A Review on Metal Nanoparticles Nucleation and Growth on/in Graphene



Abstract
:1. Introduction: Metal-Based Graphene Nanocomposites in the Nanotechnology Revolution
2. Adsorption and Diffusion of Metal Atoms on/in Graphene and Nanoparticles Nucleation and Growth
2.1. Adsorption and Diffusion of Metals Atoms on/in Graphene: Theoretical Results
2.1.1. General Considerations
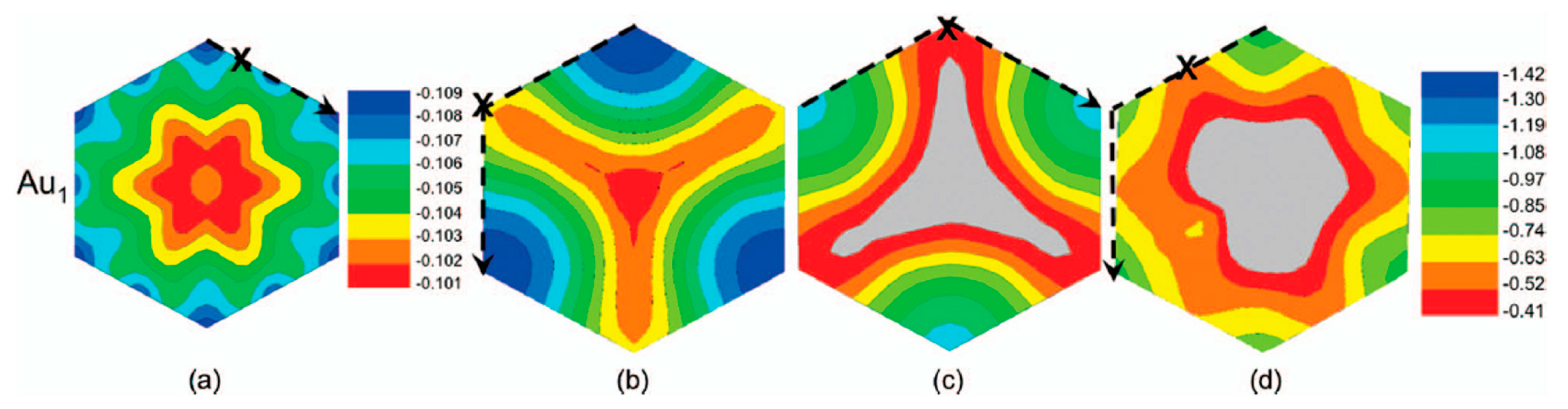
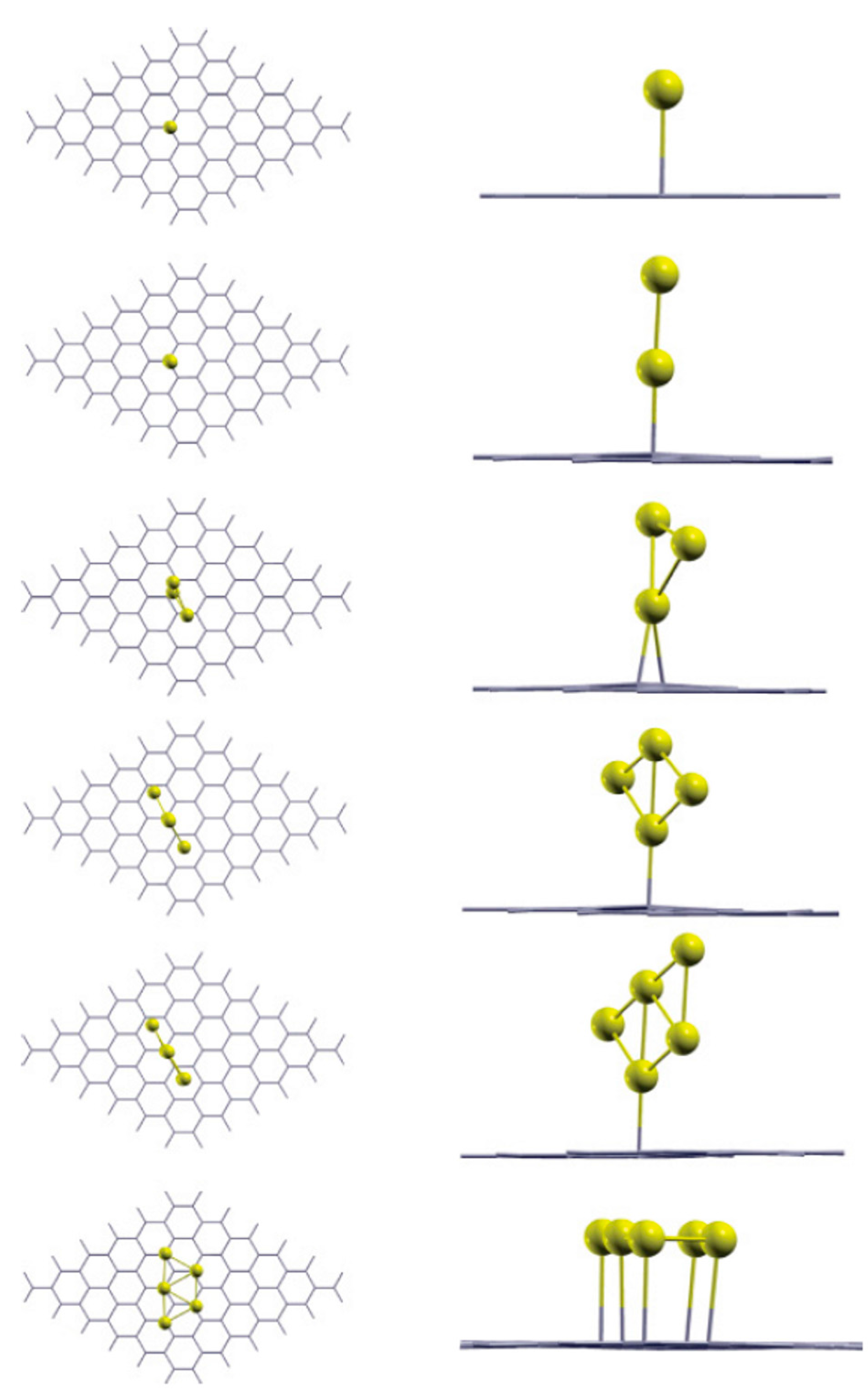
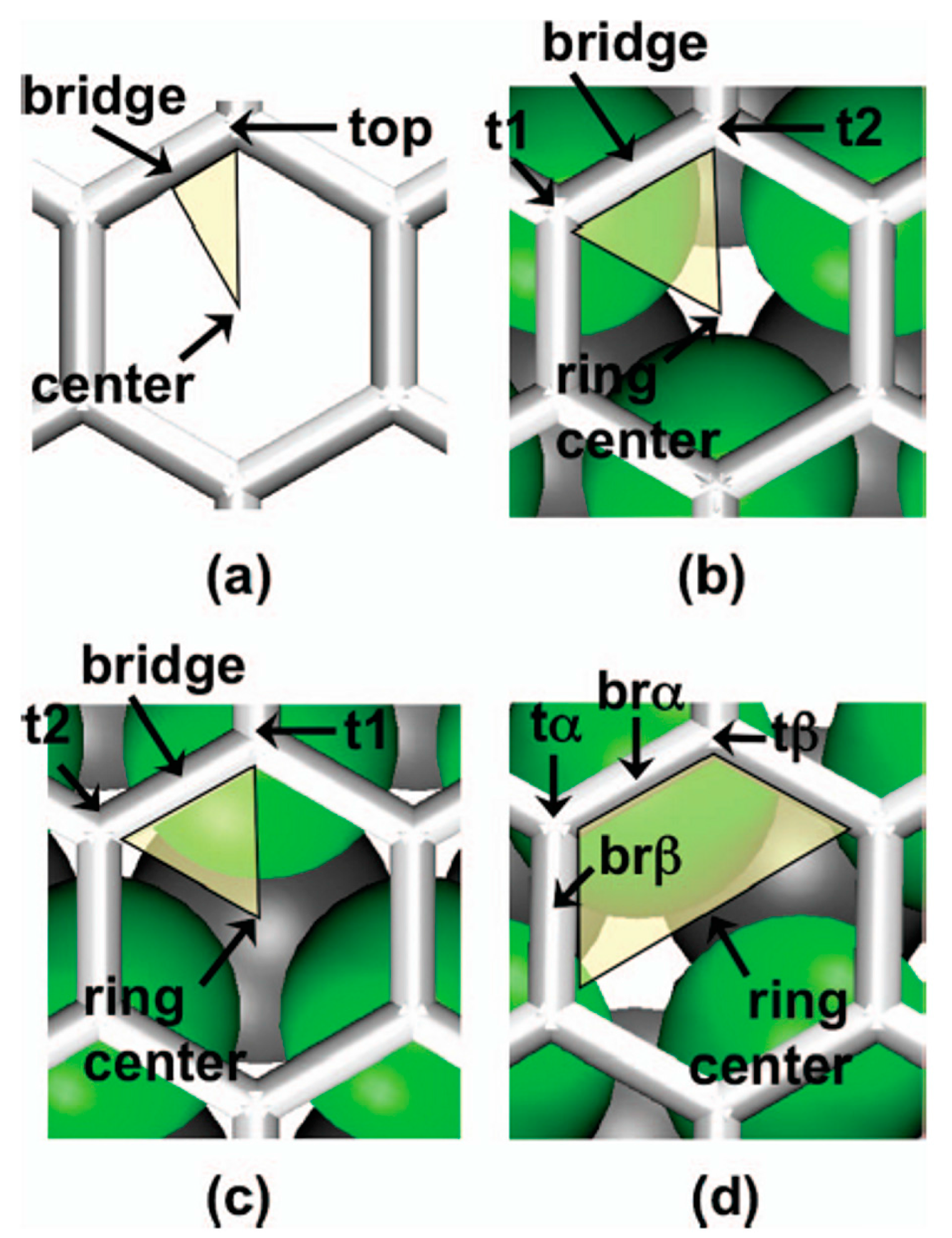
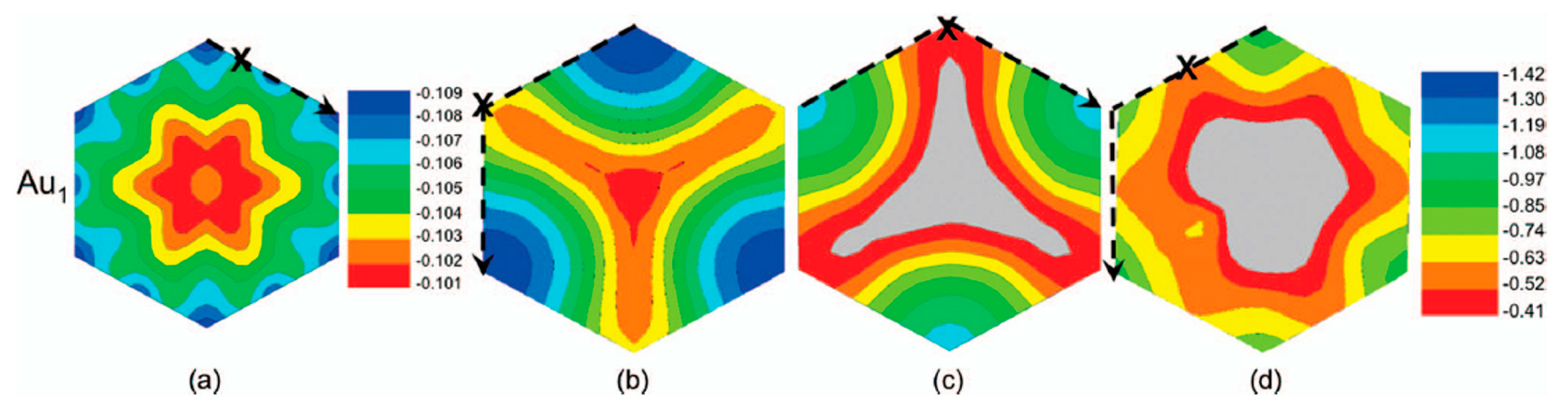
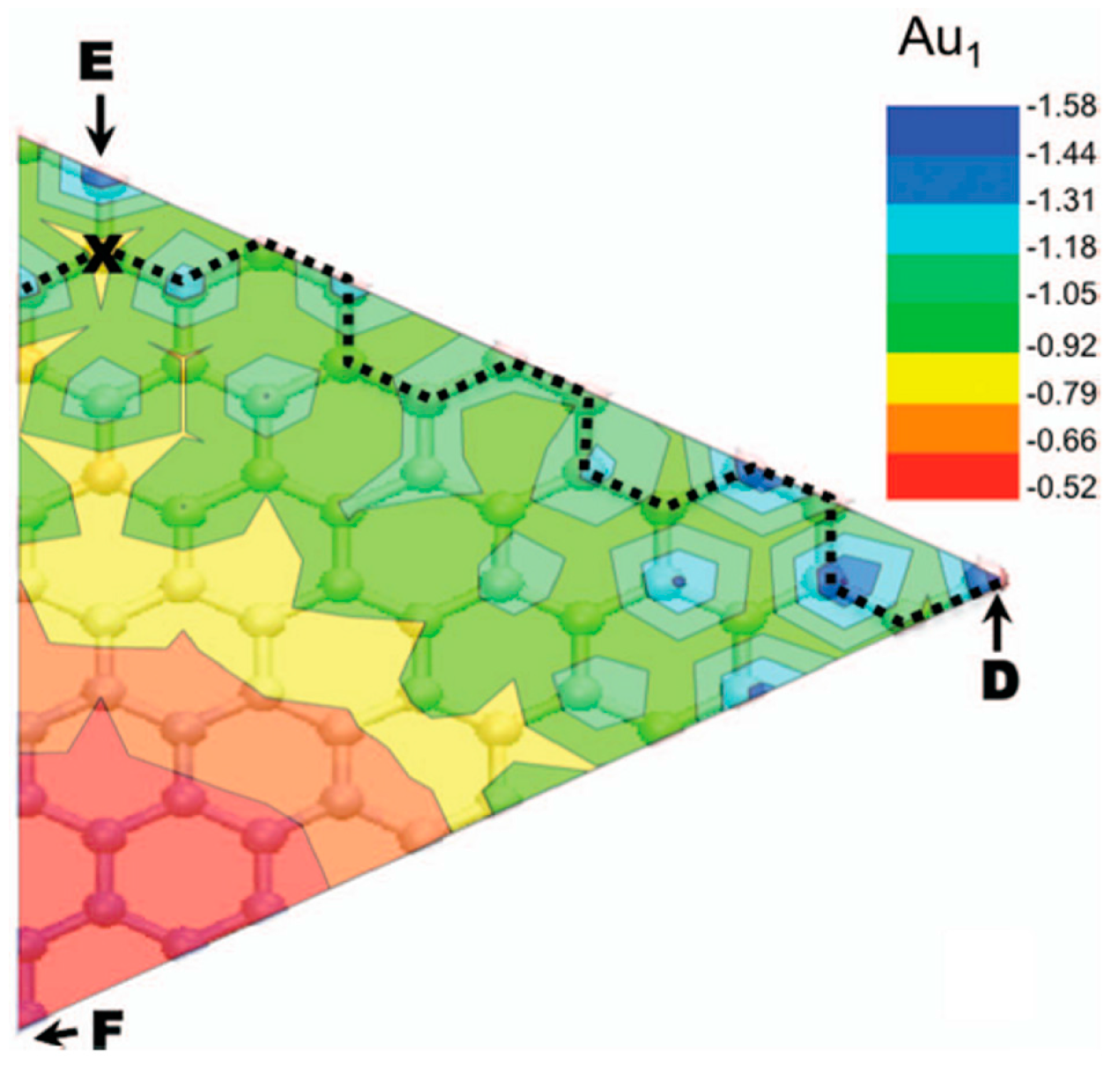
2.1.2. Mobility and Clustering of Au on Graphene
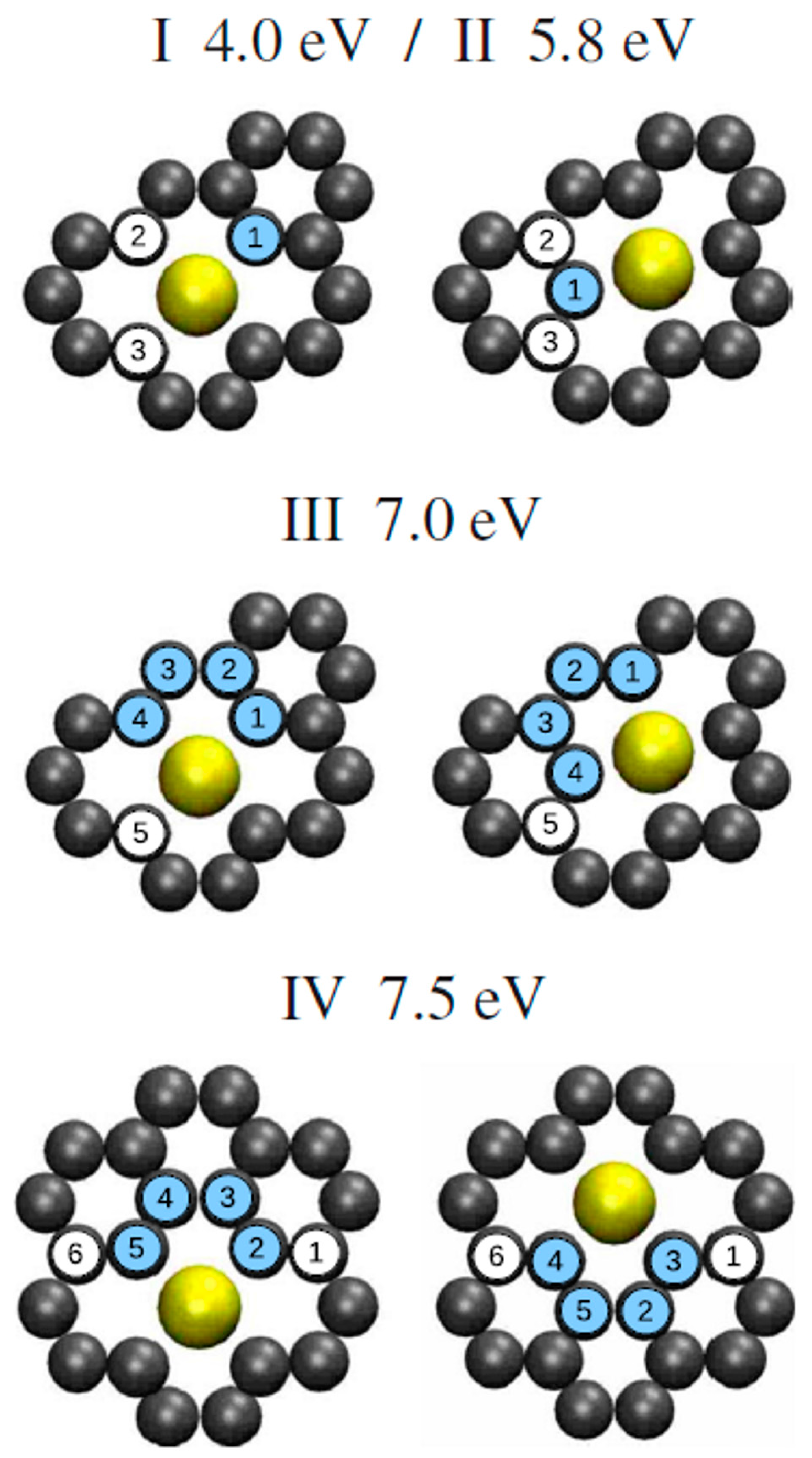
2.1.3. Adsorption and Diffusion of Au on Graphene/Ru(0001)
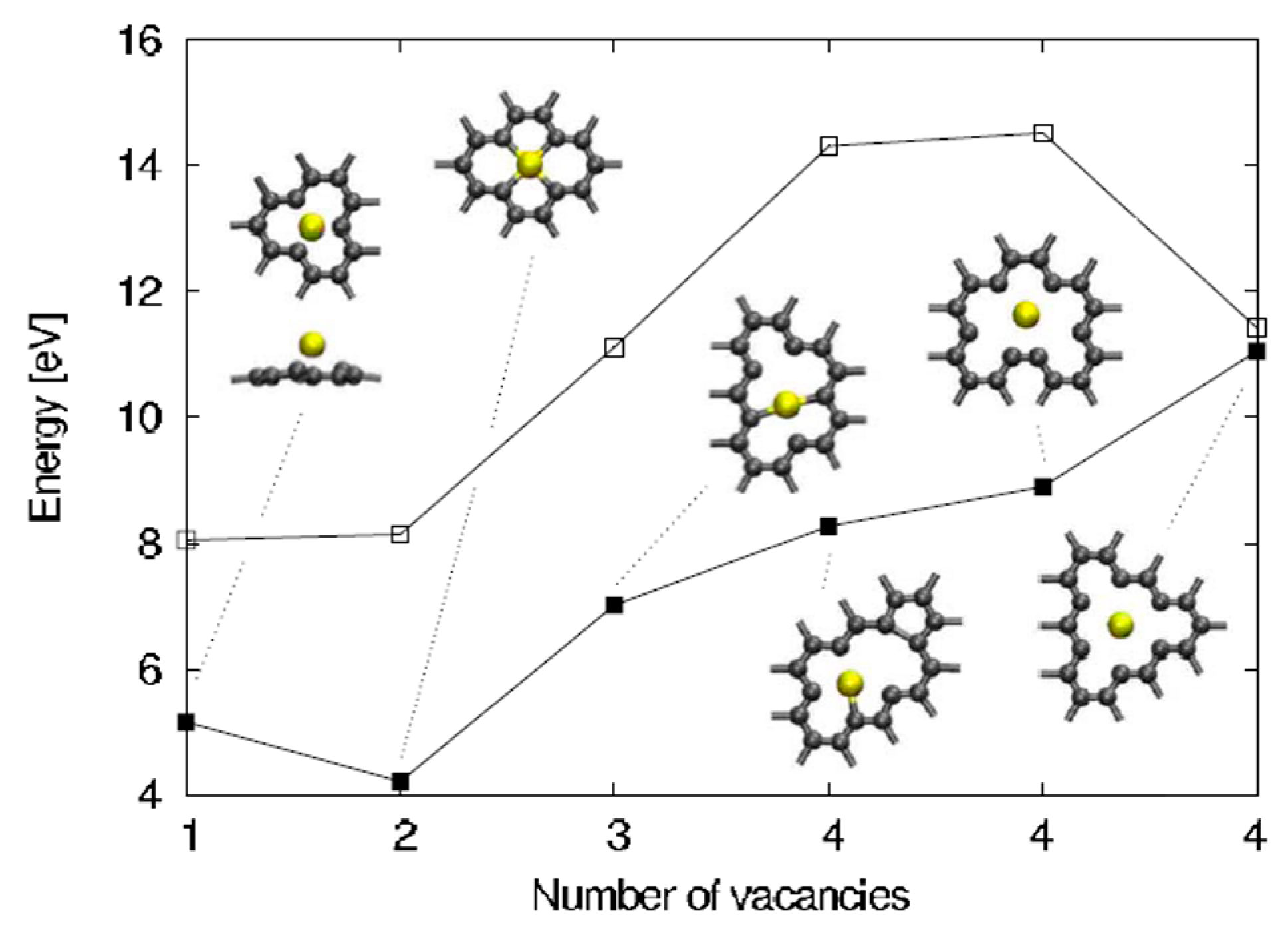
2.1.4. In-Plane Adsorption and Diffusion of Au in Graphene
2.2. Adsorption, Diffusion, Nucleation and Growth of Metal Atoms on/in Graphene: Experimental Results
2.2.1. General Considerations
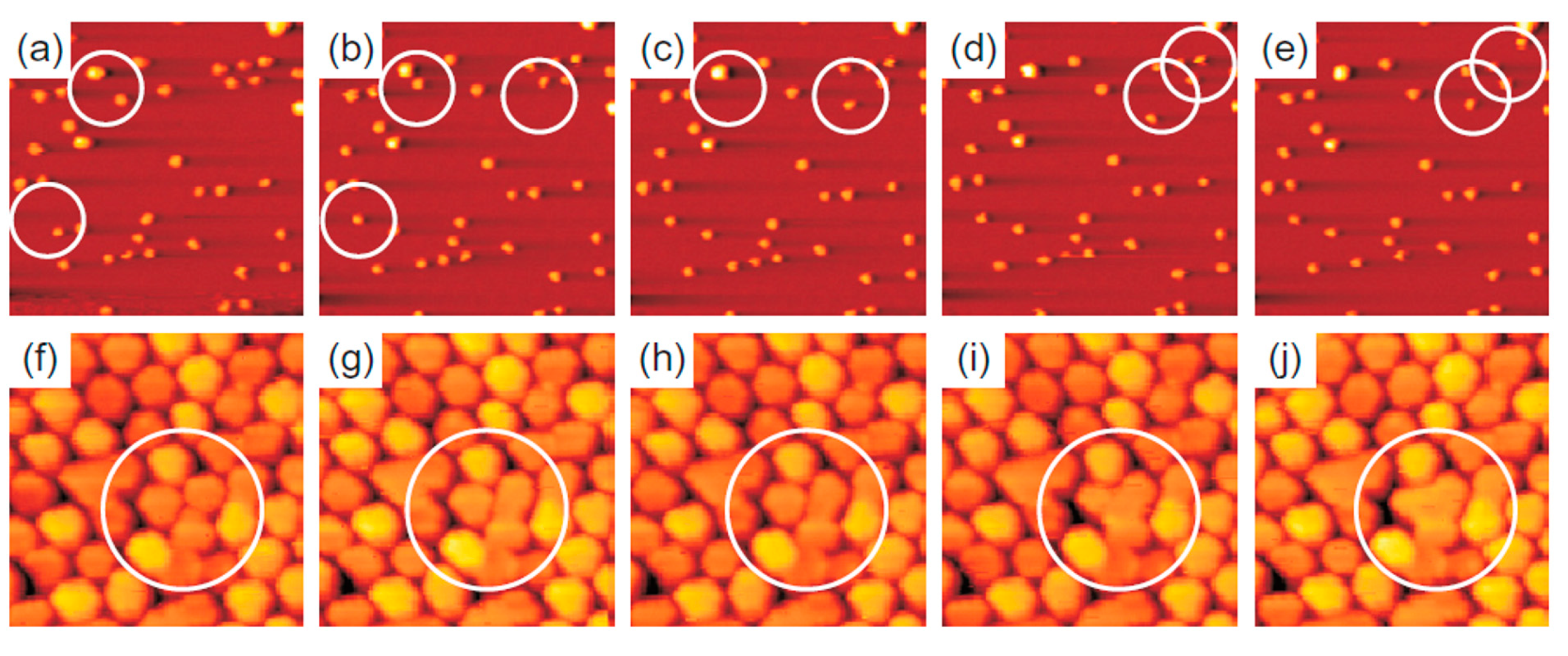
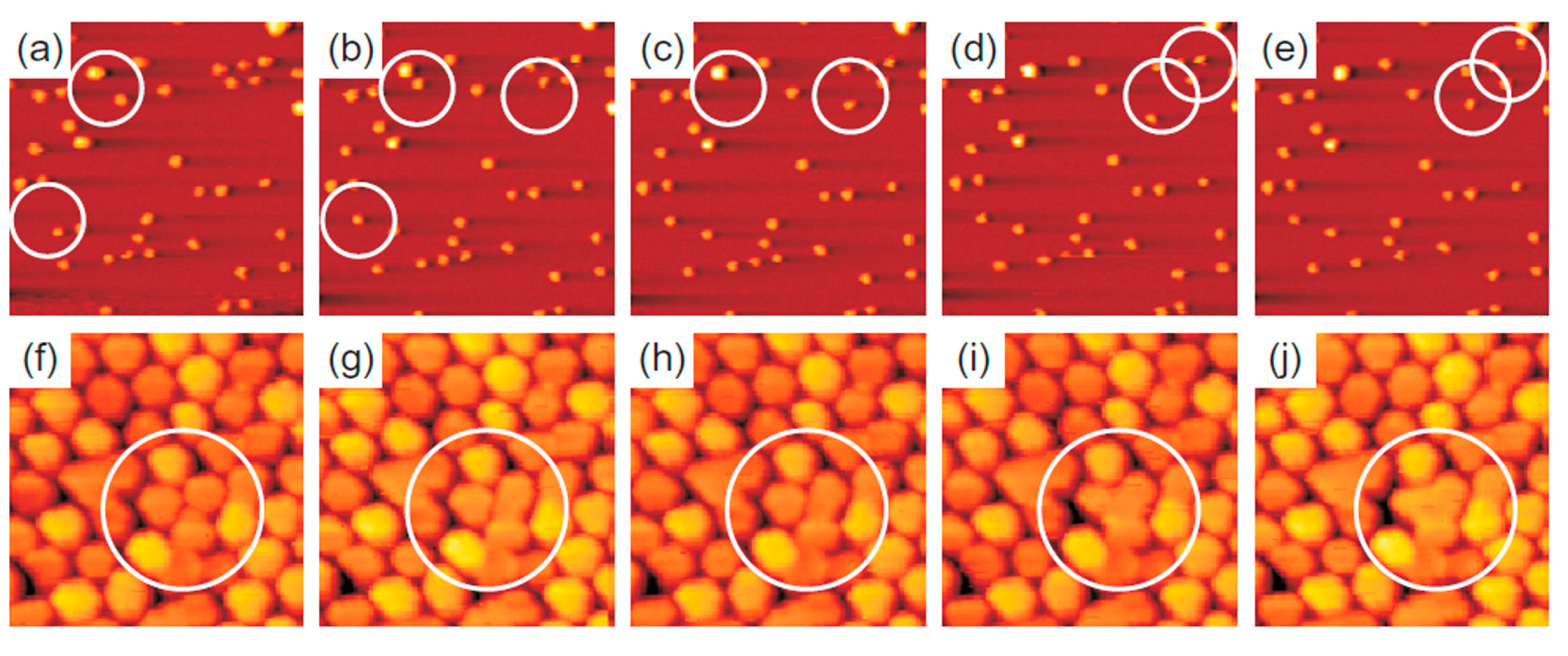
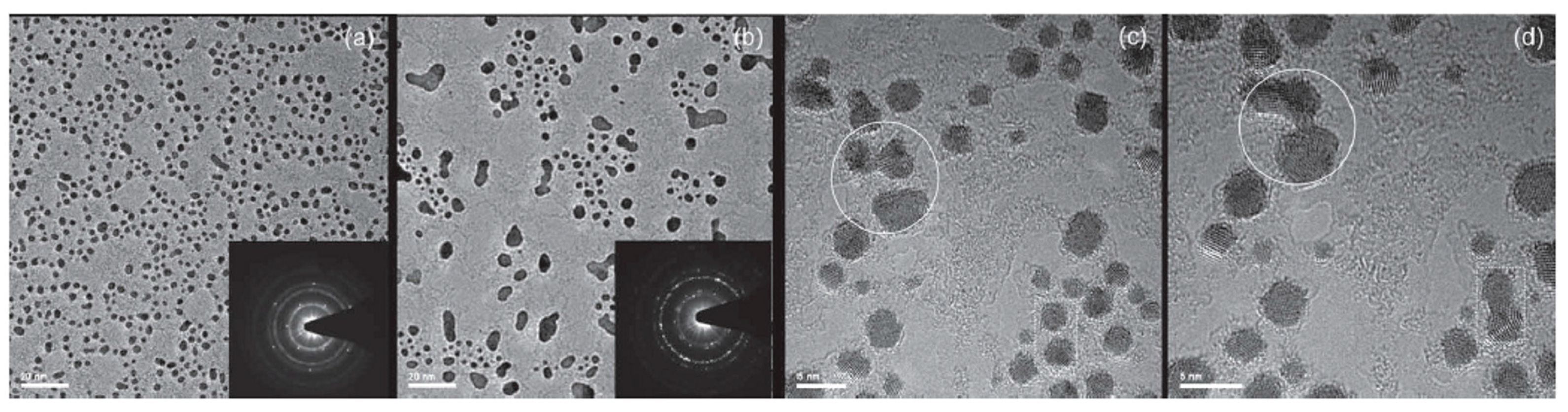
2.2.2. Au Nanoparticles on Graphene
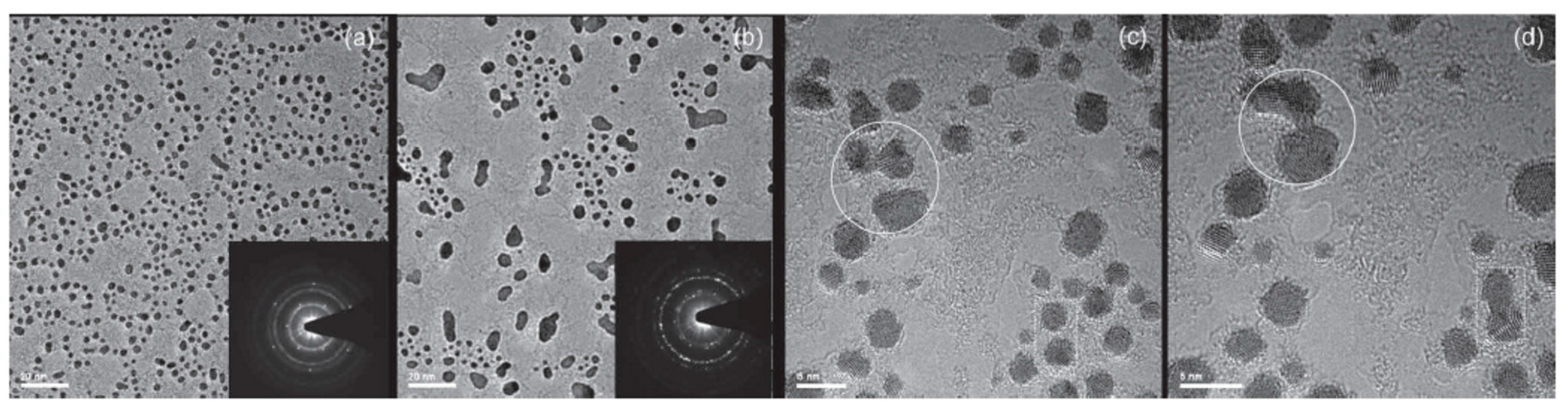
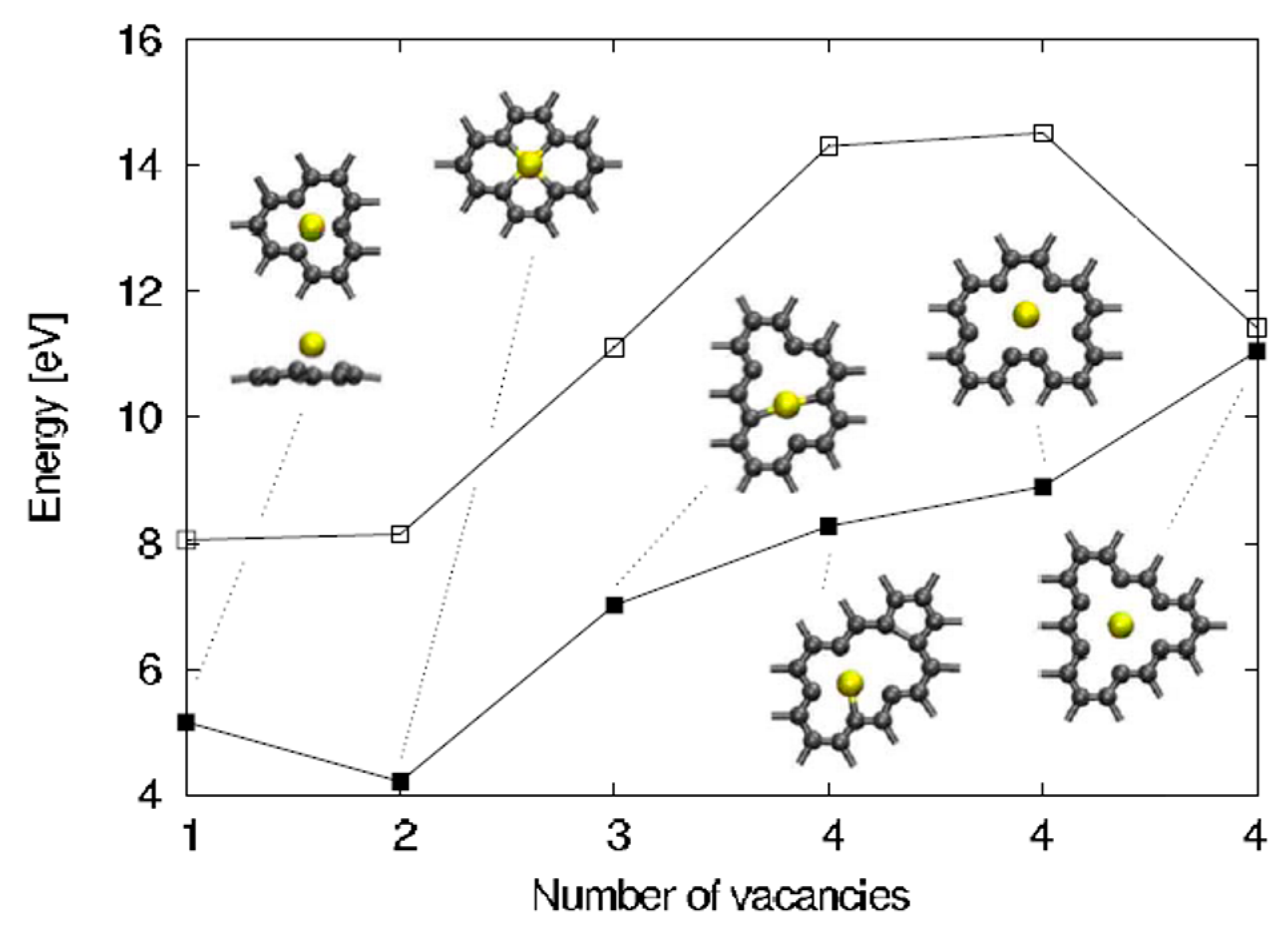
2.2.3. Au and Pt Nanoparticles in Graphene
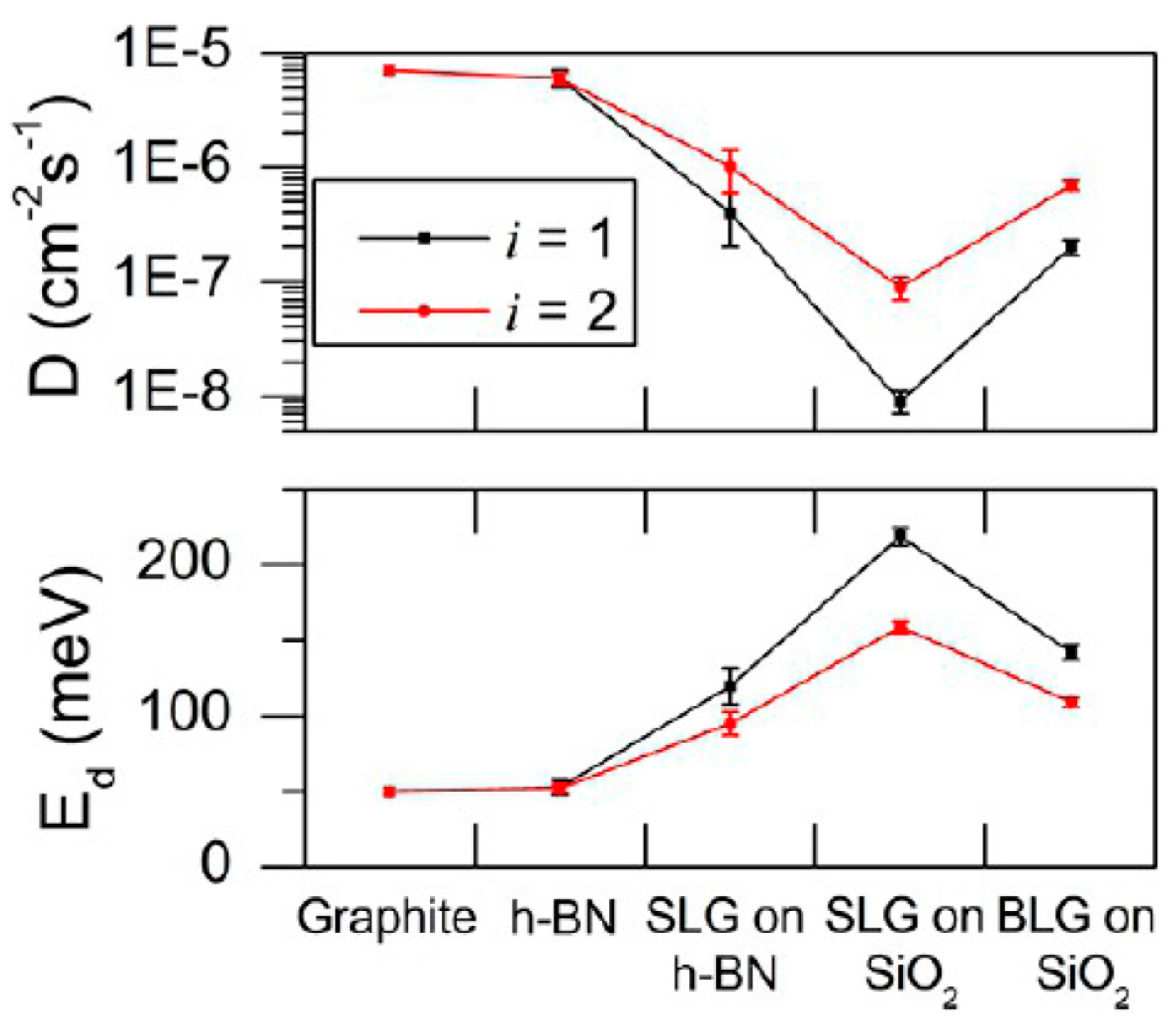
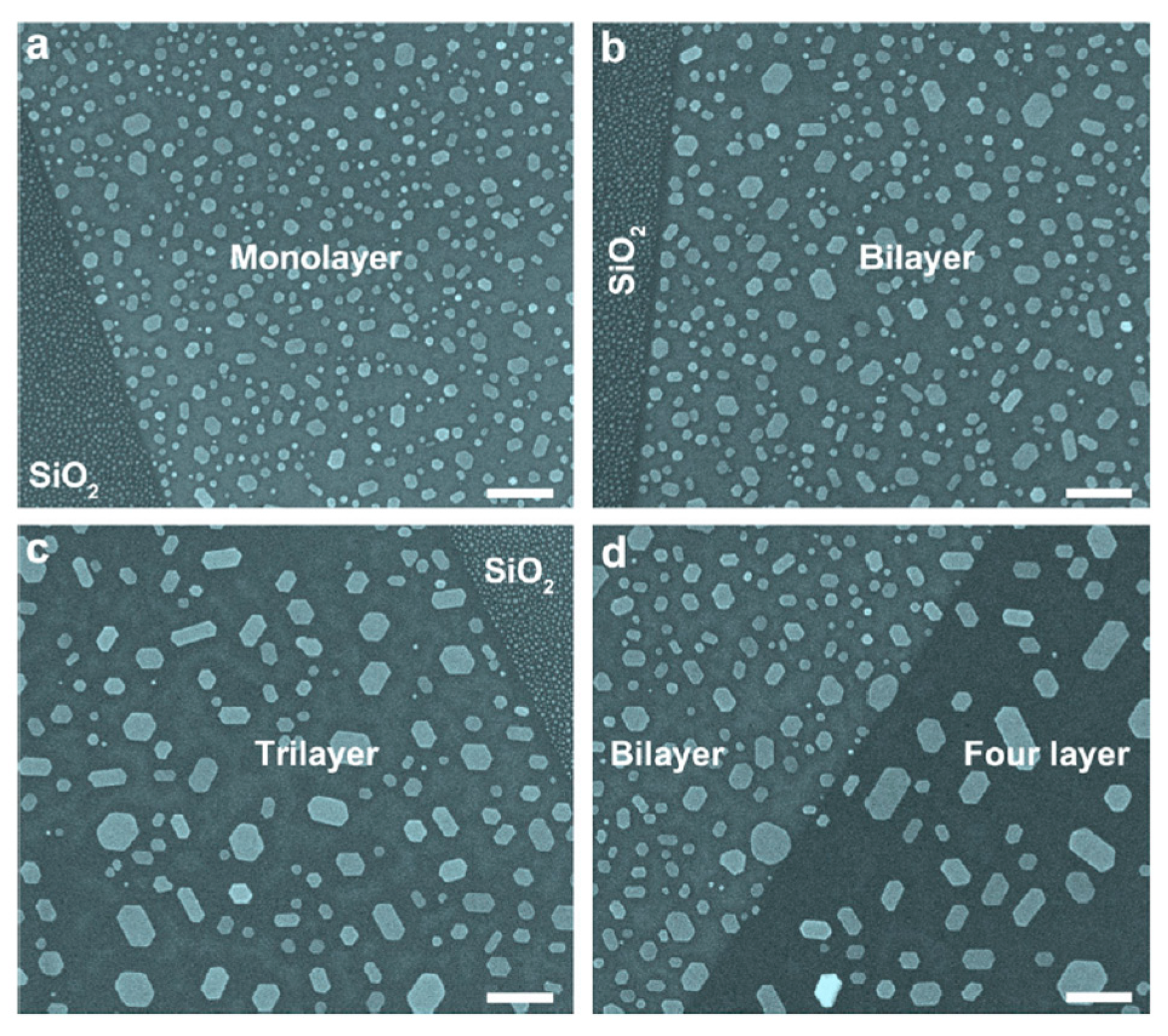
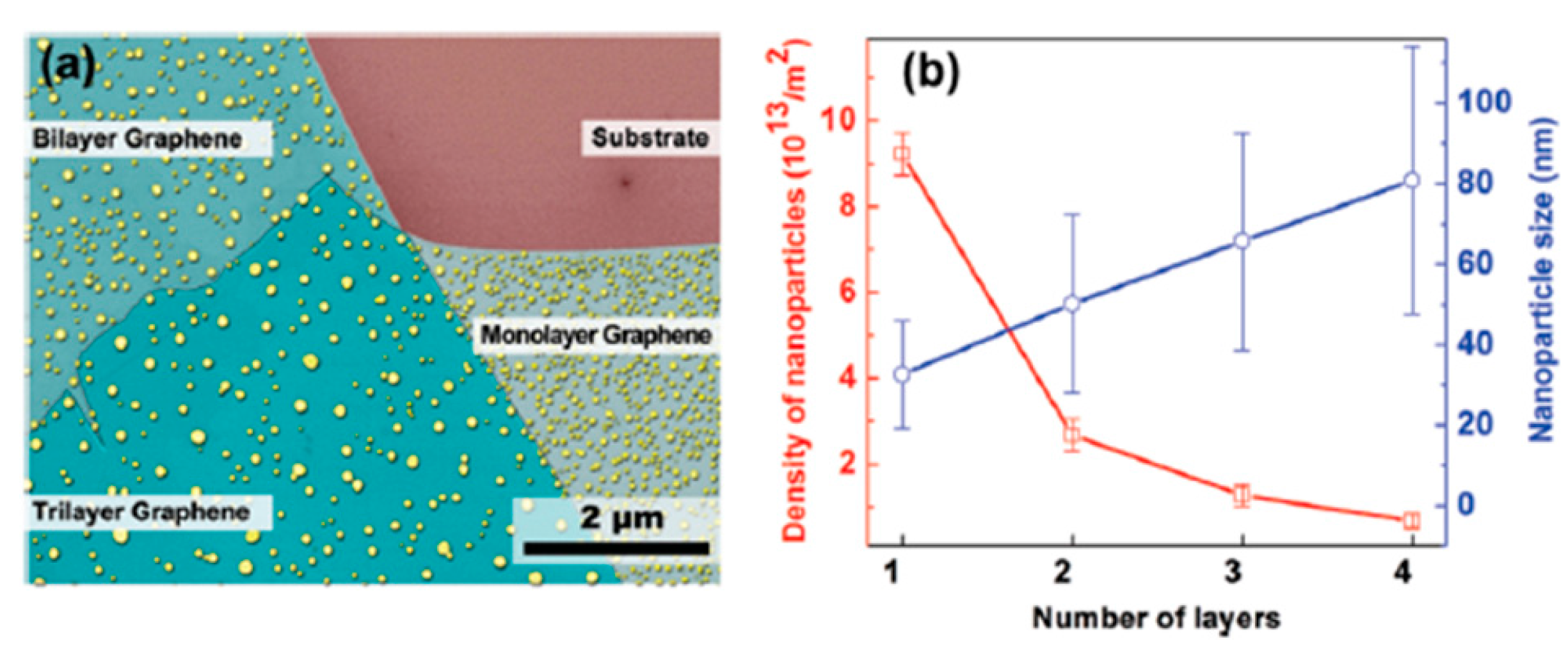
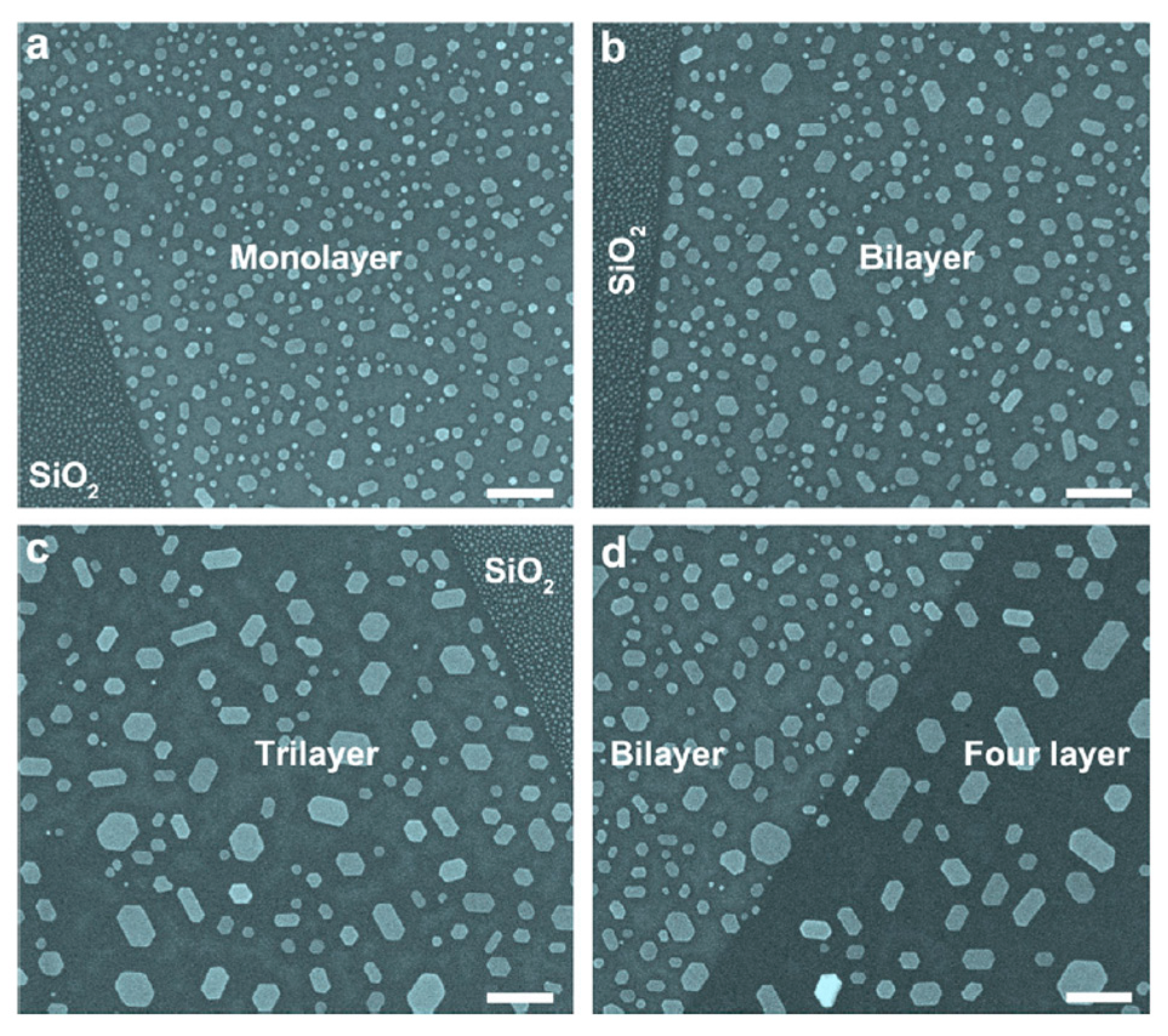
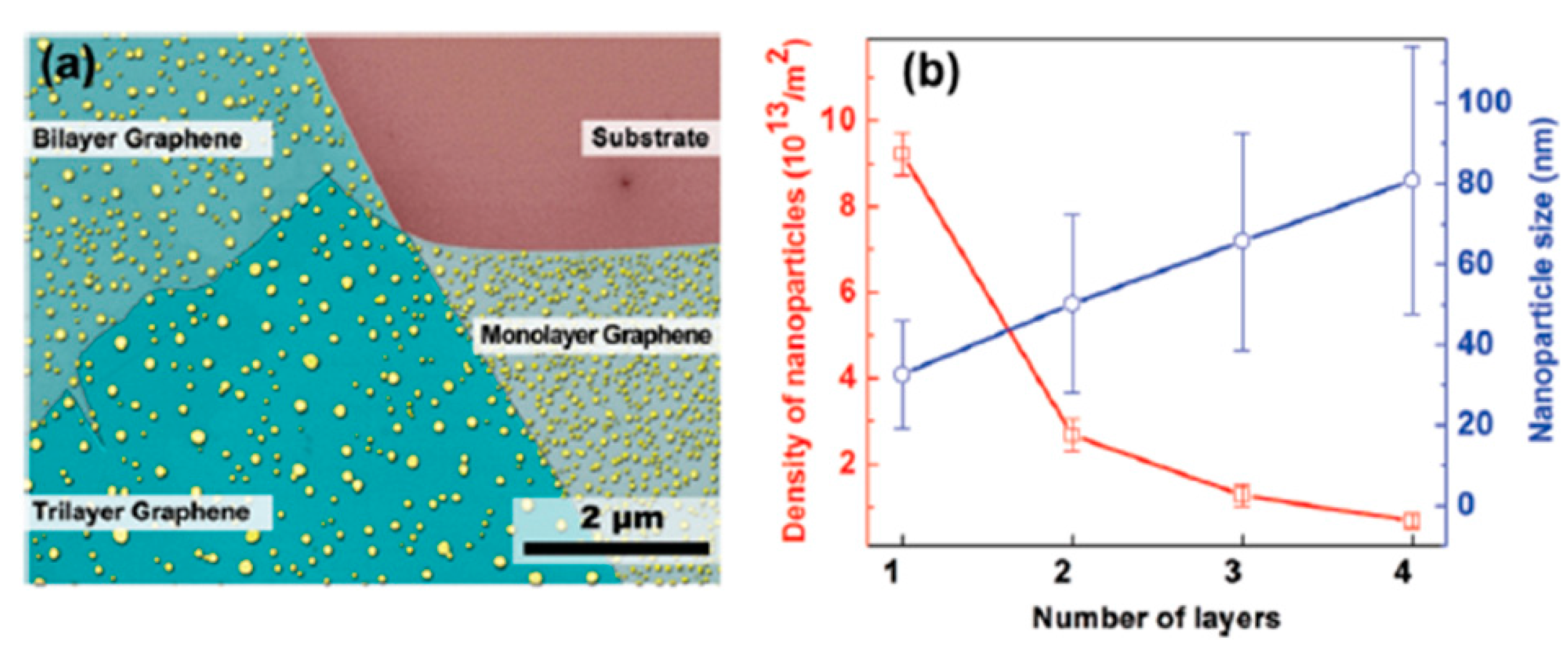
2.2.4. Au Nanoparticles on Graphene Supported on Different Substrates
3. Thin Metal Films Deposition on Graphene and Nanoparticles Formation by Dewetting Processes
3.1. The Dewetting Process
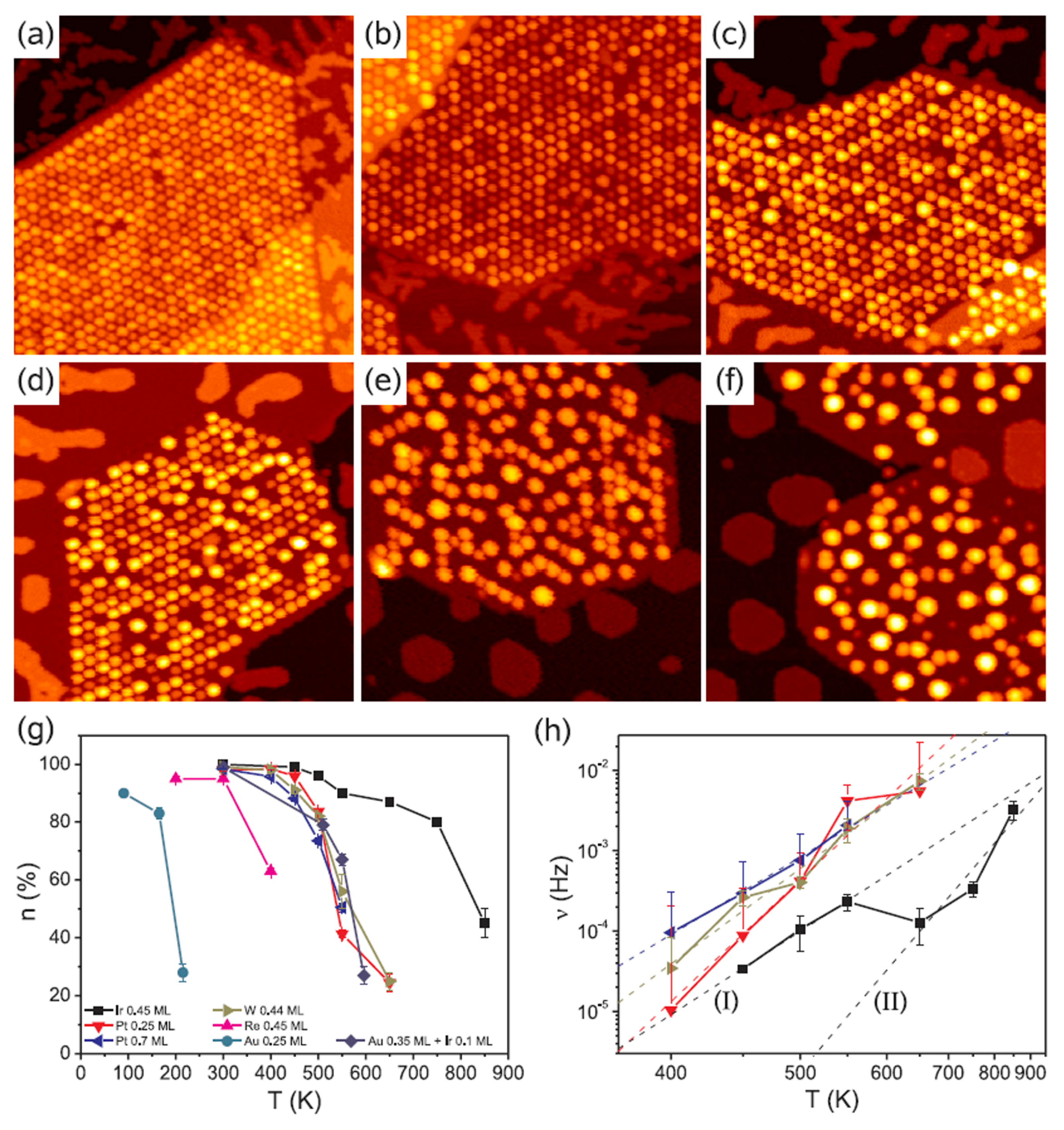
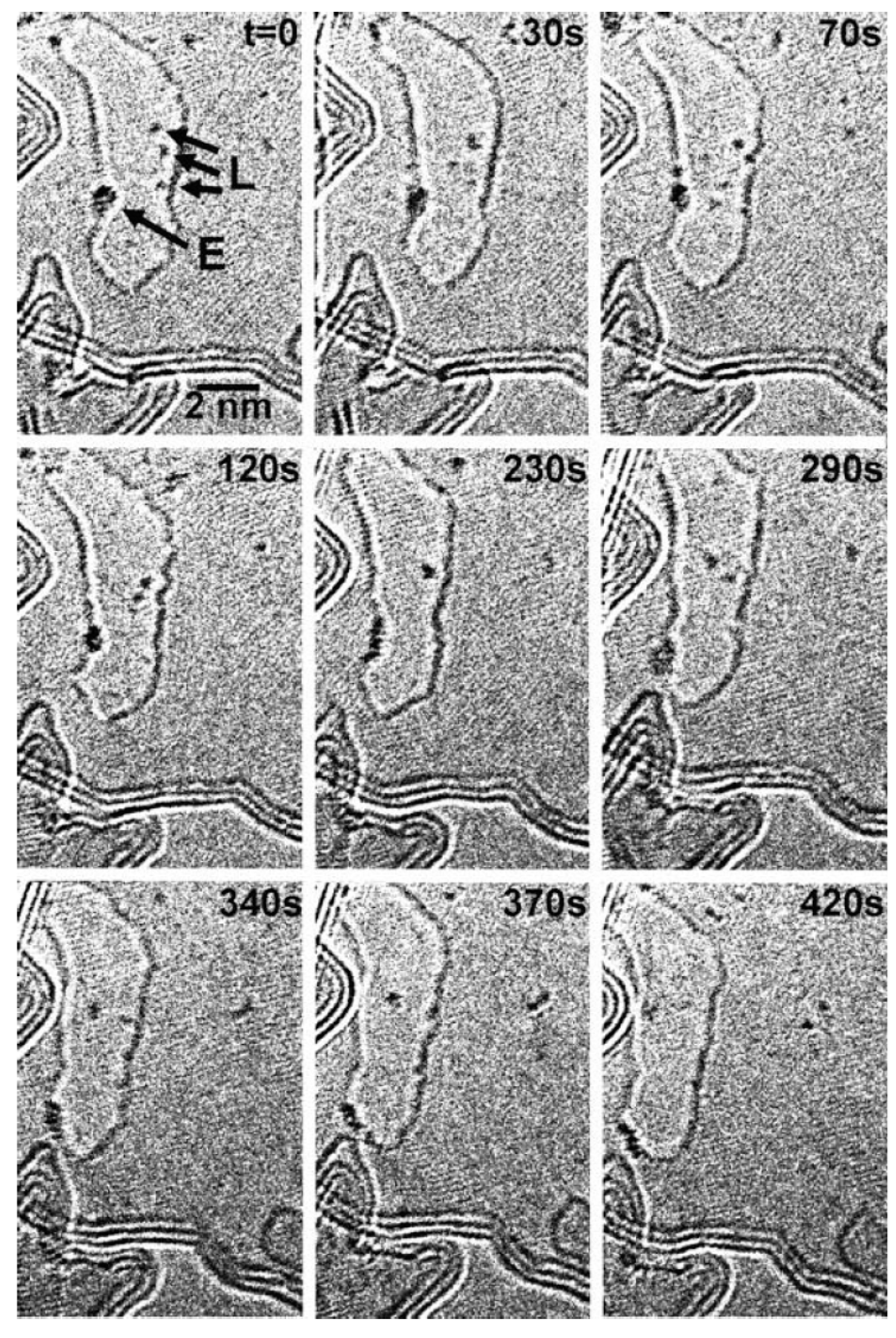
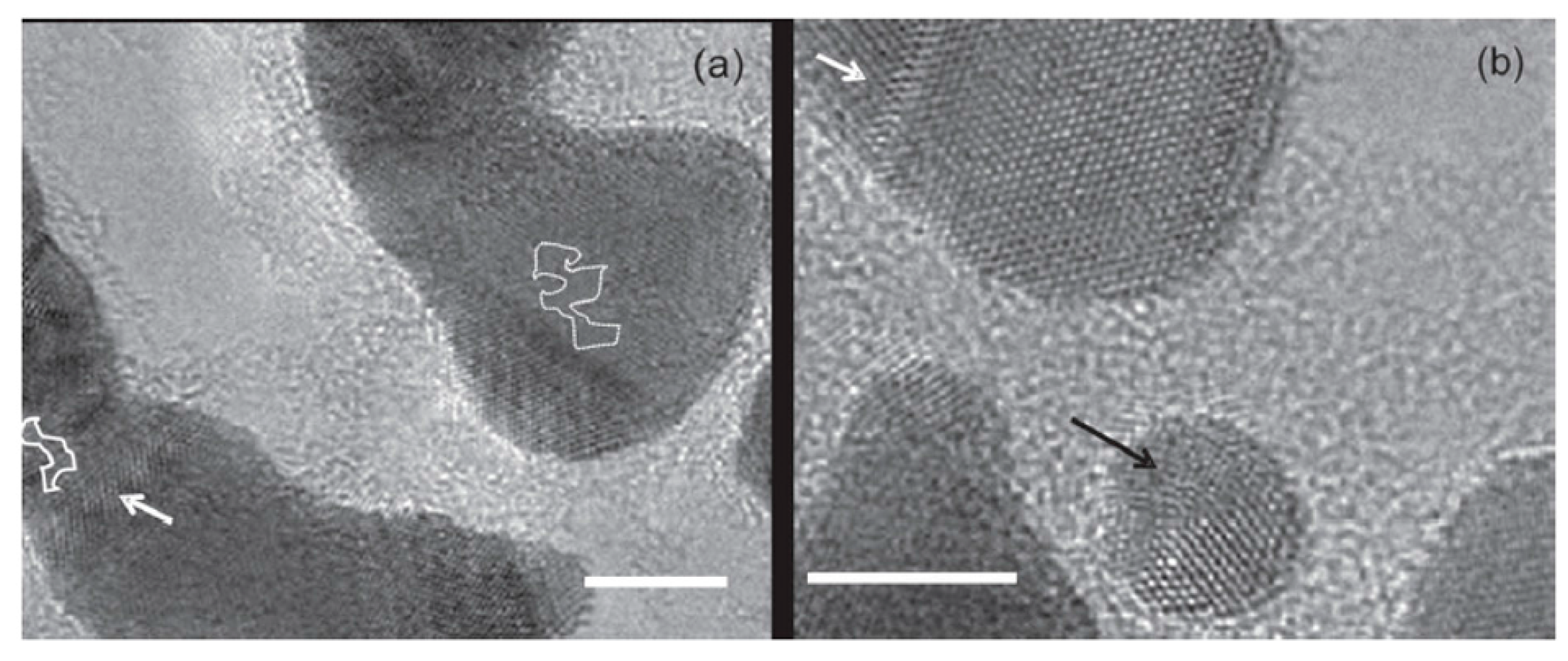
3.2. Dewetting of Au Films on Graphene
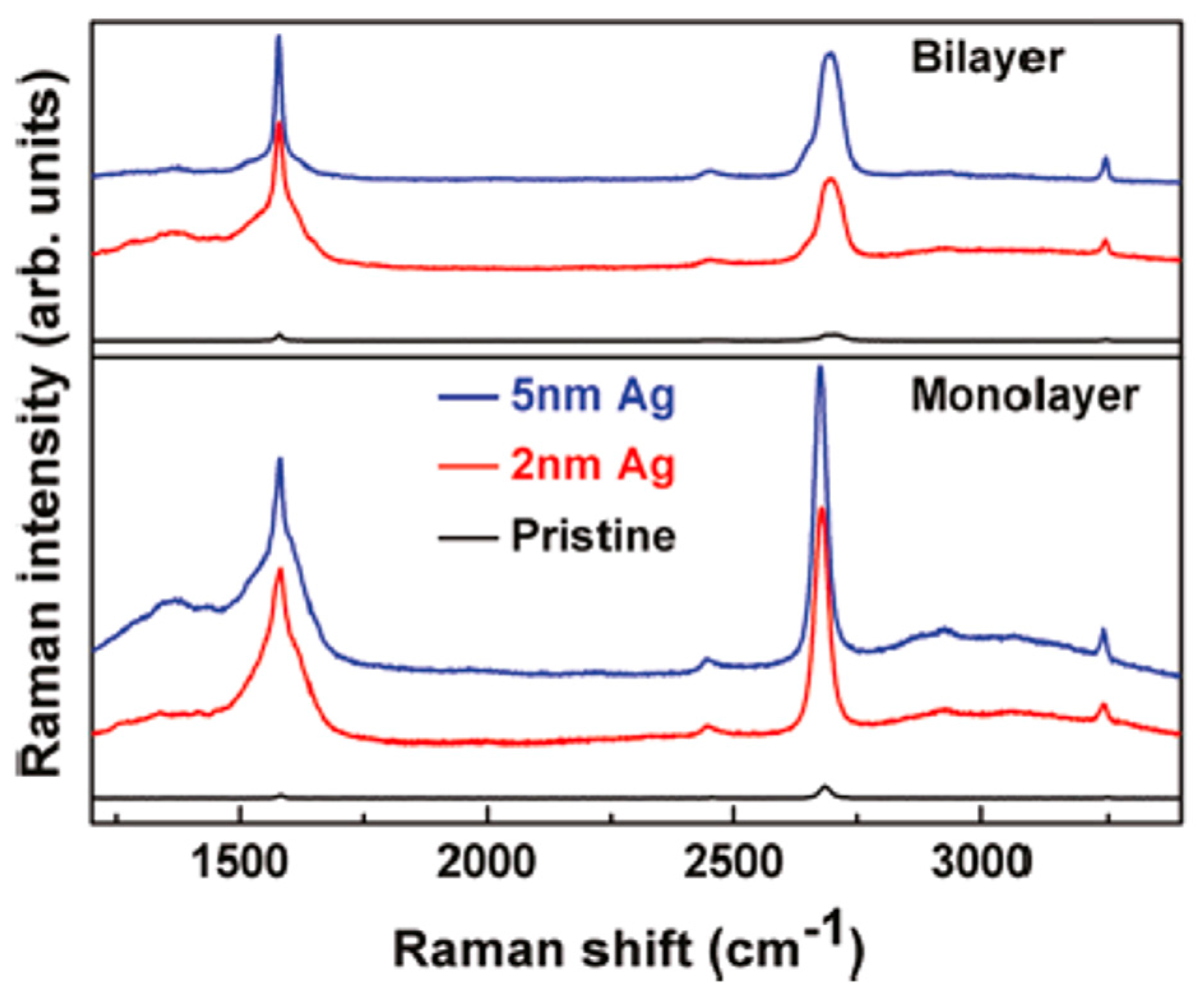
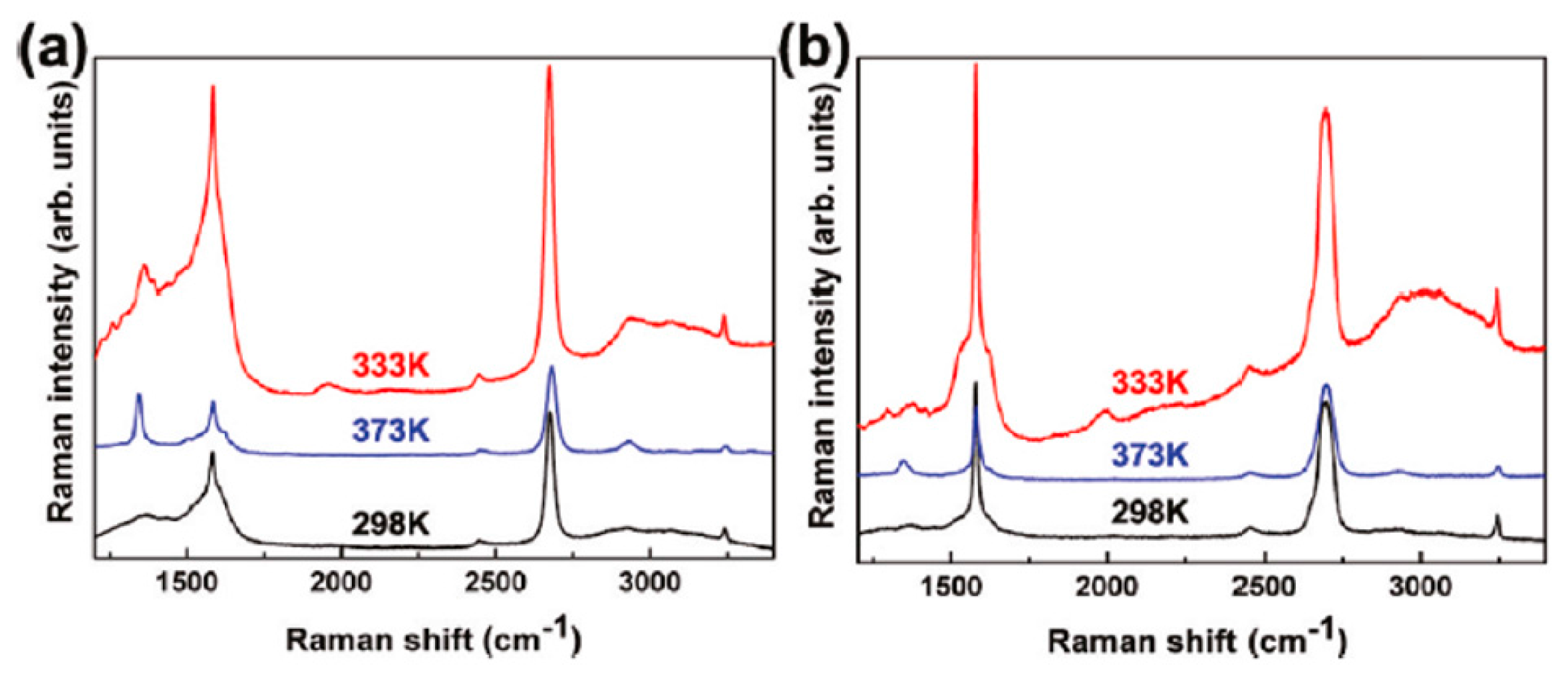
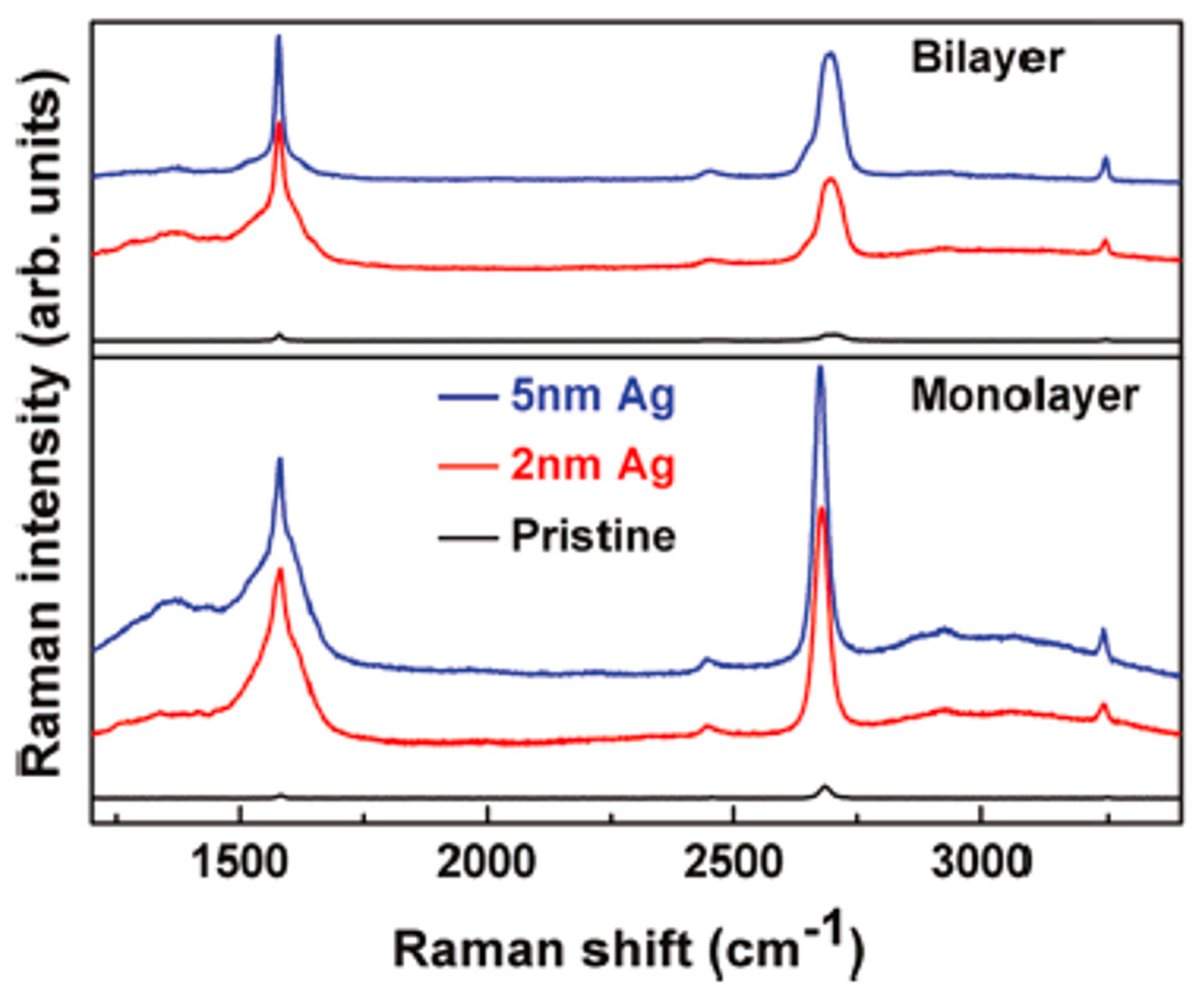
3.3. Dewetting of Ag Films on Graphene
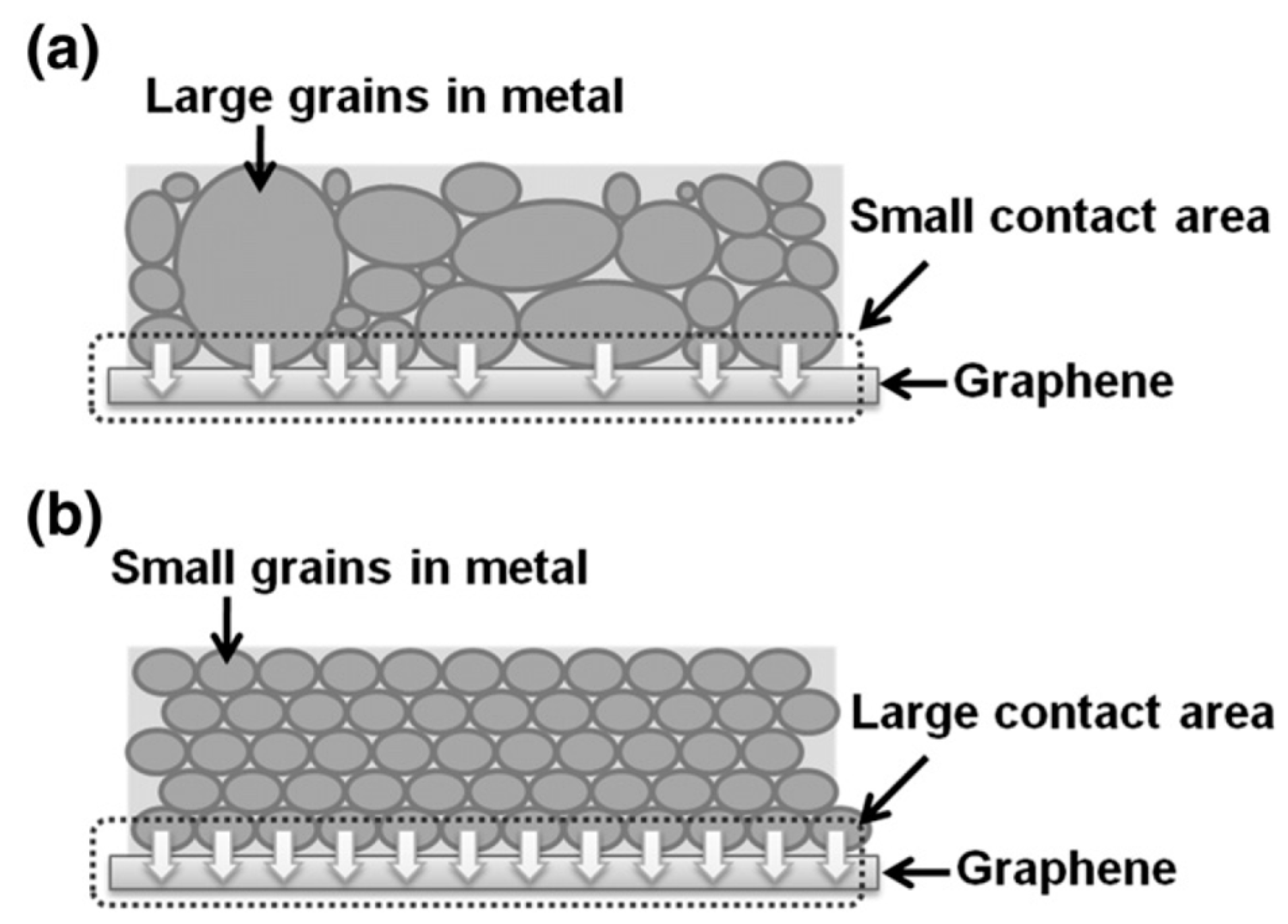
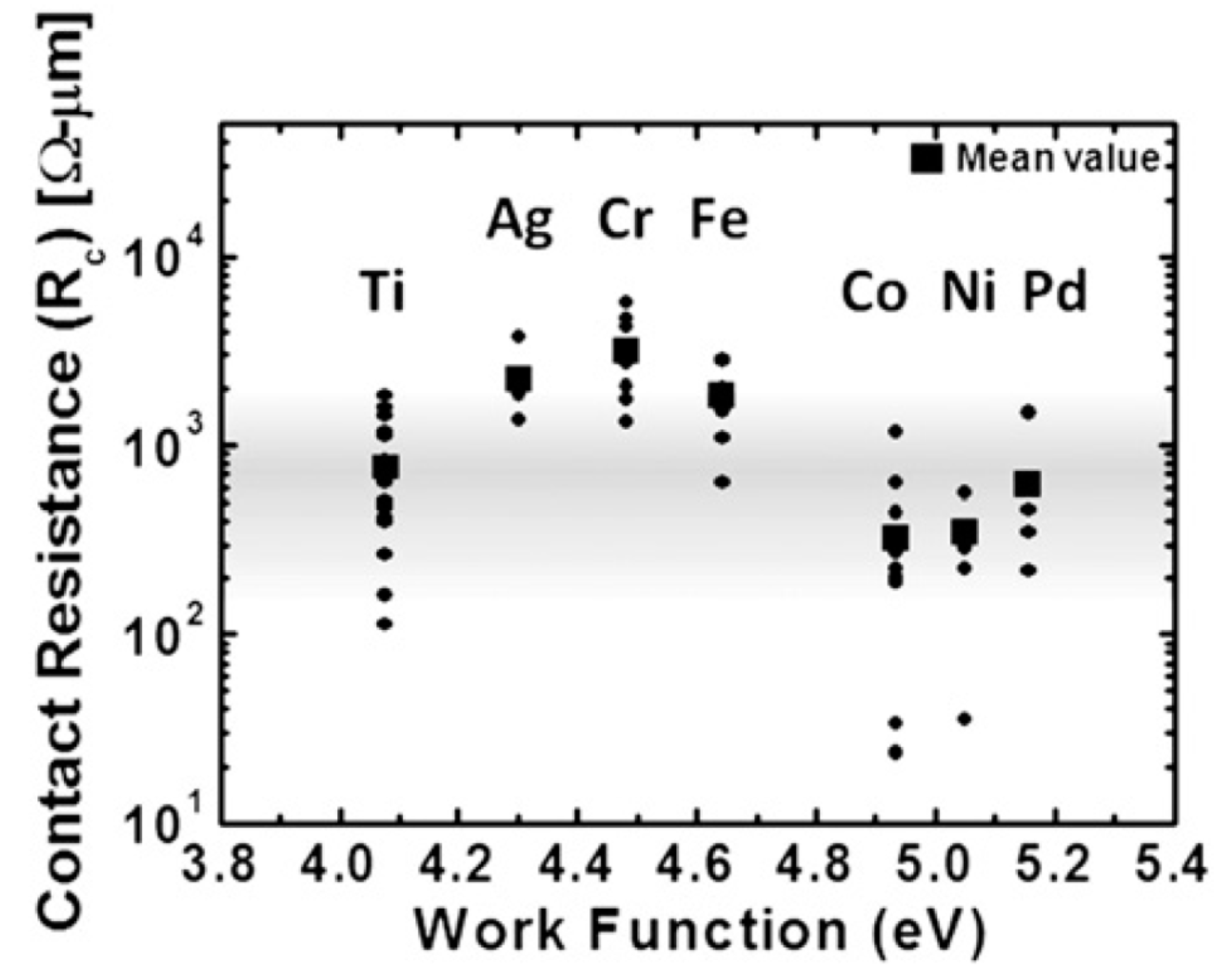
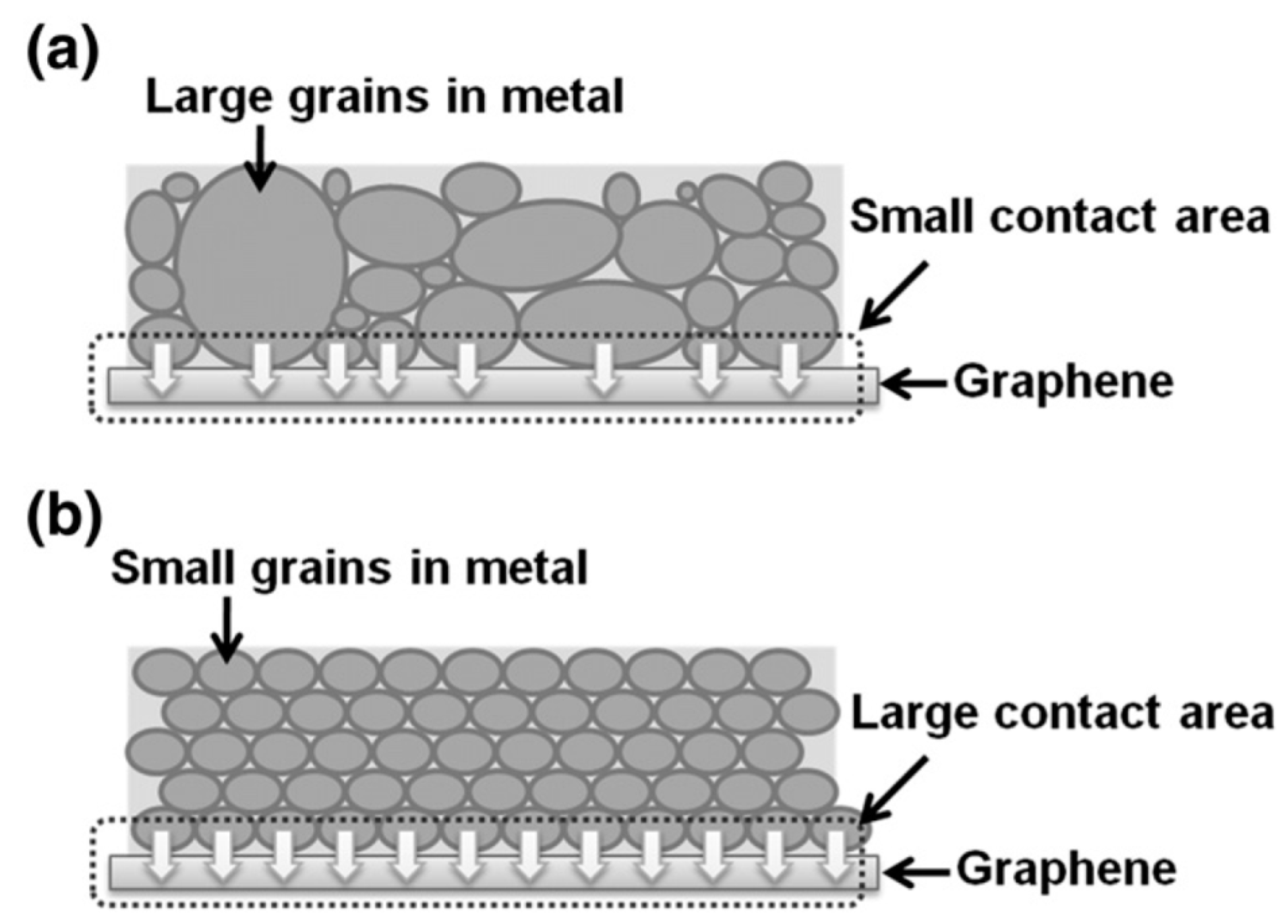
4. Some Considerations on the Electrical Behavior of Metal-Graphene Contacts
5. Conclusions, Open Points, and Perspectives
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Skachko, I.; Barker, A.; Andrei, E. Approaching ballistic transport in suspended graphene. Nat. Nanotechnol. 2008, 3, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Balandin, A.A. Thermal properties of graphene and nanostructured carbon materials. Nat. Mater. 2011, 10, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Nika, D.L.; Balandin, A.A. Two-dimensional phonon transport in graphene. J. Phys. Condens. Matter. 2012, 24, 233203. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.R.; Geim, A.K. Fine structure constant defines visual transparency of graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [PubMed]
- Stoller, M.D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R.S. Graphene-Based Ultracapacitors. Nano Lett. 2008, 8, 3498–3502. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, B.; Kotlyar, R.; Heinz, F.; Matagne, P.; Rakshit, T.; Giles, M.D.; Stettler, M.A.; Nikonov, D.E. Analysis of graphene nanoribbons as a channel material for field-effect transistors. Appl. Phys. Lett. 2006, 88, 142102. [Google Scholar] [CrossRef]
- Hass, J.; Feng, R.; Li, T.; Li, X.; Zong, Z.; de Heer, W.A.; First, P.N.; Conrad, E.H.; Jeffrey, C.A.; Berger, C. Highly ordered graphene for two dimensional electronics. Appl. Phys. Lett. 2006, 89, 143106. [Google Scholar] [CrossRef]
- Katsnelson, M.I. Graphene: Carbon in two dimensions. Mater. Today 2007, 10, 20–27. [Google Scholar] [CrossRef]
- Gilje, S.; Han, S.; Wang, M.S.; Wang, K.L.; Kaner, R.B. A Chemical Route to Graphene for Device Applications. Nano Lett. 2007, 7, 3394–3398. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.H.; Shi, Y.M.; Shi, W.H.; Lu, G.; Huang, X.; Yan, Q.Y.; Zhang, Q.; Zhang, H. Preparation of Novel 3D Graphene Networks for Supercapacitor Applications. Small 2011, 7, 3163–3168. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.Y.; Li, H.; Lam, J.W.Y.; Yuan, X.T.; Wei, J.; Tang, B.Z.; Zhang, H. Graphene Oxide as a Novel Nanoplatform for Enhancement of Aggregation-Induced Emission of Silole Fluorophores. Adv. Mater. 2012, 24, 4191–4195. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Lee, J. Graphene: Synthesis and Applications; CRC Press: New York, NY, USA, 2012. [Google Scholar]
