 Å; Al–N: 1.840–1.929 Å [26].
5.3.7. Sodium–Magnesium Amidoborane, NaMg(NH2BH3)3 [NaMg(AB)3]
NaMg(AB)3 was synthesized via a mechanochemical reaction of NaMgH3 with ammonia borane in molar ratio 1:3 by Kang et al. [42]. The milling must be followed by isothermal treatment at 45 °C overnight to complete the synthesis, according to Equation (28). During this thermal treatment 3.4 wt% of hydrogen is being released, which corresponds to 0.8 equivalents of hydrogen per formula unit thus suggesting an incomplete conversion of substrates (this was confirmed by X-ray powder diffraction [42]).
NaMgH3 + 3NH3BH3 → NaMg(NH2BH3)3 + 3H2↑
NaMg(AB)3 thermally decomposes via a slightly endothermic process (3.4 kJ/mol H2) at temperature range of 75–225 °C. The process can be divided into two overlapping steps where intense hydrogen evolution starts at 140 °C. Traces of ammonia in the hydrogen stream are seen in the temperature range 100–170 °C. Total observed mass loss is 7.4 wt% which is equal to five equivalents of hydrogen per formula unit. Interestingly, a milled composite NaMgH3/3AB which was not treated at 45 °C is able to rapidly evolve 9.8 wt% of pure hydrogen during 2 min isothermal heating at 80 °C (see S29 in Supplementary Materials) [42,43]. NaBH4 was detected in the products of the thermal decomposition of NaMg(AB)3 [42].
The X-ray powder pattern of NaMg(AB)3 was indexed by Kang et al. [42] to a monoclinic unit cell with lattice parameters: a = 17.011 Å, b = 9.432 Å, c = 9.398 Å, β = 115.99°, V = 1355.3 Å3. Possible space groups are P21 (No. 4) and P21/m (No. 11). Full crystal structure of NaMg(AB)3 including atomic coordinates has not yet been determined.
5.3.8. Potassium–Magnesium Amidoborane, KMg(NH2BH3)3 [KMg(AB)3]
KMg(AB)3 was obtained by Kang et al. [43] in reaction of KMgH3 and ammonia borane in molar ratio of 3:1. The milling needs to be followed by isothermal treatment in 45 °C for 10 h to complete reaction presented in Equation (29). Milling was performed in six 10 min intervals separated by 5 min pauses to ensure steady temperature of reaction [43].
KMgH3 + 3NH3BH3 → KMg(NH2BH3)3 + 3H2↑
KMg(AB)3 undergoes thermal decomposition in the temperature range of 75–225 °C similarly to NaMg(AB)3 [43]. A milled composite KMgH3/3AB which was not treated at 45 °C (i.e., not KMg(AB)3) evolves ca. 10 wt% of hydrogen contaminated with traces of ammonia (released in the temperature range 100–180 °C) [43]. The composite can rapidly evolve 9.3 wt% of pure hydrogen during 2 min isothermal heating at 80 °C [43]. Data for thermolysis of pure KMg(AB)3 are not available in the literature.
KMg(AB)3 is a crystalline solid. All attempts to index the X-ray diffraction pattern and to solve the crystal structure of KMg(AB)3 have failed [43].
5.3.9. Rubidium–Magnesium Amidoborane, RbMg(NH2BH3)3 [RbMg(AB)3]
Synthesis of RbMg(AB)3 was described by Kang et al. [43]. It involves a mechanochemical reaction between RbMgH3 and ammonia borane in molar ratio of 3:1. The overall milling time was 1 h, which was divided into 10 min intervals. The obtained powder was afterwards heated to 45 °C to increase reaction efficiency. The 10 h lasting thermal treatment caused evolution of 0.83 equivalent of H2 per AB molecule, which means that the synthesis was not complete. The synthesis can be described by the simplified reaction Equation (30).
RbMgH3 + 3NH3BH3 → RbMg(NH2BH3)3 + 3H2↑
RbMg(AB)3 thermally decomposes in the temperature range of 75–225 °C similarly to NaMg(AB)3 and KMg(AB)3 [43]. A milled composite RbMgH3/3AB which was not treated at 45 °C (i.e., not RbMg(AB)3) evolves ca. 8 wt% of hydrogen contaminated with traces of ammonia (released 100–180 °C) [43]. The RbMgH3/3AB composite can rapidly evolve 6.2 wt% of pure hydrogen during 2 min isothermal heating at 80 °C [43]. Data for thermolysis of pure RbMg(AB)3 are not available.
RbMg(AB)3 is a crystalline solid. All attempts to index the X-ray diffraction pattern and to solve the crystal structure of RbMg(AB)3 failed [43].
5.3.10. Disodium–Magnesium Amidoborane, Na2Mg(NH2BH3)4 [Na2Mg(AB)4]
Na2Mg(AB)4 was synthesized via a mechanochemical reaction by Wu et al. [44]. A mixture of NaH, MgH2 and ammonia borane in molar ratio 1:1:2 was ball milled for 80 min. Reaction can be presented as Equation (31). Unreacted substrates were present in the product apart from Na2Mg(AB)4.
2NaH + MgH2 + 4NH2BH3 → Na2Mg(NH2BH3)4 + 4H2↑
Na2Mg(AB)4 can be also synthesized in a mechanochemical reaction of Mg(NH2BH3)2∙NH3 and NaH in bolar ratio 1:1 according to reaction Equation (32), as reported by Chua, et al. [45]. Milling was performed for 10 h.
2Mg(NH2BH3)2∙NH3 + 2NaH → Na2Mg(NH2BH3)4 + Mg(NH2)2 + 2H2↑
Na2Mg(AB)4 thermally decomposes releasing 8.4 wt% of hydrogen contaminated with traces of ammonia and borazine (see S30, S32 in Supplementary Materials) [44,45]. The first decomposition step is seen in the temperature range of 65–150 °C and results in evolution of 2 wt% of hydrogen. The process proceeds slowly until ca. 120 °C when a rapid hydrogen evolution is observed. The second decomposition step yields 6.4 wt% mass loss in the form of hydrogen gas in the temperature range of 150–200 °C.
Na2Mg(AB)4 crystallizes in tetragonal space group I41/a. The crystal structure was solved by Wu et al. form X-ray powder data resulting a unit cell with lattice parameters a = 9.4134(17) Å, c = 12.7221(23) Å, V = 1127.33 Å3 with Z = 4 [45]. Volume of formula unit is equal to 281.83 Å3. Magnesium cations are coordinated exclusively by four amidoborate anions via their nitrogen atoms while forming the Mg(NH2BH3)42− tetrahedra. The Mg–N distance (2.106 Å) is comparable with Mg–N bond length in magnesium amide. Sodium cations are in octahedral coordination of six [BH3] groups with the Na–B distance (2.961–3.032 Å) comparable with ones seen for sodium borohydride. The B–N bond length (1.562 Å [45]) is slightly shorter than that for ammonia borane (1.597 Å [3]) and comparable to that for sodium amidoborane (1.56 Å [18]). Other interatomic distances in disodium–magnesium amidoborane: N–H: 1.027–1.028 Å; B–H: 1.236–1.237 Å [45].
5.3.11. Dipotassium–Magnesium Amidoborane, K2Mg(NH2BH3)4 [K2Mg(AB)4]
K2Mg(AB)4 was synthesized by Chua, et al. [45] via a mechanochemical reaction of Mg(NH2BH3)2∙NH3 and KH in molar ratio 1:1; the synthesis proceeds according to the reaction Equation (33). Milling was performed under argon atmosphere for 10 h. Separation of K2Mg(AB)4 from Mg(NH2)2 after milling was impossible. The post milled product contains 20.4 wt% Mg(NH2)2 as a “dead mass”.
2Mg(NH2BH3)2∙NH3 + 2KH → K2Mg(NH2BH3)4 + Mg(NH2)2 + 2H2↑
The as prepared K2Mg(AB)4/Mg(NH2)2 composite decomposes with evolution of both hydrogen and ammonia [45]. Chua, et al. reported 3.7 wt% mass loss up to 150 °C mainly related to NH3 release. Further decomposition occurs at 150–200 °C with hydrogen evolution peak at 158 °C and a concomitant mass loss of ca. 4.4 wt%. This process is endothermic (+7.0 kJ/mol H2). Total mass loss of ca. 7 wt% up to 285 °C was observed, which corresponds to 9.7 equivalents of hydrogen per formula unit of the composite K2Mg(NH2BH3)4/Mg(NH2)2 (see S32 in Supplementary Materials) [45].