- Warner, J.H.; Schäffel, F.; Bachmatiuk, A.; Rümmel, M.H. Graphene: Fundamentals and Emergent Applications; Elsevier: Oxford, UK, 2013. [Google Scholar]
- Tiwari, A.; Syväjärvi, M. Graphene Materials-Fundamental and Emerging Applications; Scrivener Publishing-Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Huang, X.; Qi, X.Y.; Boey, F.; Zhang, H. Graphene-based composites. Chem. Soc. Rev. 2012, 41, 666–686. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yin, Z.Y.; Wu, S.X.; Qi, X.Y.; He, Q.Y.; Zhang, Q.C.; Yan, Q.; Boey, F.; Zhang, H. Graphene-Based Materials: Synthesis, Characterization, Properties, and Applications. Small 2011, 7, 1876–1902. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Polymer-Graphene Nanocomposites; RSC Publishing: Cambridge, UK, 2012. [Google Scholar]
- Potts, G.R.; Dreyer, D.R.; Bielawski, C.W.; Ruoff, R.S. Graphene-based polymer nanocomposites. Polymer 2011, 52, 5–25. [Google Scholar] [CrossRef]
- Rafiee, M.A.; Lu, W.; Thomas, A.V.; Zandiatashbar, A.; Rafiee, J.; Tour, J.M.; Koratkar, N.A. Graphene nanoribbon composites. ACS Nano 2010, 4, 7415–7420. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Du, J. Facile synthesis of germanium–graphene nanocomposites and their application as anode materials for lithium ion batteries. Cryst. Eng. Commun. 2012, 14, 397–400. [Google Scholar] [CrossRef]
- Yin, Z.Y.; Wu, S.X.; Zhou, X.Z.; Huang, X.; Zhang, Q.C.; Boey, F.; Zhang, H. Electrochemical Deposition of ZnO Nanorods on Transparent Reduced Graphene Oxide Electrodes for Hybrid Solar Cells. Small 2010, 6, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-W.; Hsu, T.-K.; Sun, C.-L.; Nien, Y.-T.; Pu, N.-W.; Ger, M.-D. Synthesis of CuO/graphene nanocomposites for nonenzymatic electrochemical glucose biosensor applications. Electrochim. Acta 2012, 82, 152–157. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, J.; Meng, L.; Huang, Y.; Liu, L. A highly efficient chemical sensor material for ethanol: Al2O3/Graphene nanocomposites fabricated from graphene oxide. Chem. Commun. 2011, 47, 6350–6352. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, J.C.; Wang, Y.; Hu, Y.; Yu, X.; Zhang, G. WO3 nanorods/graphene nanocomposites for high-efficiency visible-light-driven photocatalysis and NO2 gas sensing. J. Mater. Chem. 2012, 22, 8525–8531. [Google Scholar] [CrossRef]
- Su, J.; Cao, M.; Ren, L.; Hu, C. Fe3O4–Graphene Nanocomposites with Improved Lithium Storage and Magnetism Properties. J. Phys. Chem. C 2011, 115, 14469–14477. [Google Scholar] [CrossRef]
- Williams, G.; Seger, B.; Kamat, P.V. TiO2–Graphene Nanocomposites. UV-Assisted Photocatalytic Reduction of Graphene Oxide. ACS Nano 2008, 2, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, Z.-R.; Fu, X.; Xu, Y.-J. TiO2−Graphene Nanocomposites for Gas-Phase Photocatalytic Degradation of Volatile Aromatic Pollutant: Is TiO2−Graphene Truly Different from Other TiO2−Carbon Composite Materials? ACS Nano 2010, 4, 7303–7314. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wu, W.; Wang, D.; Wang, D.; Shan, J.; Qing, Y.; Zhang, Z. Nanogold-enwrapped graphene nanocomposites as trace labels for sensitivity enhancement of electrochemical immunosensors in clinical immunoassays: Carcinoembryonic antigen as a model. Biosens. Bioelectron. 2010, 25, 2379–2383. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Cheng, J.-S.; Liu, X.-F.; Bai, H.-T.; Jiang, J.-H. Palladium nanoparticle/chitosan-grafted graphene nanocomposites for construction of a glucose biosensor. Biosens. Bioelectron. 2011, 26, 3456–3463. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, J.; Amatore, C.; Chen, Y.; Jiang, H.; Wang, X.-M. Gold Nanoclusters and Graphene Nanocomposites for Drug Delivery and Imaging of Cancer Cells. Angew. Chem. Int. Ed. 2011, 50, 11644–11648. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xiao, F.; Ching, C.B.; Duan, H. One-Step Electrochemical Synthesis of PtNi Nanoparticle-Graphene Nanocomposites for Nonenzymatic Amperometric Glucose Detection. ACS Appl. Mater. Interfaces 2011, 3, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Wang, L.; Qin, X.; Tian, J.; Lu, W.; Chang, G.; Sun, X. One-pot green synthesis of Ag nanoparticles-graphene nanocomposites and their applications in SERS, H2O2, and glucose sensing. RSC Adv. 2012, 2, 538–545. [Google Scholar] [CrossRef]
- Johnson, J.L.; Behnam, A.; Pearton, S.J.; Ural, A. Hydrogen Sensing Using Pd-Functionalized Multi-Layer Graphene Nanoribbon Networks. Adv. Mater. 2010, 22, 4877–4880. [Google Scholar] [CrossRef] [PubMed]
- Goyal, V.; Balandin, A.A. Thermal properties of the hybrid graphene-metal nano-micro-composites: Applications in thermal interface materials. Appl. Phys. Lett. 2012, 100, 073113. [Google Scholar] [CrossRef]
- Wang, Z.J.; Zhang, J.; Yin, Z.Y.; Wu, S.X.; Mandler, D.; Zhang, H. Fabrication of nanoelectrode ensembles by electrodepositon of Au nanoparticles on single-layer graphene oxide sheets. Nanoscale 2012, 4, 2728–2733. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shim, S.; Kim, B.; Shin, H.S. Surface-Enhanced Raman Scattering of Single- and Few-Layer Graphene by the Deposition of Gold Nanoparticles. Chem. Eur. J. 2011, 17, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, M.H.; Shin, H.-J.; Choi, D. Control of density and LSPR of Au nanoparticles on graphene. Nanotechnology 2013, 24, 275702. [Google Scholar] [CrossRef] [PubMed]
- Gutés, A.; Hsia, B.; Sussman, A.; Mickelson, W.; Zettl, A.; Carraro, C.; Maboudian, R. Graphene decoration with metal nanoparticles: Towards easy integration for sensing applications. Nanoscale 2012, 4, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.V. Graphene-Based Nanoarchitectures. Anchoring Semiconductor and Metal Nanoparticles on a Two-Dimensional Carbon Support. J. Phys. Chem. Lett. 2010, 1, 520–527. [Google Scholar] [CrossRef]
- Vedala, H.; Sorescu, D.C.; Kotchey, G.P.; Star, A. Chemical Sensitivity of Graphene Edges Decorated with Metal Nanoparticles. Nano Lett. 2011, 11, 2342–2347. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, H.; Li, S.Z.; Wu, S.X.; Boey, F.; Ma, J.; Zhang, H. Synthesis of gold square-like plates from ultrathin gold square sheets: the evolution of structure phase and shape. Angew. Chem. Int. Ed. 2011, 50, 12245–12248. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, H.; Qiu, C.; Liu, Z.; Yu, F.; Chen, M.; Hu, L.; Xia, X.; Yang, H.; Gu, C.; et al. Experimental evidence of local magnetic moments at edges of n-layer graphenes and graphite. J. Phys. Chem. C 2011, 115, 15785–15792. [Google Scholar] [CrossRef]
- Huang, X.; Li, S.Z.; Wu, S.X.; Huang, Y.Z.; Boey, F.; Gan, C.L.; Zhang, H. Graphene Oxide-Templated Synthesis of Ultrathin or Tadpole-Shaped Au Nanowires with Alternating hcp and fcc Domains. Adv. Mater. 2012, 24, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Zaniewski, A.M.; Schriver, M.; Lee, J.G.; Crommie, M.F.; Zettl, A. Electronic and optical properties of metal-nanoparticle filled graphene sandwiches. Appl. Phys. Lett. 2013, 102, 023108. [Google Scholar] [CrossRef]
- Jin, Z.; Nackashi, D.; Lu, W.; Kittrell, C.; Tour, J.M. Decoration, Migration, and Aggregation of Palladium Nanoparticles on Graphene Sheets. Chem. Mater. 2010, 22, 5695–5699. [Google Scholar] [CrossRef]
- Huang, X.; Zhou, X.Z.; Wu, S.X.; Wei, Y.Y.; Qi, X.Y.; Zhang, J.; Boey, F.; Zhang, H. Reduced Graphene Oxide-Templated Photochemical Synthesis and in situ Assembly of Au Nanodots to Orderly Patterned Au Nanodot Chains. Small 2010, 6, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, S.Z.; Huang, Y.Z.; Wu, S.X.; Zhou, X.Z.; Li, S.Z.; Gan, C.L.; Boey, F.; Mirkin, C.A.; Zhang, H. Synthesis of hexagonal close-packed gold nanostructures. Nat. Commun. 2011, 2, 292. [Google Scholar] [CrossRef] [PubMed]
- Muszynski, R.; Seger, B.; Kamat, P.V. Decorating Graphene Sheets with Gold Nanoparticles. J. Phys. Chem. C 2008, 112, 5263–5266. [Google Scholar] [CrossRef]
- Sidorov, A.N.; Sławiński, G.W.; Jayatissa, A.H.; Zamborini, F.P.; Sumanasekera, G.U. A surface-enhanced Raman spectroscopy study of thin graphene sheets functionalized with gold and silver nanostructures by seed-mediated growth. Carbon 2012, 50, 699–705. [Google Scholar] [CrossRef]
- Subrahmanyam, K.S.; Manna, A.K.; Pati, S.K.; Rao, C.N.R. A study of graphene decorated with metal nanoparticles. Chem. Phys. Lett. 2010, 497, 70–75. [Google Scholar] [CrossRef]
- Lee, J.; Novoselov, K.S.; Shin, H.S. Interaction between Metal and Graphene: Dependence on the Layer Number of Graphene. ACS Nano 2011, 5, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, F.; Yang, H.; Qiu, C.; Chen, M.; Hu, L.; Guo, Y.; Yang, H.; Gu, C.; Sun, L. Layer-dependent morphologies and charge transfer of Pd on n-layer graphenes. Chem. Commun. 2011, 47, 9408–9410. [Google Scholar] [CrossRef] [PubMed]