K2Mg(AB)4 was determined by Chua, et al. to crystallize in tetragonal space group I41/a [45]. K2Mg(AB)4 is isostructural to Na2Mg(AB)4. The lattice constants are: a = b = 9.5974(17) Å, c = 13.581(4) Å, V = 1250.95 Å3, Z = 4. Mg2+ cations was found to be tetrahedrally bound to four N atoms from amidoborate anions while forming the isolated [Mg(NH2BH3)4]2− dianions. Interatomic distances in dipotassium–magnesium amidoborane: N–H: 1.029 Å; B–H: 1.237–1.241 Å; B–N: 1.562 Å; K–N: 3.342–3.456 Å; and Mg–B: 2.208 Å [45].
6. Conclusions
In this review paper, we have presented an overview of mono- and bimetallic amidoboranes. We have described their crystal structures, lattice connectivity, and crystal packing, as well as their thermal analysis, and the vibrational and NMR spectra. These compounds are hydrogen-rich and they evolve substantial amounts of hydrogen upon mild heating. As such, they are promising candidates for solid state hydrogen source for automotive use. However, their thermal decomposition is usually exothermic or only mildly endothermic, so their on-board regeneration using compressed H2 gas is not possible, and they require off-board chemical recharging. The issue of impurities of H2 gas also requires solving before these systems find any practical applications.
A large number of quasi-binary metal amidoboranes including those of Be, Ba, Sc, Ga, and many transition metals still awaits synthesis. Moreover, modifications of the amidoborane anion are possible (such as substituting hydridic H atom to halogens, and protic H atoms to alkyl or aryl substituents), and only some of these have been attempted in order to modify the chemical properties of the resulting compounds. It seems that the forthcoming years will bring further developments in this field, and possibly the concomitant improvement of selected key parameters related to hydrogen storage properties of these compounds.
Supplementary Materials
The following are available online at http://www.mdpi.com/2073-4352/6/8/88/s1, Selected figures illustrating properties of: α-LiAB [18,19,26], NaAB [18,22,23,24,26], KAB [25,26], RbAB [26], CsAB [26], Mg(AB)2 [28,29], Ca(AB)2 [19,31,32], Sr(AB)2 [33], Zn(AB)2 [34], Al(AB)3 [35], Y(AB)3 [38], LiNa(AB)2 [24,39], LiAl(AB)4 [40], NaAl(AB)4 [41], NaMg(AB)3 [42], Na2Mg(AB)3 [44,45], K2Mg(AB)3 [45].
Acknowledgments
Karol J. Fijalkowski dedicates this paper to his parents on their 40th wedding anniversary. This research was funded from grants of the Polish National Science Centre: 2012/05/N/ST5/01375 (Rafał Owarzany) and 2014/15/B/ST5/05012 (Karol J. Fijalkowski, Piotr J. Leszczyński, Wojciech Grochala).
Author Contributions
Rafał Owarzany, Piotr J. Leszczyński and Karol J. Fijalkowski were responsible for collecting the literature. Rafał Owarzany, Piotr J. Leszczyński, Karol J. Fijalkowski and Wojciech Grochala were responsible for data analysis. Rafał Owarzany, Karol J. Fijalkowski and Wojciech Grochala were responsible for preparation of the manuscript and the supplementary materials. Karol J. Fijalkowski was responsible for processing the images, submission and revision of the manuscript and supplementary materials. Karol J. Fijalkowski was responsible for carrying out correspondence with the Editors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shore, S.G.; Parry, R.W. The crystalline compound ammonia borane, H3NBH3. J. Am. Chem. Soc. 1955, 77, 6084–6085. [Google Scholar] [CrossRef]
- Baumann, J.; Baitalow, F.; Wolf, G. Thermal decomposition of polymeric aminoborane (H2BNH2)x under hydrogen release. Thermochim. Acta 2005, 430, 9–14. [Google Scholar] [CrossRef]
- Bowden, M.E.; Gainsford, G.J.; Robinson, W.T. Room-Temperature Structure of Ammonia Borane. Aust. J. Chem. 2007, 60, 149–153. [Google Scholar] [CrossRef]
- Klooster, W.T.; Koetzle, T.F.; Siegbahn, P.E.M.; Richardson, T.B.; Crabtree, R.H. Study of the N–H∙∙∙H–B Dihydrogen Bond Including the Crystal Structure of BH3NH3 by Neutron Diffraction. J. Am. Chem. Soc. 1999, 121, 6337–6343. [Google Scholar] [CrossRef]
- Chua, Y.S.; Chen, P.; Wu, G.; Xiong, Z. Development of amidoboranes for hydrogen storage. Chem. Comm. 2011, 47, 5116–5129. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.L.; Dixon, D.A.; Garner, E.B.; Gordon, J.C.; Matus, M.H.; Scott, B.; Stephens, F.H. Efficient Regeneration of Partially Spent Ammonia Borane Fuel. Angew. Chem. Int. Ed. 2009, 48, 6812–6816. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.G.; Geanangel, R.A.; Wendlandt, W.W. The thermal decomposition of ammonia borane. Thermochim. Acta 1978, 23, 249–255. [Google Scholar] [CrossRef]
- Frueh, S.; Kellett, R.; Mallery, C.; Molter, T.; Willis, W.S.; King’ondu, C.; Suib, S.L. Pyrolytic Decomposition of Ammonia Borane to Boron Nitride. Inorg. Chem. 2011, 50, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Hess, N.J.; Schenter, G.K.; Hartman, M.R.; Daemen, L.L.; Proffen, T.; Kathmann, S.M.; Mundy, C.J.; Hartl, M.; Heldebrant, D.J.; Stowe, A.C.; et al. Neutron Powder Diffraction and Molecular Simulation Study of the Structural Evolution of Ammonia Borane from 15 to 340 K. J. Phys. Chem. A 2009, 113, 5723–5735. [Google Scholar] [CrossRef] [PubMed]
- Stephens, F.H.; Pons, V.; Baker, R.T. Ammonia-borane: The hydrogen source par excellence? Dalton Trans. 2007, 25, 2613–2626. [Google Scholar] [CrossRef] [PubMed]
- Staubitz, A.; Robertson, A.P.M.; Manners, I. Ammonia-Borane and Related Compounds as Dihydrogen Sources. Chem. Rev. 2010, 110, 4079–4124. [Google Scholar] [CrossRef] [PubMed]
- Grochala, W.; Edwards, P.P. Thermal Decomposition of the Non-Interstitial Hydrides for the Storage and Production of Hydrogen. Chem. Rev. 2004, 104, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, H.I.; Burg, A.B. Hydrides of Boron. VIII. The Structure of the Diammoniate of Diborane and its Relation to the Structure of Diborane. J. Am. Chem. Soc. 1938, 60, 290–299. [Google Scholar] [CrossRef]
- Niedenzu, P.M. Studies on Polyboron Hydride Anions and Ammine-Borane. Ph.D. Thesis, The Ohio State University, Columbus, OH, USA, 1990. [Google Scholar]
- Salupo, T. Preparations of Ytterbium and Europium Borides from Yb(II) and Eu(II) Boron Hydride Precursors. Ph.D. Thesis, The Ohio State University, Columbus, OH, USA, 1993. [Google Scholar]
- DeGraffenreid, A.L. Studies on Boron—Nitrogen and Boron—Gadolinium Compounds. Ph.D. Thesis, The Ohio State University, Columbus, OH, USA, 1995. [Google Scholar]
- Myers, A.G.; Yang, B.H.; Kopecky, D.J. Lithium Amidotrihydroborate, a Powerful New Reductant. Transformation of Tertiary Amides to Primary Alcohols. Tetrahedron Lett. 1996, 37, 3623–3626. [Google Scholar] [CrossRef]
- Xiong, Z.; Yong, C.K.; Wu, G.; Chen, P.; Shaw, W.; Karkamkar, A.; Autrey, T.; Jones, M.O.; Johnson, S.R.; Edwards, P.P.; et al. High-capacity hydrogen storage in lithium and sodium amidoboranes. Nat. Mater. 2008, 7, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, W.; Ylidirim, T. Alkali and Alkaline-Earth Metal Amidoboranes: Structure, Crystal Chemistry and Hydrogen Storage Properties. J. Am. Chem. Soc. 2008, 130, 14834–14839. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wu, G.; Xiong, Z.; David, W.I.F.; Ryan, K.R.; Jones, M.O.; Edwards, P.P.; Chu, H.; Chen, P. Stepwise Phase Transition in the Formation of Lithium Amidoborane. Inorg. Chem. 2010, 49, 4319–4323. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.R. A Study of Ammonia Borane and its Derivatives. Ph.D. Thesis, University of Oxford, Oxford, UK, 2011. [Google Scholar]
- Xiong, Z.; Wu, G.; Chua, Y.S.; Hu, J.; He, T.; Xu, W.; Chen, P. Synthesis of sodium amidoborane (NaNH2BH3) for hydrogen production. Energy Environ. Sci. 2008, 1, 360–363. [Google Scholar] [CrossRef]