- Zan, R.; Bangert, U.; Ramasse, Q.; Novoselov, K.S. Metal−Graphene Interaction Studied via Atomic Resolution Scanning Transmission Electron Microscopy. Nano Lett. 2011, 11, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.-Z.; Hupalo, M.; Lin, H.-Q.; Ho, K.-M.; Tringides, M.C. Metals on Graphene: Interactions, Growth Morphology, and Thermal Stability. Crystals 2013, 3, 79–111. [Google Scholar] [CrossRef]
- Gan, Y.; Sun, L.; Banhart, F. One- and Two-Dimensional Diffusion of Metal Atoms in Graphene. Small 2008, 4, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.A.; Bell, G.R.; Rourke, J.P.; Sanchez, A.M.; Elkin, M.D.; Hickey, B.J.; Wilson, N.R. Physical vapor deposition of metal nanoparticles on chemically modified graphene: observations on metal-graphene interactions. Small 2011, 7, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, Z.; Wang, L.; Polyakova, E.; Taniguchi, T.; Watanabe, K.; Hone, J.; Flynn, G.W.; Brus, L.E. Slow Gold Adatom Diffusion on Graphene: Effect of Silicon Dioxide and Hexagonal Boron Nitride Substrates. J. Phys. Chem. B 2013, 117, 4305–4312. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, A.T.; Bleikamp, S.; Feibelman, P.J.; Michely, T. Two-dimensional Ir cluster lattice on a graphene moiré on Ir(111). Phys. Rev. Lett. 2006, 97, 215501. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Gao, M.; Huang, L.; Liu, F.; Gao, H.J. Directed self-assembly of monodispersed platinum nanoclusters on graphene Moiré template. Appl. Phys. Lett. 2009, 95, 093106. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, Q.; Cui, Y.; Tan, D.L.; Bao, X.H. Fabrication of metal nanoclusters on graphene grown on Ru(0001). Chin. Sci. Bull. 2009, 54, 2446–2450. [Google Scholar] [CrossRef]
- N’Diaye, A.T.; Gerber, T.; Busse, C.; Myslivecek, J.; Coraux, J.; Michely, T. A versatile fabrication method for cluster superlattices. New J. Phys. 2009, 11, 103045. [Google Scholar] [CrossRef]
- Zhou, Z.; Gao, F.; Goodman, D.W. Deposition of metal clusters on single-layer graphene/Ru(0001): Factors that govern cluster growth. Surf. Sci. 2010, 604, L31–L38. [Google Scholar] [CrossRef]
- Zan, R.; Bangert, U.; Ramasse, Q.; Novoselov, K.S. Evolution of Gold Nanostructures on Graphene. Small 2011, 7, 2868–2872. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yoon, B.; König, M.; Fukamori, Y.; Esch, F.; Heiz, U.; Landman, U. Size-Selected Monodisperse Nanoclusters on Supported Graphene: Bonding, Isomerism, and Mobility. Nano Lett. 2012, 12, 5907–5912. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, F.; Chen, M.; Qiu, C.; Yang, H.; Wang, G.; Yu, T.; Sun, L. The transformation of a gold film on few-layer graphene to produce either hexagonal or triangular nanoparticles during annealing. Carbon 2013, 52, 379–387. [Google Scholar] [CrossRef]
- Zhou, H.; Qiu, C.; Liu, Z.; Yang, H.; Hu, L.; Liu, J.; Yang, H.; Gu, C.; Sun, L. Thickness-Dependent Morphologies of Gold on N-Layer Graphenes. J. Am. Chem. Soc. 2010, 132, 944–946. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Somers, L.A.; Dan, Y.; Ly, T.; Kybert, N.J.; Mele, E.J.; Johnson, A.T.C. Size-Selective Nanoparticle Growth on Few-Layer Graphene Films. Nano Lett. 2010, 10, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Qiu, C.; Yu, F.; Yang, H.; Chen, M.; Hu, L.; Sun, L. Thickness-Dependent Morphologies and Surface-Enhanced Raman Scattering of Ag Deposited on n-Layer Graphenes. J. Phys. Chem. C 2011, 115, 11348–11354. [Google Scholar] [CrossRef]
- Feldheim, D.L.; Foss, C.A., Jr. Metal Nanoparticles-Synthesis, Characterizations, and Applications; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Johnston, R.L.; Wilcoxon, J. Metal Nanoparticles and Nanoalloys; Elsevier: Oxford, UK, 2012. [Google Scholar]
- Sau, T.K.; Rogach, A.L. Complex-Shaped Metal Nanoparticles-Bottom-up Syntheses and Applications; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Knoch, J.; Chen, Z.; Appenzeller, J. Properties of Metal–Graphene Contacts. IEEE Trans. Nanotechnol. 2012, 11, 513–519. [Google Scholar] [CrossRef]
- Russo, S.; Craciun, M.F.; Yamamoto, M.; Morpurgo, A.F.; Tarucha, S. Contact resistance in graphene-based devices. Phys. E 2010, 42, 677–679. [Google Scholar] [CrossRef]
- Venugopal, A.; Colombo, L.; Vogel, E.M. Contact resistance in few and multilayer graphene devices. Appl. Phys. Lett. 2010, 96, 013512. [Google Scholar] [CrossRef]
- Nagashio, K.T.; Nishimura, T.; Kita, K.; Toriumi, A. Contact resistivity and current flow path at metal/graphene contact. Appl. Phys. Lett. 2010, 97, 143514. [Google Scholar] [CrossRef]
- Schwierz, F. Graphene transistors. Nature Nanotechnol. 2010, 5, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Fiori, G.; Bonaccorso, F.; Iannaccone, G.; Palacios, T.; Neumaier, D.; Seabaugh, A.; Banerjee, S.K.; Colombo, L. Electronics based on two-dimensional materials. Nat. Nanotechnol. 2014, 9, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Fisichella, G.; Schilirò, E.; Di Franco, S.; Fiorenza, P.; Lo Nigro, R.; Roccaforte, F.; Ravesi, S.; Giannazzo, F. Interface Electrical Properties of Al2O3 Thin Films on Graphene Obtained by Atomic Layer Deposition with an in Situ Seedlike Layer. ACS Appl. Mater. Interfaces 2017, 9, 7761–7771. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, S.; Smith, A.D.; Östling, M.; Lupina, G.; Dabrowski, J.; Lippert, G.; Mehr, W.; Driussi, F.; Venica, S.; Di Lecce, V.; et al. Going ballistic: Graphene hot electron transistors. Solid State Commun. 2015, 224, 64–75. [Google Scholar] [CrossRef]
- Giannazzo, F.; Fisichella, G.; Greco, G.; La Magna, A.; Roccaforte, F.; Pecz, B.; Yakimova, R.; Dagher, R.; Michon, A.; Cordier, Y. Graphene integration with nitride semiconductors for high power and high frequency electronics. Phys. Status Solidi A 2017, 214, 1600460. [Google Scholar] [CrossRef]
- Wei, W.; Pallecchi, E.; Haque, S.; Borini, S.; Avramovic, V.; Centeno, A.; Amaia, Z.; Happy, H. Mechanically robust 39 GHz cut-off frequency graphene field effect transistors on flexible substrates. Nanoscale 2016, 8, 14097–14103. [Google Scholar] [CrossRef] [PubMed]
- Fisichella, G.; Lo Verso, S.; Di Marco, S.; Vinciguerra, V.; Schilirò, E.; Di Franco, S.; Lo Nigro, R.; Roccaforte, F.; Zurutuza, A.; Centeno, A.; et al. Advances in the fabrication of graphene transistors on flexible substrates. Beilstein J. Nanotechnol. 2017, 8, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Antcliffe, M.; Seo, H.C.; Curtis, D.; Lin, S.; Schmitz, A.; Milosavljevic, I.; Kiselev, A.A.; Ross, R.S.; Gaskill, D.K.; et al. Ultra-low resistance ohmic contacts in graphene field effect transistors. Appl. Phys. Lett. 2012, 100, 203512. [Google Scholar] [CrossRef]
- Politou, M.; Asselberghs, I.; Radu, I.; Conard, T.; Richard, O.; Lee, C.S.; Martens, K.; Sayan, S.; Huyghebaert, C.; Tokei, Z.; et al. Transition metal contacts to graphene. Appl. Phys. Lett. 2015, 107, 153104. [Google Scholar] [CrossRef]
- Smith, J.T.; Franklin, A.D.; Farmer, D.B.; Dimitrakopoulos, C.D. Reducing Contact Resistance in Graphene Devices through Contact Area Patterning. ACS Nano 2013, 7, 3661–3667. [Google Scholar] [CrossRef] [PubMed]
- Feibelman, P.J. Pinning of graphene to Ir(111) by flat Ir dots. Phys. Rev. B 2008, 77, 165419. [Google Scholar] [CrossRef]
- Feibelman, P.J. Onset of three-dimensional Ir islands on a graphene/Ir(111) template. Phys. Rev. B 2009, 80, 085412. [Google Scholar] [CrossRef]
- Varns, R.; Strange, P. Stability of gold atoms and dimers adsorbed on graphene. J. Phys. Condens. Matter 2008, 20, 225005. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Wang, Y.; Kemper, A.F.; Cheng, H.-P. Density functional study of gold and iron clusters on perfect and defected graphene. Phys. Rev. B 2012, 85, 165444. [Google Scholar] [CrossRef]
- Amft, M.; Sanyal, B.; Eriksson, O.; Skorodumova, N.V. Small gold clusters on graphene, their mobility and clustering: a DFT study. J. Phys. Condens. Matter 2011, 23, 205301. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yuan, J.; Zhong, J. Density functional calculation of transition metal adatom adsorption on graphene. J. Phys. Condens. Matter 2008, 20, 115209. [Google Scholar] [CrossRef] [PubMed]
- Sevinçli, H.; Topsakal, M.; Durgun, E.; Ciraci, S. Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys. Rev. B 2008, 77, 195434. [Google Scholar] [CrossRef]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 2008, 77, 235430. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, L.; Xu, Z.; Krasheninnikov, A.V.; Huai, P.; Zhu, Z.; Banhart, F. Migration of gold atoms in graphene ribbons: Role of the edges. Phys. Rev. B 2010, 81, 125425. [Google Scholar] [CrossRef]
- Malola, S.; Häkkinen, H.; Koskinen, P. Gold in graphene: In-plane adsorption and diffusion. Appl. Phys. Lett. 2009, 94, 043106. [Google Scholar] [CrossRef]
- Semidey-Flecha, L.; Teng, D.; Habenicht, B.F.; Sholl, D.S.; Xu, Y. Adsorption and Diffusion of the Rh and Au Adatom on Graphene Moiré/Ru(0001). J. Chem. Phys. 2013, 138, 184710. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.Z.; Hupalo, M.; Lu, W.C.; Tringides, M.C.; Yao, Y.X.; Ho, K.M. Metals on graphene: Correlation between adatom adsorption behavior and growth morphology. Phys. Chem. Chem. Phys. 2012, 14, 9157–9166. [Google Scholar] [CrossRef] [PubMed]