- Fijalkowski, K.J.; Grochala, W. Substantial emission of NH3 during thermal decomposition of sodium amidoborane, NaNH2BH3. J. Mater. Chem. 2009, 19, 2043–2050. [Google Scholar] [CrossRef]
- Fijalkowski, K.J.; Jurczakowski, R.; Kozminski, W.; Grochala, W. Insights from impedance spectroscopy into the mechanism of thermal decomposition of M(NH2BH3), M = H, Li, Na, Li0.5Na0.5, hydrogen stores. Phys. Chem. Chem. Phys. 2012, 14, 5778–5784. [Google Scholar] [CrossRef] [PubMed]
- Diyabalanage, H.V.K.; Nakagawa, T.; Shrestha, R.P.; Semelsberger, T.A.; Davis, B.L.; Scott, B.L.; Burrell, A.K.; David, W.I.F.; Ryan, K.R.; Jones, M.O.; et al. Potassium(I) Amidotrihydroborate: Structure and Hydrogen Release. J. Am. Chem. Soc. 2010, 132, 11836–11837. [Google Scholar] [CrossRef] [PubMed]
- Owarzany, R.; Fijalkowski, K.J.; Palasyuk, T.; Jaroń, T.; Grochala, W. Heavy alkali metal amidoboranes: RbNH2BH3 and CsNH2BH3. Unpublished work. 2016. [Google Scholar]
- Haider, S. Borazane (H3BNH3) Based Hydrogen Storage Materials. Ph.D. Thesis, Queen Mary, University of London, London, UK, 2005. [Google Scholar]
- Kang, X.; Ma, L.; Fang, Z.; Gao, L.; Luo, J.; Wang, S.; Wang, P. Promoted hydrogen release from ammonia borane by mechanically milling with magnesium hydride: A new destabilizing approach. Phys. Chem. Chem. Phys. 2009, 11, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Kang, X.; Wang, P. Synthesis, formation mechanism, and dehydrogenation properties of the long-sought Mg(NH2BH3)2 compound. Energy Environ. Sci. 2013, 6, 1018–1025. [Google Scholar] [CrossRef]
- Zhang, Y.; Wolverton, C. Crystal Structures, Phase Stabilities, and Hydrogen Storage Properties of Metal Amidoboranes. J. Phys. Chem. C 2012, 116, 14224–14231. [Google Scholar] [CrossRef]
- Diyabalanage, H.V.K.; Shrestha, R.P.; Semelsberger, T.A.; Scott, B.L.; Bowden, M.E.; Davis, B.L.; Burrell, A.K. Calcium Amidotrihydroborate: A Hydrogen Storage Material. Angew. Chem. Int. Ed. 2007, 46, 8995–8997. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Jansen, G.; Bandmann, H.; Harder, S. Calcium Amidoborane Hydrogen Storage Materials: Crystal Structures of Decomposition Products. Angew. Chem. Int. Ed. 2008, 47, 6290–6391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, C.; Fang, C.; Fang, F.; Sun, D.; Ouyang, L.; Zhu, M. Synthesis, Crystal Structure, and Thermal Decomposition of Strontium Amidoborane. J. Phys. Chem. C 2010, 114, 1709–1714. [Google Scholar] [CrossRef]
- Owarzany, R. Synthesis and Characterisation of Zinc Amidoborane. Bachelor’s Thesis, University of Warsaw, Warsaw, Poland, 2013. [Google Scholar]
- Hawthorne, M.F.; Jalisatgi, S.S.; Safronov, A.V.; Lee, H.B.; Wu, J. Chemical Hydrogen Storage Using Polyhedral Borane Anions and Aluminum-Ammonia-Borane Complexes; Final Report; University of Missouri. 2010. Available online: http://www.osti.gov/scitech//servlets/purl/990217-xUxbgx/ (accessed on 20 April 2016). [Google Scholar]
- Jalisatgi, S.S.; Wu, J.; Hawthorne, M.F. Chemical Hydrogen Storage Using Aluminum Ammonia-borane Complexes; Report, University of Missouri. 2009. Available online: https://www.hydrogen.energy.gov/pdfs/review09/stp_20_hawthorne.pdf/ (accessed on 28 April 2016). [Google Scholar]
- Hoy, J.M. Syntheses of Aluminum Amidotrihydroborate Compounds and Ammonia Triborane as Potential Hydrogen Storage Materials. M.Sc. Thesis, The Ohio State University, Columbus, OH, USA, 2010. [Google Scholar]
- Genova, R.V.; Fijalkowski, K.J.; Budzianowski, A.; Grochala, W. Towards Y(NH2BH3)3: Probing hydrogen storage properties of YX3/MNH2BH3 (X = F, Cl; M= Li, Na) and YHx∼3/NH3BH3 composites. J. Alloys. Comp. 2010, 499, 144–148. [Google Scholar] [CrossRef]