- Khomyakov, P.A.; Giovannetti, G.; Rusu, P.C.; Brocks, G.; van den Brink, J.; Kelly, P.J. First-principles study of the interaction and charge transfer between graphene and metals. Phys. Rev. B 2009, 79, 195425. [Google Scholar] [CrossRef]
- Giovannetti, G.; Khomyakov, P.A.; Brocks, G.; Karpan, V.M.; van den Brink, J.; Kelly, P.J. Doping graphene with metal contacts. Phys. Rev. Lett. 2008, 101, 026803. [Google Scholar] [CrossRef] [PubMed]
- Barraza-Lopez, S.; Vanević, M.; Kindermann, M.; Chou, M.Y. Effects of Metallic Contacts on Electron Transport through Graphene. Phys. Rev. Lett. 2010, 104, 076807. [Google Scholar] [CrossRef] [PubMed]
- Sławińska, J.; Wlasny, I.; Dabrowski, P.; Klusek, Z.; Zasada, I. Doping domains in graphene on gold substrates: First-principles and scanning tunneling spectroscopy studies. Phys. Rev. B 2012, 85, 235430. [Google Scholar] [CrossRef]
- Watanabe, E.; Conwill, A.; Tsuya, D.; Koide, Y. Low contact resistance metals for graphene based devices. Diam. Relat. Mater. 2012, 24, 171–174. [Google Scholar] [CrossRef]
- Leong, W.S.; Gong, H.; Thong, J.T.L. Low-contact-resistance graphene devices with nickel-etched-graphene contacts. ACS Nano 2014, 8, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Nagashio, K.; Toriumi, A. Density-of-States Limited Contact Resistance in Graphene Field-Effect Transistors. Jpn. J. Appl. Phys. 2011, 50, 070108. [Google Scholar] [CrossRef]
- Liu, W.; Wei, J.; Sun, X.; Yu, H. A Study on Graphene—Metal Contact. Crystals 2013, 3, 257–274. [Google Scholar] [CrossRef]
- Wang, X.; Xie, W.; Du, J.; Wang, C.; Zhao, N.; Xu, J.-B. Graphene/Metal Contacts: Bistable States and Novel Memory Devices. Adv. Mater. 2012, 24, 2614–2619. [Google Scholar] [CrossRef] [PubMed]
- Kathami, Y.; Li, H.; Xu, C.; Banerjee, K. Metal-to-multilayer-graphene contact-Part I: Contact resistance modeling. IEEE Trans. Nanotechnol. 2012, 59, 2444–2452. [Google Scholar] [CrossRef]
- Robinson, J.A.; LaBella, M.; Zhu, M.; Hollander, M.; Kasarda, R.; Hughes, Z.; Trumbull, K.; Cavalero, R.; Snyder, D. Contacting graphene. Appl. Phys. Lett. 2011, 98, 053103. [Google Scholar] [CrossRef]
- Xia, F.; Perebeinos, V.; Lin, Y.-M.; Wu, Y.; Avouris, P. The origins and limits of metal–graphene junction resistance. Nat. Nanotechnol. 2011, 6, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, R.S.; Steiner, M.; Chiu, H.-Y.; Engel, M.; Bol, A.A.; Krupke, R.; Burghard, M.; Kern, K.; Avouris, P. The Graphene–Gold Interface and Its Implications for Nanoelectronics. Nano Lett. 2011, 11, 3833–3837. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.A. Introduction to Surface and Thin Film Processes; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Lin, Y.; Chen, X. Advanced Nano Deposition Methods; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Campbell, C.T. Ultrathin metal films and particles on oxide surfaces: Structural, electronic and chemisorptive properties. Surf. Sci. Rep. 1997, 27, 1–111. [Google Scholar] [CrossRef]
- Ruffino, F.; Torrisi, V.; Marletta, G.; Grimaldi, M.G. Effects of the embedding kinetics on the surface nano-morphology of nano-grained Au and Ag films on PS and PMMA layers annealed above the glass transition temperature. Appl. Phys. A 2012, 107, 669–683. [Google Scholar] [CrossRef]
- Ruffino, F.; De Bastiani, R.; Grimaldi, M.G.; Bongiorno, C.; Giannazzo, F.; Roccaforte, F.; Spinella, C.; Raineri, V. Self-organization of Au nanoclusters on the SiO2 surface induced by 200 keV-Ar+ irradiation. Nucl. Instrum. Meth. Phys. Res. B 2007, 257, 810–814. [Google Scholar] [CrossRef]
- Ruffino, F.; Torrisi, V.; Marletta, G.; Grimaldi, M.G. Atomic force microscopy investigation of the kinetic growth mechanisms of sputtered nanostructured Au film on mica: Towards a nanoscale morphology control. Nanoscale Res. Lett. 2011, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Ruffino, F.; Crupi, I.; Irrera, A.; Grimaldi, M.G. Pd/Au/SiC Nanostructured Diodes for Nanoelectronics: Room Temperature Electrical Properties. IEEE Trans. Nanotechnol. 2010, 9, 414–421. [Google Scholar] [CrossRef]
- Ruffino, F.; Grimaldi, M.G. Island-to-percolation transition during the room-temperature growth of sputtered nanoscale Pd films on hexagonal SiC. J. Appl. Phys. 2010, 107, 074301. [Google Scholar] [CrossRef]