- Fijalkowski, K.J.; Genova, R.V.; Filnchuk, Y.; Budzianowski, A.; Derzsi, M.; Jaron, T.; Leszczynski, P.J.; Grochala, W. Na[Li(NH2BH3)2]—The first mixed-cation amidoborane with unusual crystal structure. Dalton Trans. 2010, 40, 4407–4413. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Tan, Y.; Chen, X.; Guo, Z.; Liu, H.; Yu, X. Mixed-metal (Li, Al) amidoborane: synthesis and enhanced hydrogen storage properties. J. Mater. Chem. A. 2013, 1, 1810–1820. [Google Scholar] [CrossRef]
- Dovgaliuk, I.; Jepsen, L.H.; Safin, D.A.; Łodziana, Z.; Dyadkin, V.; Jensen, T.R.; Devillers, M.; Filinchuk, Y. A Composite of Complex and Chemical Hydrides Yields the First Al-Based Amidoborane with Improved Hydrogen Storage Properties. Chem. Eur. J. 2015, 21, 14562–14570. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Luo, J.; Zhang, Q.; Wang, P. Combined formation and decomposition of dual-metal amidoborane NaMg(NH2BH3)3 for high-performance hydrogen storage. Dalton Trans. 2011, 40, 3799–3801. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.-D.; Luo, J.-H.; Wang, P. Efficient and highly rapid hydrogen release from ball-milled 3NH3BH3/MMgH3 (M [Na, K, Rb]) mixtures at low temperatures. Int. J. Hydrog. Energy 2012, 37, 4259–4266. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Pinkerton, F.E.; Meyer, M.S.; Yao, Q.; Gadipelli, S.; Udovic, T.J.; Yildirim, T.; Rush, J.J. Sodium magnesium amidoborane: the first mixed-metal amidoborane. Chem. Commun. 2011, 47, 4102–4104. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Li, W.; Wu, G.; Xiong, Z.; Chen, P. From Exothermic to Endothermic Dehydrogenation—Interaction of Monoammoniate of Magnesium Amidoborane and Metal Hydrides. Chem. Mater. 2012, 24, 3574–3581. [Google Scholar] [CrossRef]
- He, T.; Wang, J.; Chen, Z.; Wu, A.; Wu, G.; Yin, J.; Chu, H.; Xiong, Z.; Zhang, T.; Chen, P. Metathesis of alkali-metal amidoborane and FeCl3 in THF. J. Mater. Chem. 2012, 22, 7478–7483. [Google Scholar] [CrossRef]
- Xia, G.; Yu, X.; Guo, Y.; Wu, Z.; Yang, C.; Liu, H.; Dou, S. Amminelithium Amidoborane Li(NH3)NH2BH3: A New Coordination Compound with Favorable Dehydrogenation Characteristics. Chem. Eur. J. 2010, 16, 3763–3769. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Wu, G.; Xiong, Z.; Karkamkar, A.; Guo, J.; Jian, M.; Wong, M.W.; Autrey, T.; Chen, P. Synthesis, structure and dehydrogenation of magnesium amidoborane monoammoniate. Chem. Commun. 2010, 46, 5752–5754. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Kang, X.; Fang, Z.; Wang, P. Promotion of hydrogen release from ammonia borane with magnesium nitride. Dalton Trans. 2011, 40, 6469–6474. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Wu, H.; Zhou, W.; Udovic, T.J.; Wu, G.; Xiong, Z.; Wong, M.W.; Chen, P. Monoammoniate of Calcium Amidoborane: Synthesis, Structure, and Hydrogen-Storage Properties. Inorg. Chem. 2012, 51, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Wu, G.; Xiong, Z.; He, T.; Chen, P. Calcium Amidoborane Ammoniate; Synthesis, Structure, and Hydrogen Storage Properties. Chem. Mater. 2009, 21, 4899–4904. [Google Scholar] [CrossRef]
- Yang, J.; Beaumont, P.R.; Humphries, T.D.; Jensen, C.M.; Li, X. Efficient Synthesis of an Aluminum Amidoborane Ammoniate. Energies 2015, 8, 9107–9116. [Google Scholar] [CrossRef]
- He, T.; Wu, H.; Wu, G.; Li, Z.; Zhou, W.; Juz, X.; Xie, D.; Chen, P. Lithium amidoborane hydrazinates: synthesis, structure and hydrogen storage properties. J. Chem. Mater. A 2015, 3, 10100–10106. [Google Scholar] [CrossRef]
- Li, Z.; He, T.; Wu, G.; Chen, W.; Chua, Y.S.; Guo, J.; Xie, D.; Ju, X.; Chen, P. Synthesis, structure and the dehydrogenation mechanism of calcium amidoborane hydrazinates. Phys. Chem. Chem. Phys. 2016, 18, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wu, G.; Xiong, Z.; Han, X.; Chu, H.; He, T.; Chen, P. LiNH2BH3∙NH3BH3: Structure and Hydrogen Storage Properties. Chem. Mater. 2010, 22, 3–5. [Google Scholar] [CrossRef]
- Nöth, H.; Thomas, S.; Schmidt, M. Solvates of Lithium (Dimethylamino)trihydroborate. Chem. Ber. 1996, 129, 451–458. [Google Scholar] [CrossRef]
- Keller, P.C. Reactions of Lithium Dimethylamide with Some Borane Derivatives. Evidence for the Displacement of Lithium Hydride. Inorg. Chem. 1975, 14, 438–440. [Google Scholar] [CrossRef]
- Mountford, A.J.; Clegg, W.; Coles, S.J.; Harrington, R.W.; Horton, P.N.; Humphrey, S.M.; Hursthouse, M.B.; Wright, J.A.; Lancaster, S.J. The Synthesis, Structure and Reactivity of B(C6F5)3-Stabilised Amide (M-NH2) Complexes of the Group 4 Metals. Chem. Eur. J. 2007, 13, 4535–4547. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Blacque, O.; Fox, T.; Frech, C.M.; Berke, H. Development of Rhenium Catalysts for Amine Borane Dehydrocoupling and Transfer Hydrogenation of Olefins. Organometallics 2009, 28, 5493–5504. [Google Scholar] [CrossRef]
- Wolstenholme, D.J.; Flogeras, J.; Che, F.N.; Decken, A.; McGrady, G.S. Homopolar Dihydrogen Bonding in Alkali Metal Amidoboranes: Crystal Engineering of Low-Dimensional Molecular Materials. J. Am. Chem. Soc. 2013, 135, 2439–2442. [Google Scholar] [CrossRef] [PubMed]
- Bellham, P.; Hill, M.S.; Kociok-Köhn, G. Alkali metal-mediated dehydrocoupling of Me2NH·BH3. Dalton Trans. 2015, 44, 12078–12081. [Google Scholar] [CrossRef] [PubMed]
- Stennett, T.E.; Harder, S. s-Block amidoboranes: Syntheses, structures, reactivity and applications. Chem. Soc. Rev. 2016, 45, 1112–1128. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Bolte, M.; Harder, S. Synthesis and structure of a magnesium-amidoborane complex and its role in catalytic formation of a new bis-aminoborane ligand. Chem. Commun. 2009, 45, 6934–6936. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Piesik, D.F.-J.; Harder, S. Thermal Decomposition of Mono- and Bimetallic Magnesium Amidoborane Complexes. Chem. Eur. J. 2010, 16, 8307–8318. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Harder, S. Hydrogen Elimination in Bulky Calcium Amidoborane Complexes: Isolation of a Calcium Borylamide Complex. J. Am. Chem. Soc. 2009, 131, 5064–5065. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Piesik, D.; Wittkamp, B.; Jansen, G.; Harder, S. Convenient synthesis and crystal structure of a monomeric zinc hydride complex with a three-coordinate metal center. Chem. Commun. 2009, 23, 3455–3456. [Google Scholar] [CrossRef] [PubMed]
- Forster, T.D.; Tuononen, H.M.; Parvez, M.; Roesler, R. Characterization of β-B-Agostic Isomers in Zirconocene Amidoborane Complexes. J. Am. Chem. Soc. 2009, 131, 6689–6691. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, A.N.; Jorgensen, S.W. Scaffolded Borazane-Lithium Hydride Hydrogen Storage Materials. U.S. Patent US 7166150 B2 (appl. 2005), 23 January 2007. [Google Scholar]
- Burrell, A.K.; Davis, B.J.; Thorn, D.L.; Gordon, J.C.; Baker, R.T.; Semelsberger, T.A.; Tumas, W.; Diyabalanage, H.V.K.; Shrestha, R.P. Metal Aminoboranes. U.S. Patent US 7713506 B2 (appl. 2008), 11 May 2010. [Google Scholar]
- Balema, V.; Josyula, K.; Xu, G.; Wallock, N.; Batcheller, S.; Gao, P.; Jasty, S. Metal Amidoborane Compositions and Processes for Their Preparation. U.S. Patent US 8920760 B2 (appl. 2010), 30 December 2014. [Google Scholar]
- Kim, D.Y.; Singh, N.J.; Lee, H.M.; Kim, K.S. Hydrogen-Release Mechanisms in Lithium Amidoboranes. Chem. Eur. J. 2009, 15, 5598–5604. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.L.; McKee, M.L. Mechanistic Study of LiNH2BH3 Formation from (LiH)4 + NH3BH3 and Subsequent Dehydrogenation. Inorg. Chem. 2009, 48, 7564–7575. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, S.; Nguyen, V.S.; Nguyen, M.T. Potential hydrogen storage of lithium amidoboranes and derivatives. Chem. Phys. Lett. 2010, 489, 148–153. [Google Scholar] [CrossRef]