- Ruffino, F.; Torrisi, V.; Marletta, G.; Grimaldi, M.G. Kinetic growth mechanisms of sputter-deposited Au films on mica: From nanoclusters to nanostructured microclusters. Appl. Phys. A 2010, 100, 7–13. [Google Scholar] [CrossRef]
- Herminghaus, S.; Brinkmann, M.; Seemann, R. Wetting and Dewetting of Complex Surface Geometries. Annu. Rev. Mater. Res. 2008, 38, 101–121. [Google Scholar] [CrossRef]
- Thompson, C.V. Solid-State Dewetting of Thin Films. Annu. Rev. Mater. Res. 2012, 42, 399–434. [Google Scholar] [CrossRef]
- Giermann, A.L.; Thompson, C.V. Solid-state dewetting for ordered arrays of crystallographically oriented metal particles. Appl. Phys. Lett. 2005, 86, 121903. [Google Scholar] [CrossRef]
- Henley, S.J.; Carrey, J.D.; Silva, S.R.P. Pulsed-laser-induced nanoscale island formation in thin metal-on-oxide films. Phys. Rev. B 2005, 72, 195408. [Google Scholar] [CrossRef]
- Ye, J.; Thompson, C.V. Templated Solid-State Dewetting to Controllably Produce Complex Patterns. Adv. Mater. 2011, 23, 1567–1571. [Google Scholar] [CrossRef] [PubMed]
- Ruffino, F.; Grimaldi, M.G. Self-organized patterned arrays of Au and Ag nanoparticles by thickness-dependent dewetting of template-confined films. J. Mater. Sci. 2014, 49, 5714–5729. [Google Scholar] [CrossRef]
- Ruffino, F.; Grimaldi, M.G. Template-confined dewetting of Au and Ag nanoscale films on mica substrate. Appl. Surf. Sci. 2013, 270, 697–706. [Google Scholar] [CrossRef]
- Ruffino, F.; Pugliara, A.; Carria, E.; Bongiorno, C.; Spinella, C.; Grimaldi, M.G. Formation of nanoparticles from laser irradiated Au thin film on SiO2/Si: Elucidating the Rayleigh-instability role. Mater. Lett. 2012, 84, 27–30. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, H.G.; Shi, D.X.; Sun, J.T.; Du, S.X.; Liu, F.; Gao, H.J. Highly Ordered, Millimeter-Scale, Continuous, Single-Crystalline Graphene Monolayer Formed on Ru (0001). Adv. Mater. 2009, 21, 2777–2780. [Google Scholar] [CrossRef]
- Xu, Y.; Semidey-Flecha, L.; Liu, L.; Zhou, Z.; Goodman, D.W. Exploring the structure and chemical activity of 2-D gold islands on graphene moiré/Ru(0001). Faraday Discuss. 2011, 152, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, M.; Eelbo, T.; Wasniowska, M.; Wiesendanger, R. Fe adatoms on graphene/Ru(0001): Adsorption site and local electronic properties. Phys. Rev. B 2011, 84, 113403. [Google Scholar] [CrossRef]
- Donner, K.; Jakob, P. Structural properties and site specific interactions of Pt with the graphene/Ru(0001) moiré overlayer. J. Chem. Phys. 2009, 131, 164701. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jia, C.; Ma, B.; Wang, W.; Fang, Z.; Zhang, G.; Guo, X. Substrate-induced interfacial plasmonics for photovoltaic conversion. Sci. Rep. 2015, 5, 14497. [Google Scholar] [CrossRef] [PubMed]
- Maiti, R.; Sinha, T.K.; Mukherjee, S.; Adhikari, B.; Ray, S.M. Enhanced and Selective Photodetection Using Graphene-Stabilized Hybrid Plasmonic Silver Nanoparticles. Plasmonics 2016, 11, 1297–1304. [Google Scholar] [CrossRef]
- Chen, X.; Jia, B.; Zhang, Y.; Gu, M. Exceeding the limit of plasmonic light trapping in textured screen-printed solar cells using Al nanoparticles and wrinkle-like graphene sheets. Light Sci. Appl. 2013, 2, e92. [Google Scholar] [CrossRef]
- Li, Y.; Fan, X.; Qi, J.; Ji, J.; Wang, S.; Zhang, G.; Zhang, F. Gold nanoparticles–graphene hybrids as active catalysts for Suzuki reaction. Mater. Res. Bull. 2010, 45, 1413–1418. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Y.; Zeng, X.; Peng, S. Synergetic effect of metal nickel and graphene as a cocatalyst for enhanced photocatalytic hydrogen evolution via dye sensitization. Sci. Rep. 2015, 5, 10589. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Szpunar, J.A. Hydrogen Storage Performance in Pd/Graphene Nanocomposites. ACS Appl. Mater. Interface 2016, 8, 25933–25940. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lu, C.; Ding, D.; Zhao, M.; Wang, D.; Hu, C.; Qiu, J.; Xie, G.; Tang, Z. Capping nanoparticles with graphene quantum dots for enhanced thermoelectric performance. Chem. Sci. 2015, 6, 4103–4108. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
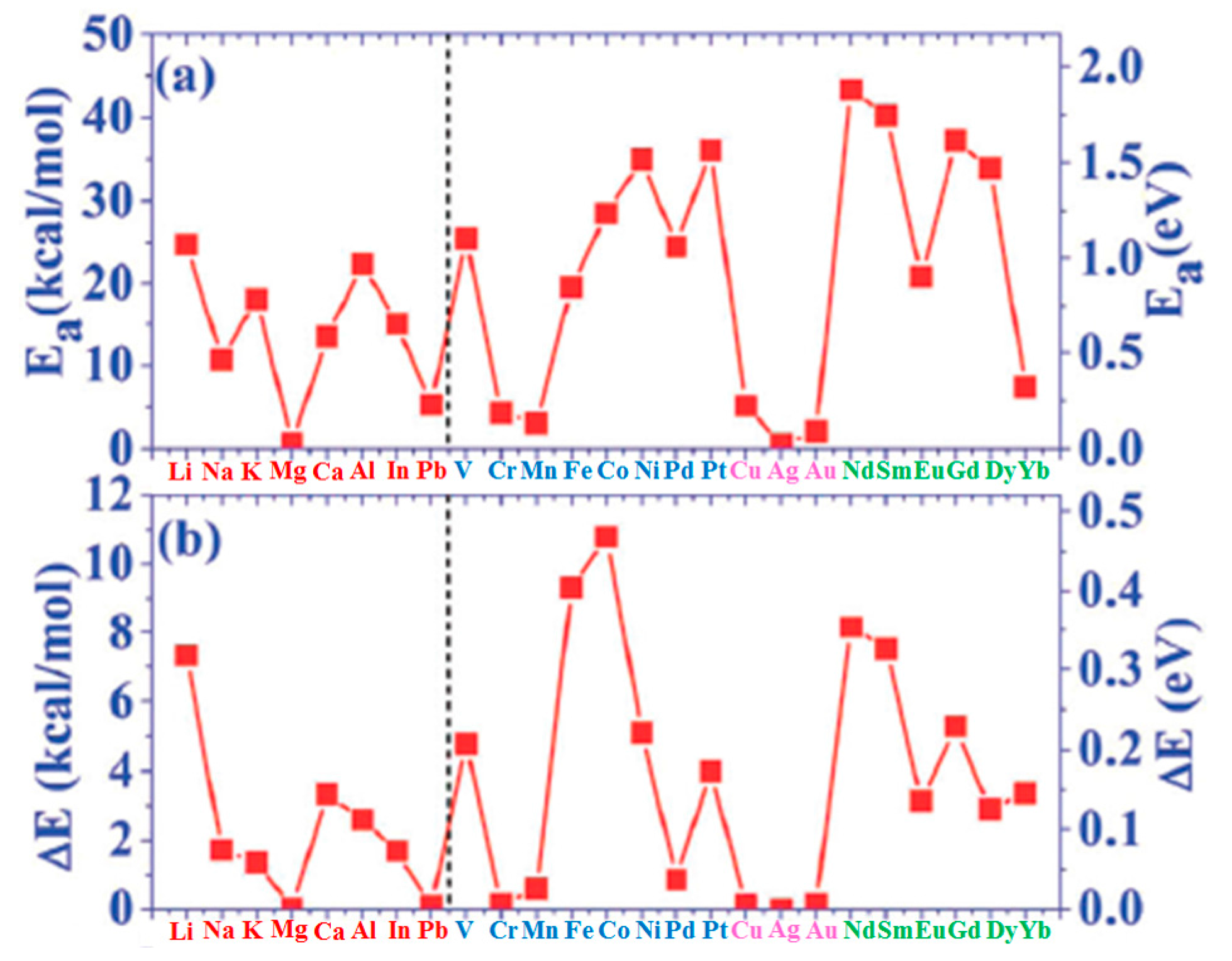
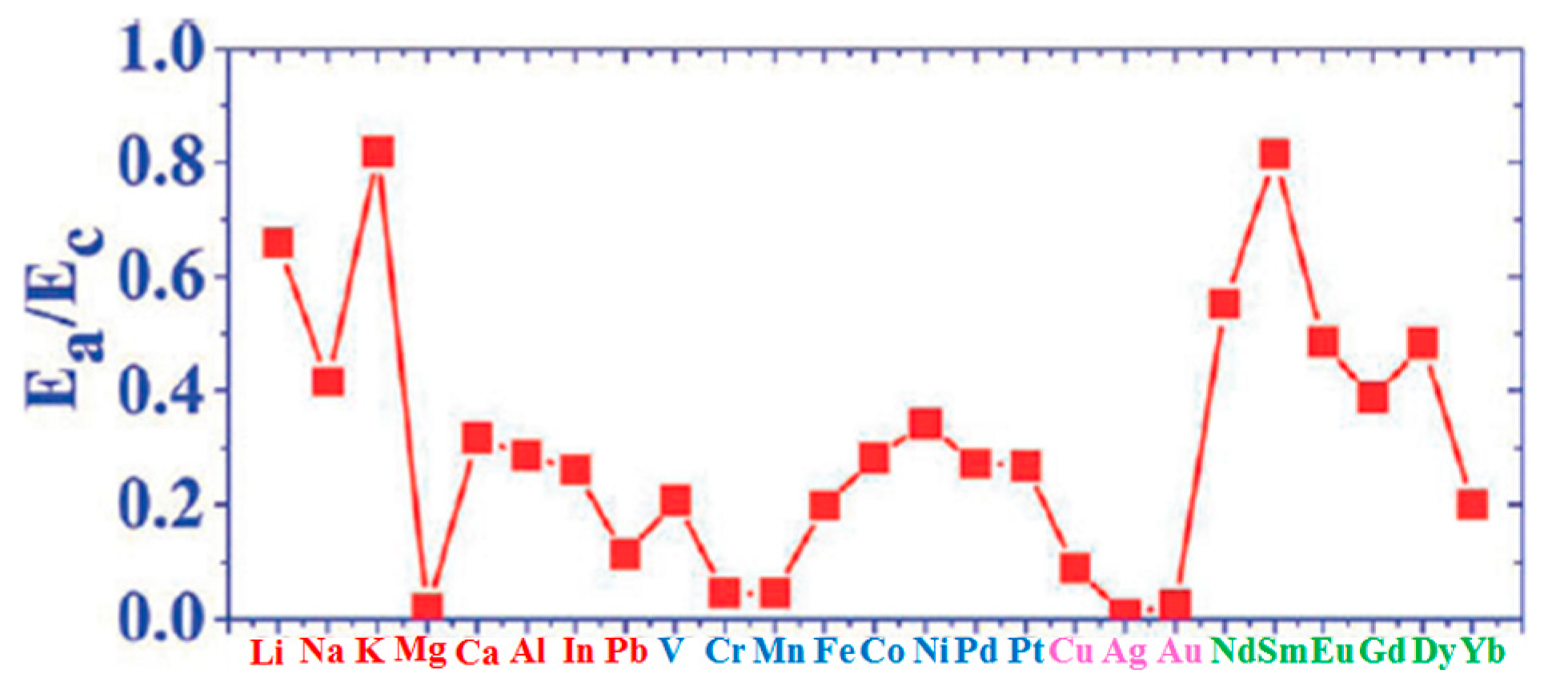
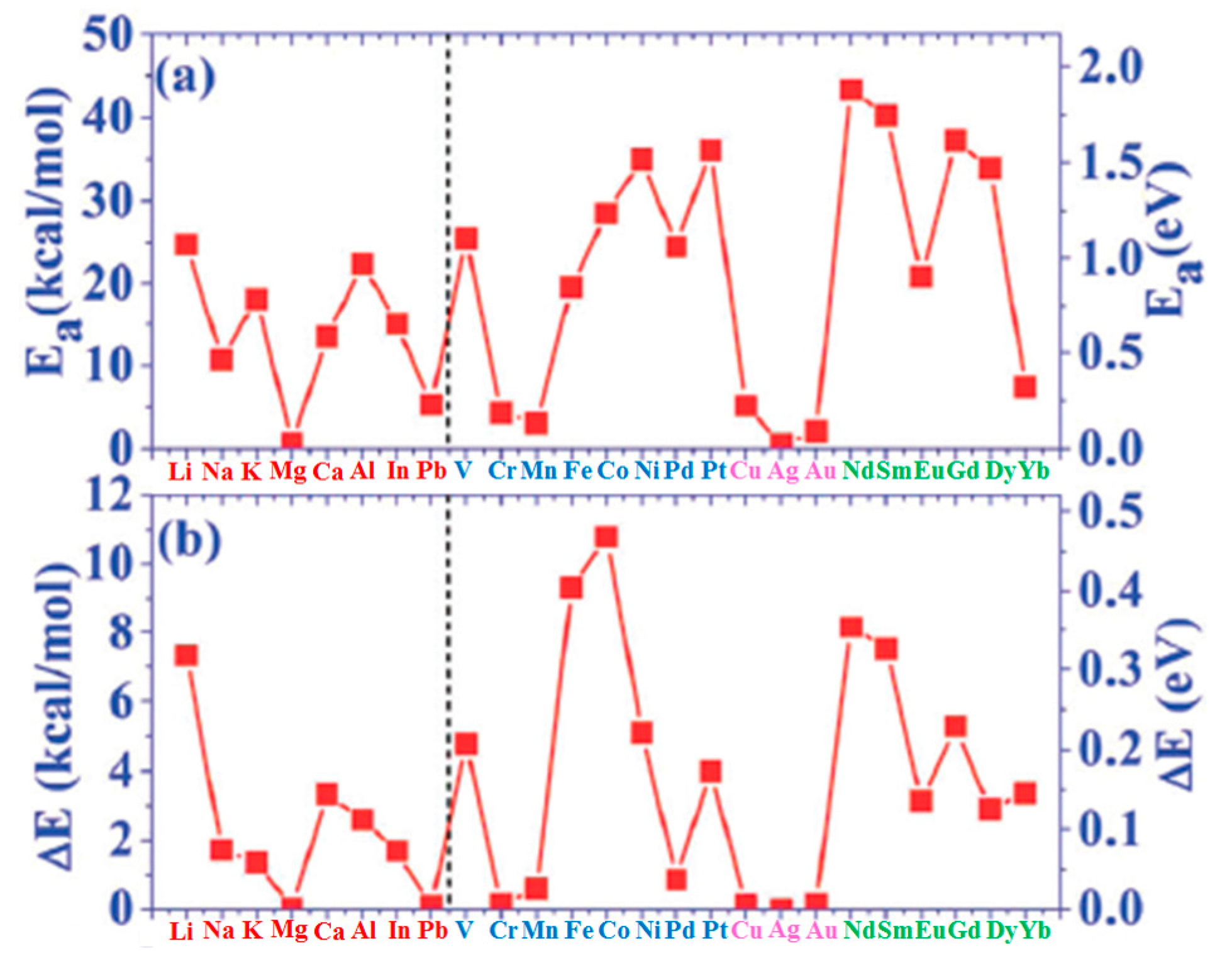
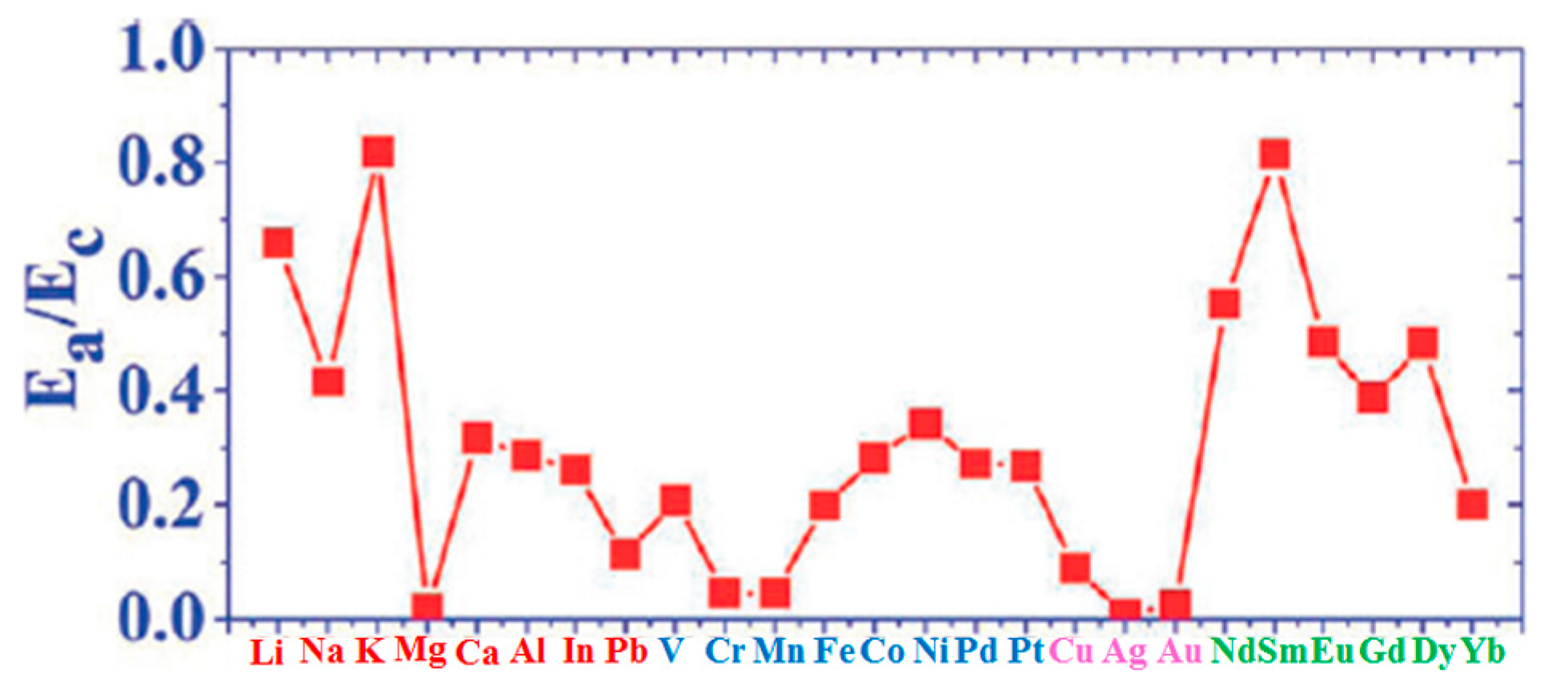
| Adatoms | Sites | Ea | ΔE | Ea/Ec | Ec−Ea |
|---|---|---|---|---|---|
| Li | H | 24.77 | 7.33 | 0.659 | 12.82 |
| Na | H | 10.70 | 1.71 | 0.417 | 14.97 |