- Chittari, B.L.; Tewari, S.P. First principles calculations of LiNH2BH3, LiNH3BH4, and NaNH2BH3. Phys. Status Solidi B 2014, 251, 898–906. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, J.; Zhang, T. Crystal and electronic structures of solid M(NH2BH3)n (M = Li, Na, K) and the decomposition mechanisms. Int. J. Hydrog. Energ. 2014, 39, 21372–21379. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, J.G.; He, P. Theoretical study on the structure and dehydrogenation mechanism of mixed metal amidoborane, Na[Li(NH2BH3)]2. J. Alloys Comp. 2013, 581, 59–65. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, J.G. Structural study and dehydrogenation mechanisms of a novel mixed metal amidoborane: Sodium magnesium amidoborane. Chem. Phys. Lett. 2013, 590, 27–34. [Google Scholar] [CrossRef]
- Chittari, B.L.; Tewari, S.P. Structural, bonding and elastic properties of Mg(NH2BH3)2, Ca(NH2BH3)2 and Sr(NH2BH3)2. Mater. Chem. Phys. 2014, 148, 364–370. [Google Scholar] [CrossRef]
- Ryan, K.R.; Ramirez-Cuesta, A.J.; Refson, K.; Jones, M.O.; Edwards, P.P.; David, W.I.F. A combined experimental inelastic neutron scattering, Raman and ab initio lattice dynamics study of a-lithium amidoborane. Phys. Chem. Chem. Phys. 2011, 13, 12249–12253. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.J.; Titah, J.T.; Che, F.N.; Traboulsee, K.T.; Flogeras, J.; McGrady, G.S. Homopolar Dihydrogen Bonding in Alkali-Metal Amidoboranes and Its Implications for Hydrogen Storage. J. Am. Chem. Soc. 2011, 133, 16598–16604. [Google Scholar] [CrossRef] [PubMed]
- Leardini, F.; Ares, J.R.; Bodega, J.; Valero-Pedraza, M.J.; Bañares, M.A.; Fernández, J.F.; Sánchez, C. Hydrogen Desorption Behavior of Calcium Amidoborane Obtained by Reactive Milling of Calcium Hydride and Ammonia Borane. J. Phys. Chem. C 2012, 116, 24430–24435. [Google Scholar] [CrossRef]
- Banu, T.; Debnath, T.; Ash, T.; Das, A.K. Hydrolysis of ammonia borane and metal amidoboranes: A comparative study. J. Chem. Phys. 2015, 143, 194305. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.C.; Kalnajs, J. The Lattice Constants of the Alkali Borohydrides and the Low-Temperature Phase of Sodium Borohydride. J. Chem Phys. 1954, 22, 434–436. [Google Scholar]
- Juza, R.; Jacobs, H.; Klose, W. Die Kristallstrukturen der Tieftemperaturmodifikationen von Kalium- und Rubidiumamid. Z. Anorg. Allg. Chem. 1965, 338, 171–178. [Google Scholar] [CrossRef]
- Juza, R.; Mehne, A. Zur Kristallstruktur der Alkalimetallamide. Z. Anorg. Allg. Chem. 1965, 299, 33–40. [Google Scholar] [CrossRef]
- Luedtke, A.T.; Autrey, T. Hydrogen Release Studies of Alkali Metal Amidoboranes. Inorg. Chem. 2010, 49, 3905–3910. [Google Scholar] [CrossRef] [PubMed]
- Fijalkowski, K.J.; Jaron, T.; Leszczynski, P.J.; Magos-Palasyuk, E.; Palasyuk, T.; Cyranski, M.K.; Grochala, W. M(BH3NH2BH2NH2BH3)—The missing link in the mechanism of the thermal decomposition of light alkali metal amidoboranes. Phys. Chem. Chem. Phys. 2014, 16, 23340–23346. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Zhang, Y.; Ichikawa, T.; Miyaoka, H.; Kojima, Y. Solid state NMR study on the thermal decomposition pathway of sodium amidoborane NaNH2BH3. J. Mater. Chem. 2011, 21, 2609–2615. [Google Scholar] [CrossRef]
- Shimoda, K.; Doi, K.; Nakagawa, T.; Zhang, Y.; Miyaoka, H.; Ichikawa, T.; Tansho, M.; Shimizu, T.; Burrel, A.K.; Kojima, Y. Comparative Study of Structural Changes in NH3BH3, LiNH2BH3, and KNH2BH3 During Dehydrogenation Process. J. Phys. Chem. C 2012, 116, 5957–5964. [Google Scholar] [CrossRef]
- Kang, K.; Fang, Z.; Kong, L.; Cheng, H.; Yao, X.; Lu, G.; Wang, P. Ammonia Borane Destabilized by Lithium Hydride: An Advanced On-Board Hydrogen Storage Material. Adv. Mater. 2008, 20, 2756–2759. [Google Scholar] [CrossRef] [PubMed]
- Fijalkowski, K.J. Synthesis and Characterization of Amidoboranes of Selected Elements in a Context of Their Ability of Hydrogen Storage. Ph.D. Thesis, University of Warsaw, Warsaw, Poland, 2012. [Google Scholar]
- Owarzany, R.; Fijalkowski, K.J.; Jaron, T.; Leszczynski, P.; Dobrzycki, Ł.; Cyranski, M.K.; Grochala, W. Complete Series of Alkali-Metal M(BH3NH2BH2NH2BH3) Hydrogen-Storage Salts Accessed via Metathesis in Organic Solvents. Inorg. Chem. 2016, 55, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles, U.S. Department of Energy. 2015. Available online: http://energy.gov/sites/prod/files/2015/05/f22/fcto_targets_onboard_hydro_storage_explanation.pdf/ (accessed on 28 April 2016).
- Baitalow, F.; Baumann, J.; Wolf, G.; Jaenicke-Roessler, K.; Leitner, G. Thermal decomposition of B–N–H compounds investigated by using combined thermoanalytical methods. Termochim. Acta 2002, 391, 159–168. [Google Scholar] [CrossRef]
- Bowden, M.; Autrey, T.; Brown, I.; Ryan, M. The thermal decomposition of ammonia borane: A potential hydrogen storage material. Curr. Appl. Phys. 2008, 8, 498–500. [Google Scholar] [CrossRef]
- Gutowska, A.; Li, L.; Shin, Y.; Wang, C.M.; Li, X.S.; Linehan, J.C.; Smith, R.S.; Kay, B.D.; Schmid, B.; Shaw, W.; et al. Nanoscaffold Mediates Hydrogen Release and the Reactivity of Ammonia Borane. Angew. Chem. Int. Ed. 2005, 44, 3578–3582. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.-B.; Gao, L.-L.; Liang, Y.; Kang, X.-D.; Wang, P. Promoted hydrogen generation from ammonia borane aqueous solution using cobalt-molybdenum-boron/nickel foam catalyst. J. Power Sources 2010, 195, 307–312. [Google Scholar] [CrossRef]
- Sandra, F.P.R.; Demirci, U.B.; Chiriac, R.; Moury, R.; Miele, P. A simple preparation method of sodium amidoborane, highly efficient derivative of ammonia borane dehydrogenating at low temperature. Int. J. Hydrog. Energy 2011, 36, 7423–7430. [Google Scholar] [CrossRef]
- Stowe, A.C.; Shaw, W.J.; Linehan, J.C.; Schmid, B.; Autrey, T. In situ solid state 11B MAS-NMR studies of the thermal decomposition of ammonia borane: mechanistic studies of the hydrogen release pathways from a solid state hydrogen storage material. Phys. Chem. Chem. Phys. 2007, 9, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Evans, I.C. Metallated Derivatives of Ammonia Borane with a View to Their Potential as Hydrogen Storage Materials. Ph.D. Thesis, University of Birmingham, Birmingham, West Midlands, UK, 2011. [Google Scholar]
- Tang, Z.; Zhang, L.; Wan, L.; Huang, Z.; Liu, H.; Guo, Z.; Yu, X. Regeneration of alkaline metal amidoboranes with high purity. Int. J. Hydrog. Energy 2016, 41, 407–412. [Google Scholar] [CrossRef]
- Najiba, S.; Chen, J. High-pressure study of lithium amidoborane using Raman spectroscopy and insight into dihydrogen bonding absence. Proc. Natl. Acad. Sci. USA 2012, 109, 19140–149144. [Google Scholar] [CrossRef] [PubMed]
- Magos-Palasyuk, E.; Fijalkowski, K.J.; Palasyuk, T. Chemically driven strong negative linear compressibility in sodium amidoborane, Na(NH2BH3). Sci. Rep. 2016, 6, 28745. [Google Scholar] [CrossRef] [PubMed]
- Magos-Palasyuk, E.; Palasyuk, T.; Zaleski-Ejgierd, P.; Fijalkowski, K.J. Hydrogen-mediated affinity of ions found in compressed potassium amidoborane, K[NH2BH3]. CrystEngComm 2014, 16, 10367–10370. [Google Scholar] [CrossRef]
- Zhang, Y.; Shimoda, K.; Ichikawa, T.; Kojima, Y. Activation of Ammonia Borane Hybridized with Alkaline-Metal Hydrides: A Low-Temperature and High-Purity Hydrogen Generation Material. J. Phys. Chem. 2010, 114, 14662–14664. [Google Scholar] [CrossRef]
- Li, W.; Miao, L.; Scheicher, R.H.; Xiong, Z.; Wu, G.; Araújo, C.M.; Blomqvist, A.; Ahuja, R.; Feng, Y.P.; Chen, P. Li-Na Ternary Amidoborane for Hydrogen Storage: Experimental and First-Principles Study. Dalton Tran. 2012, 41, 4754–4764. [Google Scholar] [CrossRef] [PubMed]
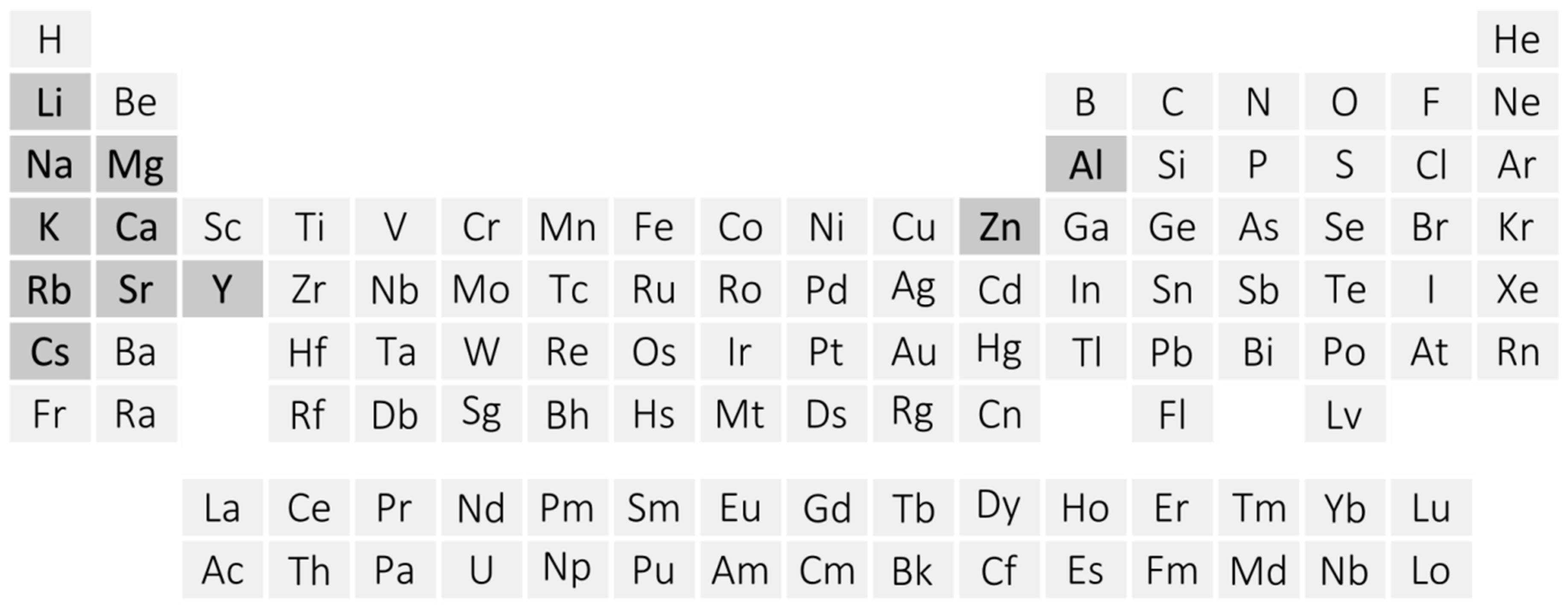
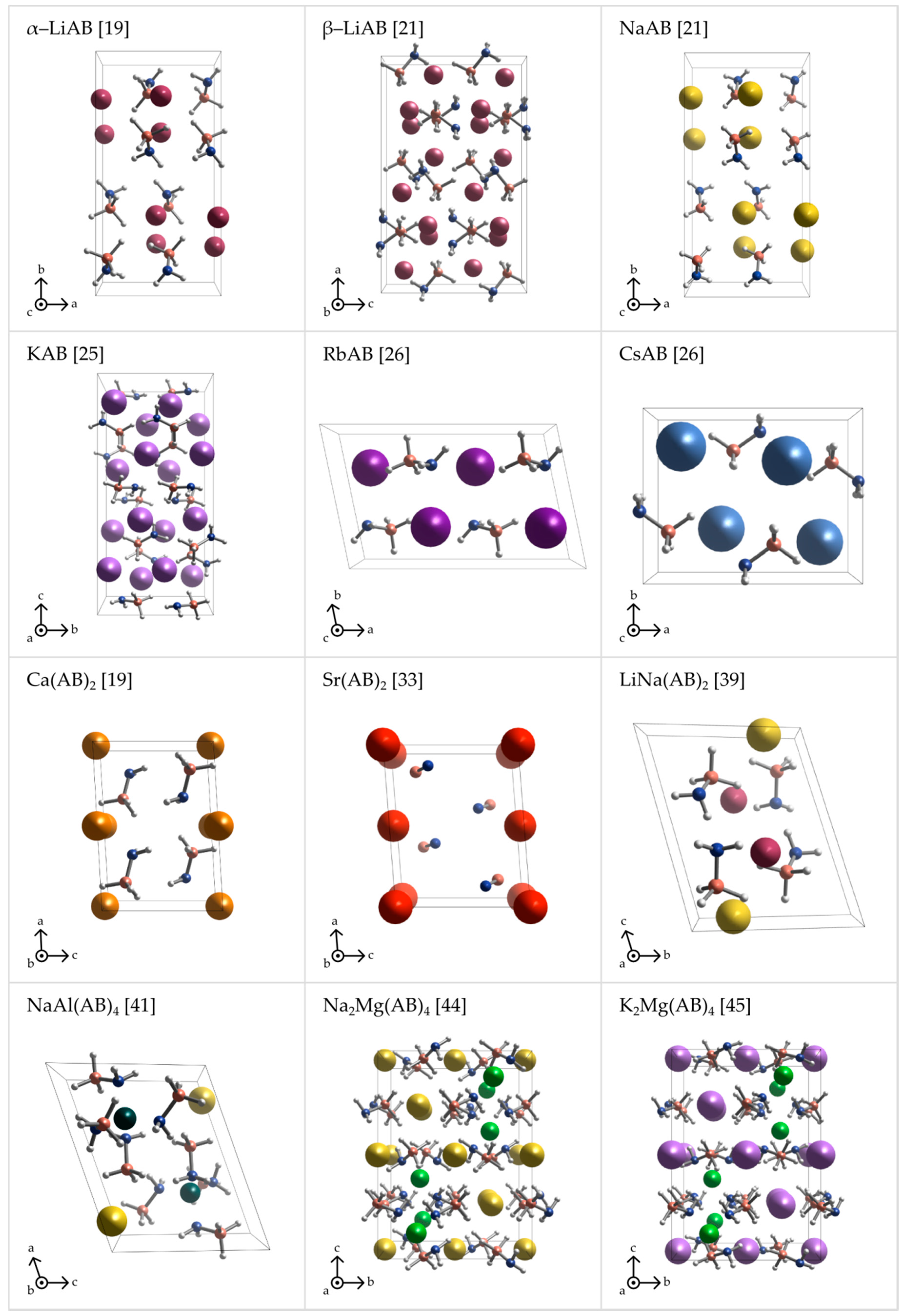
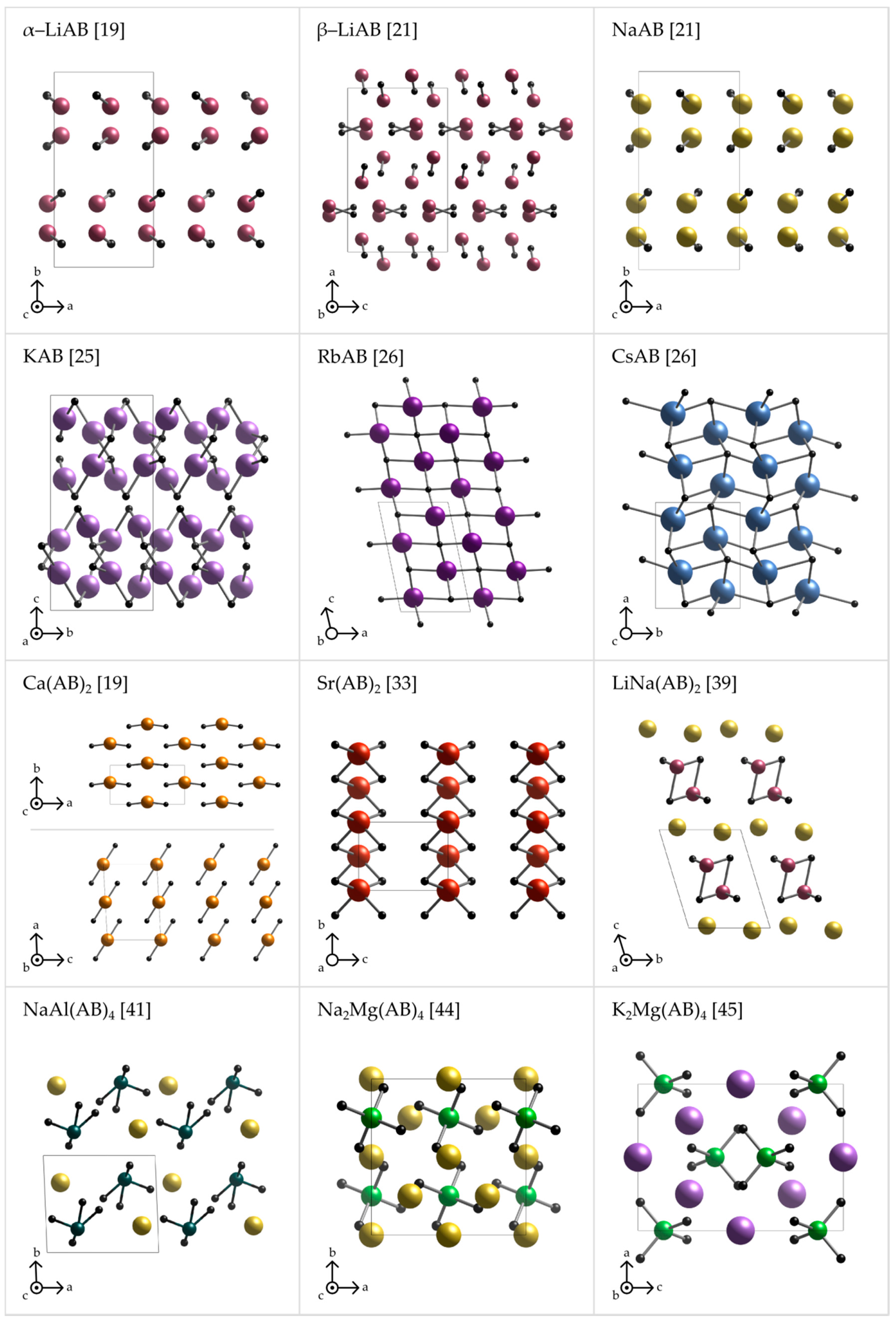
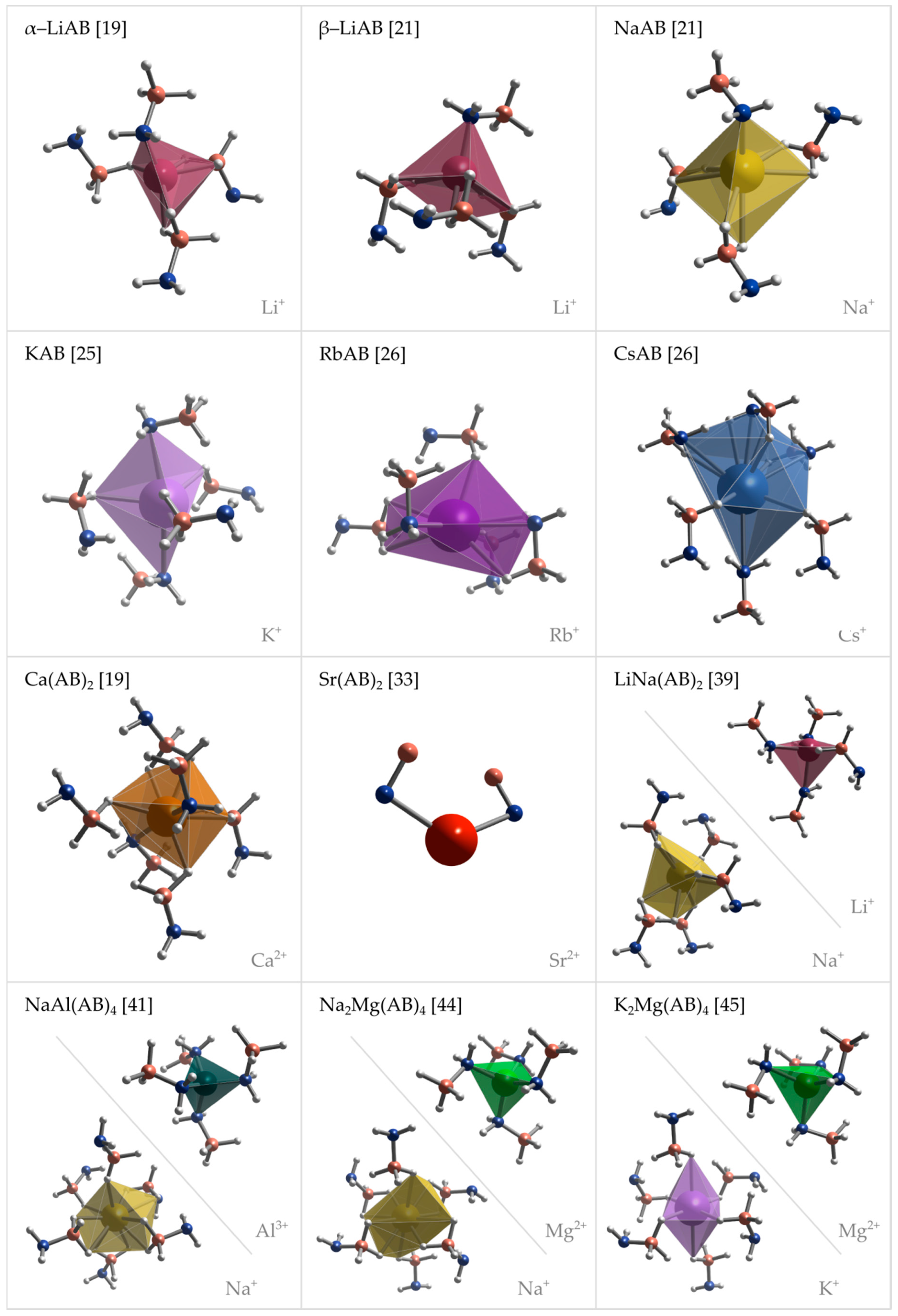
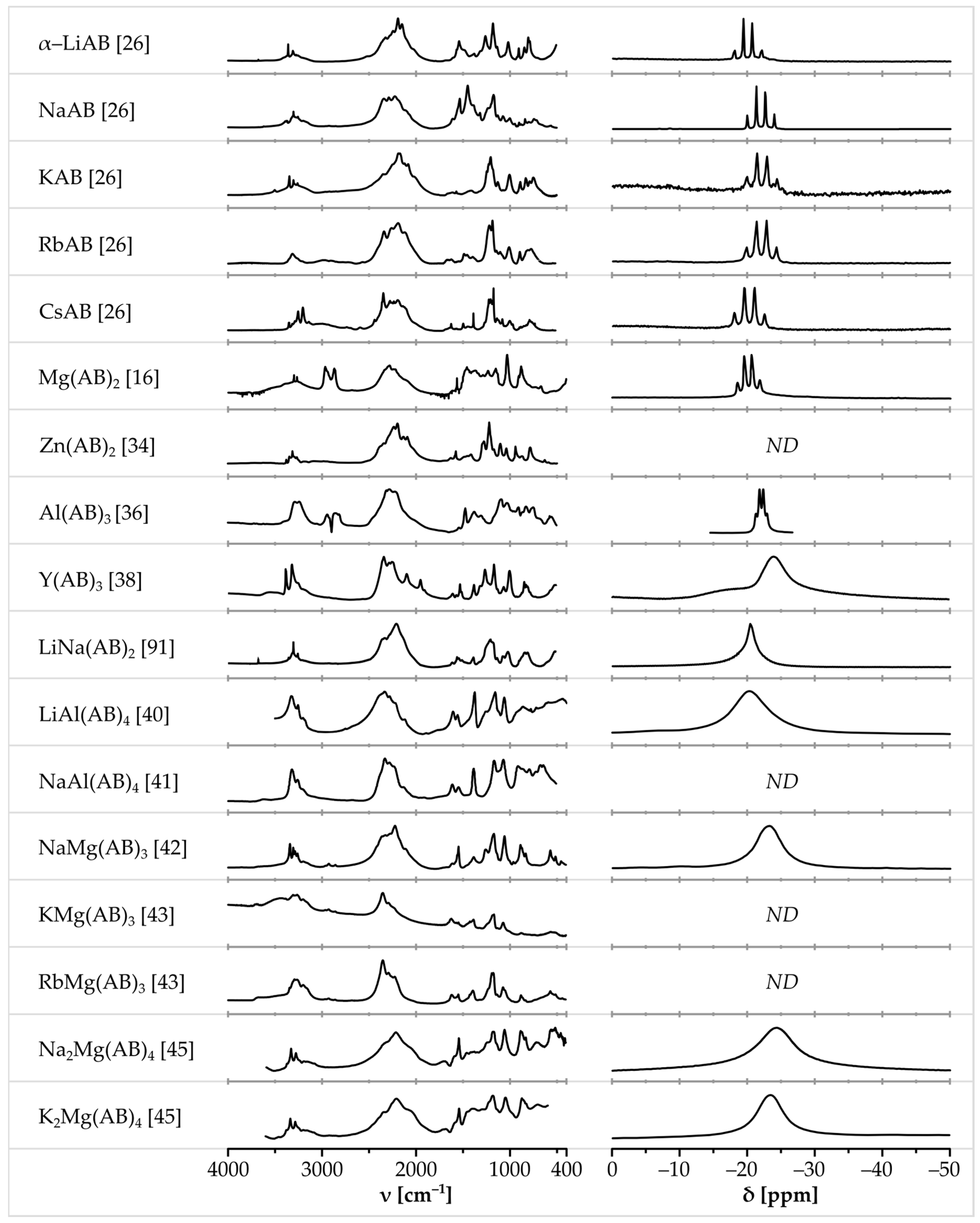
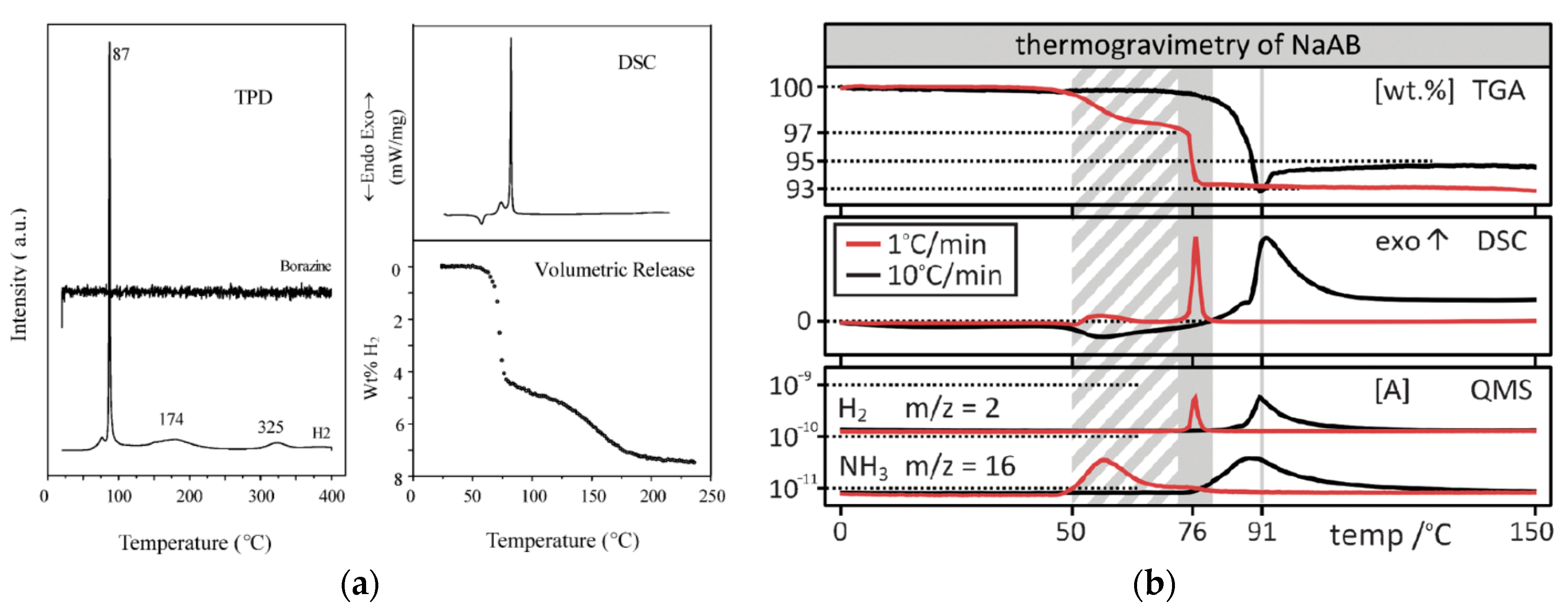
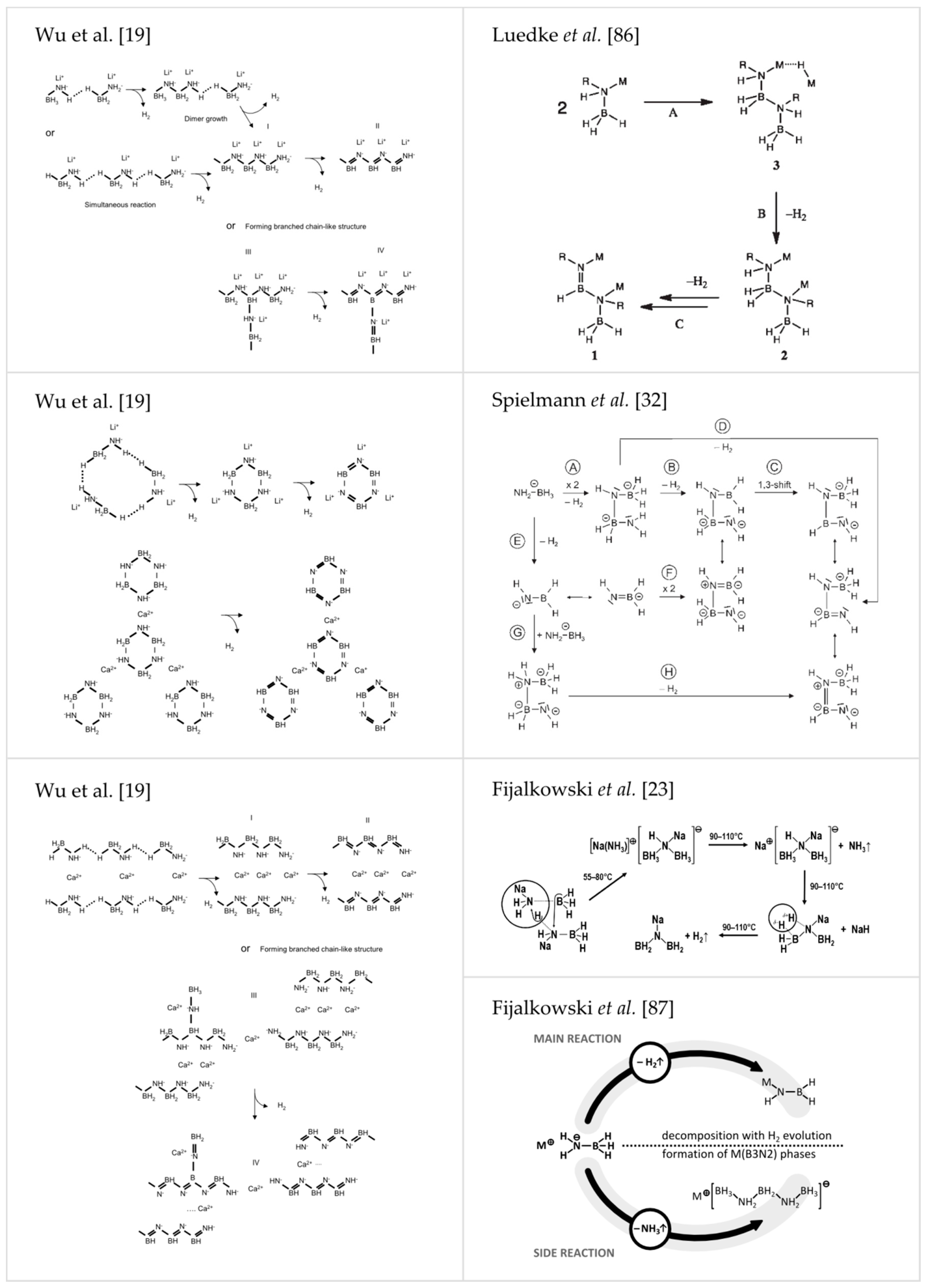
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).