| K | H | 18.10 | 1.36 | 0.818 | 4.036 |
| Mg | H | 0.65 | 0.02 | 0.019 | 34.18 |
| Ca | H | 13.44 | 3.34 | 0.317 | 28.99 |
| Al | H | 22.41 | 2.58 | 0.287 | 55.70 |
| In | H | 15.15 | 1.68 | 0.261 | 42.90 |
| Pb | T | 5.28 | 0.09 | 0.113 | 41.47 |
| V | H | 25.44 | 4.77 | 0.208 | 96.86 |
| Cr | B | 4.34 | 0.14 | 0.046 | 90.22 |
| Mn | H | 3.04 | 0.60 | 0.045 | 64.30 |
| Fe | H | 19.65 | 9.32 | 0.199 | 79.08 |
| Co | H | 28.53 | 10.79 | 0.282 | 72.65 |
| Ni | H | 35.01 | 5.12 | 0.342 | 67.37 |
| Pd | B | 24.47 | 0.85 | 0.273 | 65.24 |
| Pt | B | 36.09 | 3.99 | 0.268 | 98.59 |
| Cu | T | 5.17 | 0.12 | 0.090 | 52.26 |
| Ag | B | 0.51 | 0.00 | 0.007 | 67.53 |
| Au | T | 2.08 | 0.14 | 0.024 | 85.79 |
| Nd | H | 43.31 | 8.16 | 0.552 | 35.15 |
| Sm | H | 40.15 | 7.52 | 0.814 | 9.20 |
| Eu | H | 20.85 | 3.14 | 0.486 | 22.05 |
| Gd | H | 37.17 | 5.28 | 0.389 | 58.39 |
| Dy | H | 33.94 | 2.88 | 0.484 | 36.19 |
| Yb | H | 7.40 | 3.37 | 0.201 | 29.43 |
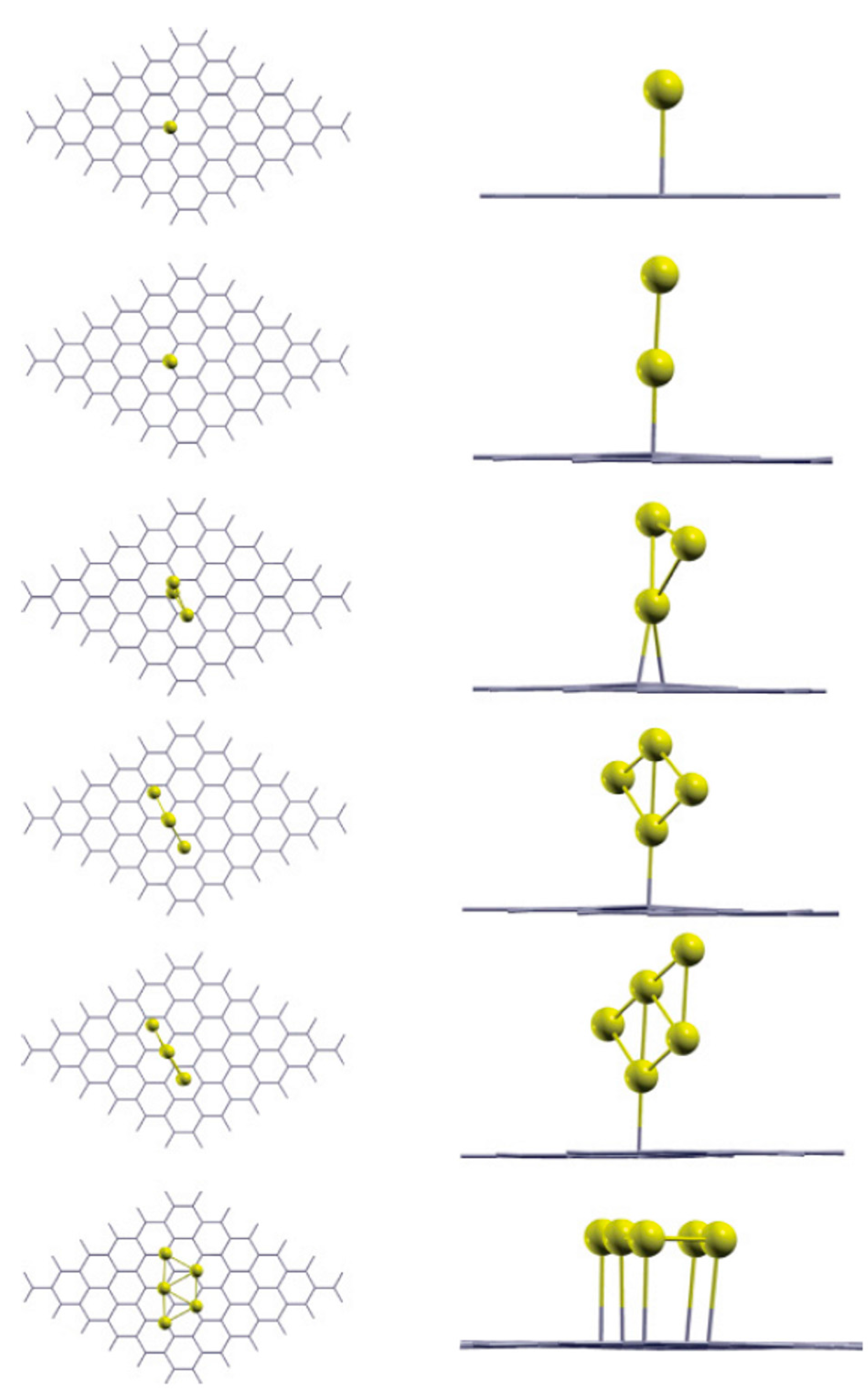
| System | ha (Å) | dac (Å) | BE1 (eV) | BE2 (eV) | BE3 (eV) |
|---|---|---|---|---|---|
| Au1 | 2.89 | 2.82 | −0.107 | −0.107 | −0.122 |
| Au2 | 2.45 | 2.32 | −1.373 | −2.639 | −0.526 |
| Au3 | 2.43 | 2.33 | −1.345 | −1.288 | −0.654 |
| Au4 | 2.49 | 2.34 | −1.608 | −2.397 | −0.515 |
| Au5 | 2.57 | 2.45 | −1.681 | −1.975 | −0.218 |
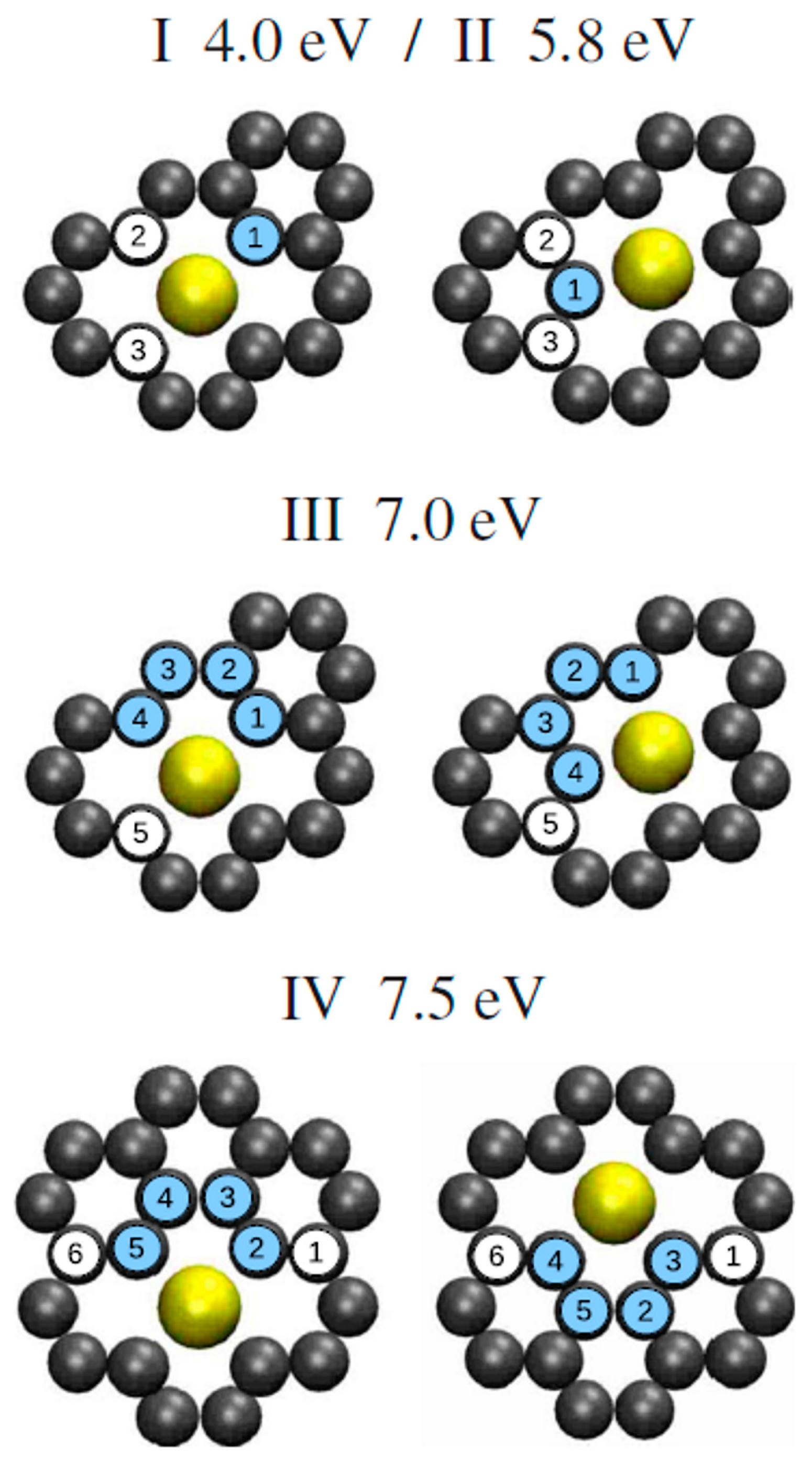
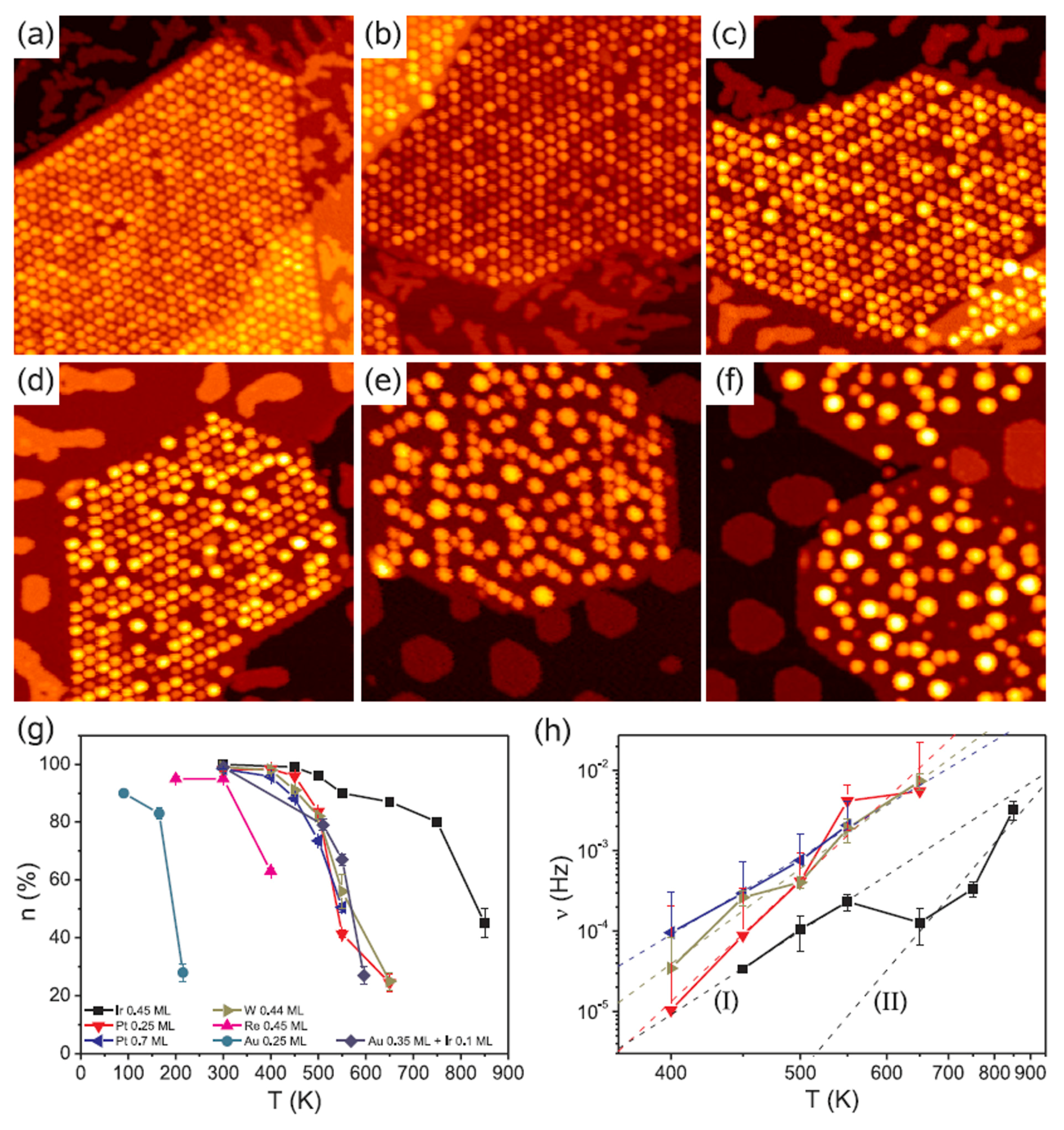
| Clusters | Ea (eV) | ΔEa (eV) | υ0 (Hz) |
|---|---|---|---|
| Ir, 0.45 ML (I) | 0.41 | 0.02 | 1.4 |
| Ir, 0.45 ML (II) | 0.75 | 0.2 | 67 |
| Ir, 0.45 ML | 0.28 | 0.08 | 0.06 |
| Pt, 0.25 ML | 0.60 | 0.08 | 500 |
| Pt, 0.70 ML | 0.38 | 0.02 | 6.2 |
| W, 0.44 ML | 0.47 | 0.04 | 33 |
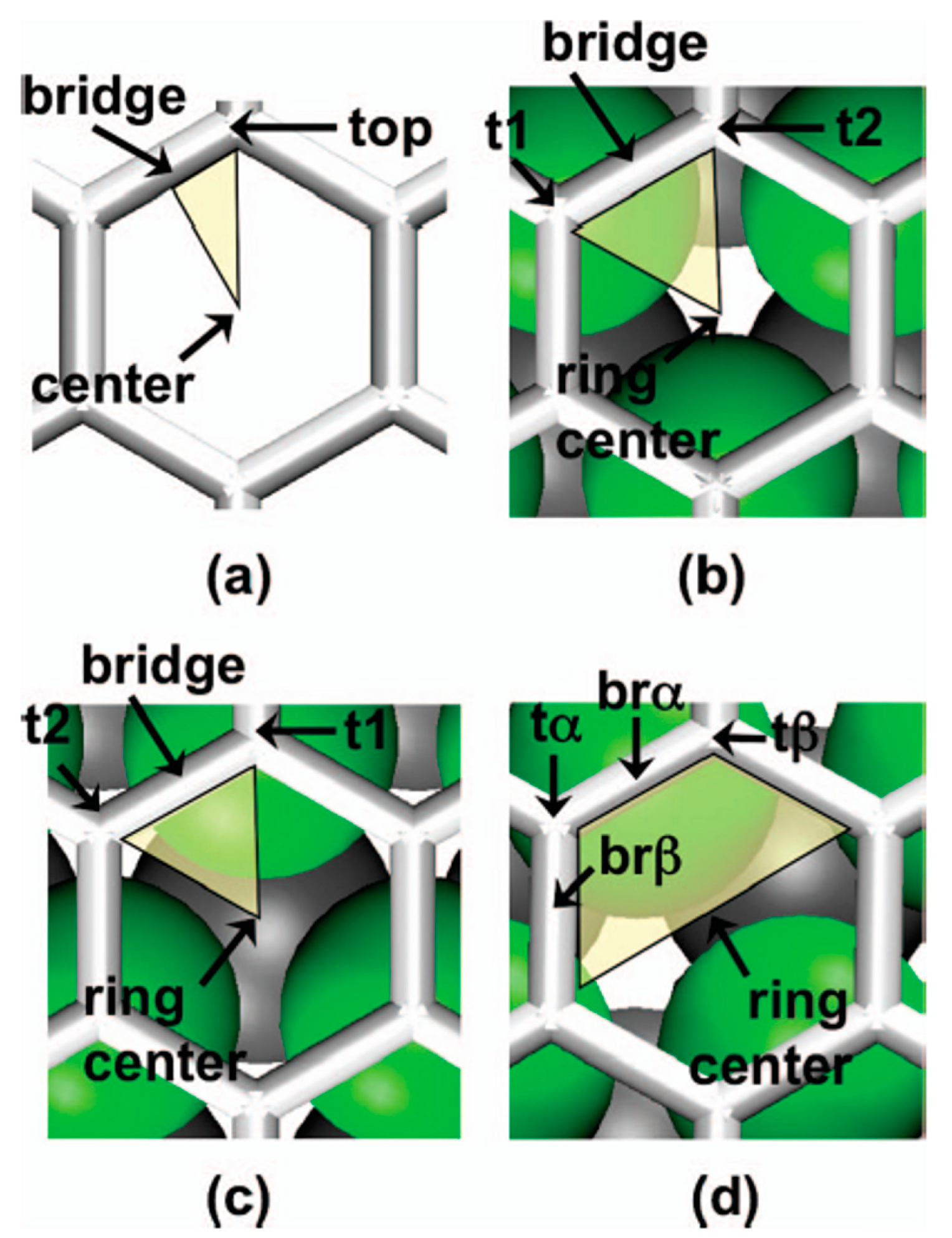
| Gr | Ni | Co | Pd | Al | Ag | Cu | Au | Pt | |
|---|---|---|---|---|---|---|---|---|---|
| deq (Å) | 2.05 | 2.05 | 2.30 | 3.41 | 3.33 | 3.26 | 3.31 | 3.30 | |
| ΔE (eV) | 0.125 | 0.160 | 0.084 | 0.027 | 0.043 | 0.033 | 0.030 | 0.038 | |
| WM (eV) | 5.47 | 5.44 | 5.67 | 4.22 | 4.92 | 5.22 | 5.54 | 6.13 | |
| W (eV) | 4.48 | 3.66 | 3.78 | 4.03 | 4.04 | 4.24 | 4.40 | 4.74 | 4.87 |
| System | Property | Application | Reference |
|---|---|---|---|
| Au NPs/Graphene | Sensitivity Enhancement | Clinical Immunoassays | [31] |
| Pd NPs/Graphene | Electrochemical Activity | Glucose Biosensor | [32] |
| Ag NPs/Graphene | Raman Scattering Electrochemical activity | Surface Enhanced Raman scattering H2O2 Sensing Glucose Sensing | [35] |
| Pd NPs/Graphene | Electrical Conduction | Hydrogen Sensing | [36] |
| Ag NPs/Graphene | Thermal Conductivity | Thermal Interface Materials | [37] |
| Au NPs/Graphene | Localized Surface Plasmon Resonance | Flexible and Transparent Optoelectronics | [40] |
| Au, Pd, Pt NPs/Graphene | Electrochemical Activity | H2S Sensing | [41] |
| Pd NPs/Graphene | - | Heterogeneous Catalysis | [48] |
| Au NPs/Graphene | Plasmon Absorption | - | [51] |
| Au, Ag NPs/Graphene | Plasmonic Properties | Surface Enhanced Raman Spectroscopy | [52] |
| Au, Ag, Pd, Pt NPs/Graphene | Plasmonic Properties | Raman Spectroscopy | [53] |
| Ag NPs/Graphene | Plasmonic Properties | Surface Enhanced Raman Spectroscopy | [54] |
| Au, Co, Pd, Pt, Rh NPs/Graphene | - | Catalysis | [65] |
| Au NPs/Graphene | - | Surface Enhanced Raman Spectroscopy | [68] |
| Ag NPs/Graphene | - | Surface Enhanced Raman Spectroscopy | [71] |
| AuAg NPs/Graphene | Plasmonic properties | Solar Cell | [135] |
| Ag NPs/Graphene | Plasmonic properties | Photodetection | [136] |
| Al NPs/Graphene | Plasmonic properties | Solar Cell | [137] |
| Au NPs/Graphene | - | Catalysis | [138] |
| Ni NPs/Graphene | - | Photocatalysis | [139] |
| Pd NPs/Graphene | - | Hydrogen Storage | [140] |
| Pb NPs/Graphene | - | Thermoelectric Devices | [141] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruffino, F.; Giannazzo, F. A Review on Metal Nanoparticles Nucleation and Growth on/in Graphene. Crystals 2017, 7, 219. https://doi.org/10.3390/cryst7070219
Ruffino F, Giannazzo F. A Review on Metal Nanoparticles Nucleation and Growth on/in Graphene. Crystals. 2017; 7(7):219. https://doi.org/10.3390/cryst7070219
Chicago/Turabian StyleRuffino, Francesco, and Filippo Giannazzo. 2017. "A Review on Metal Nanoparticles Nucleation and Growth on/in Graphene" Crystals 7, no. 7: 219. https://doi.org/10.3390/cryst7070219
APA StyleRuffino, F., & Giannazzo, F. (2017). A Review on Metal Nanoparticles Nucleation and Growth on/in Graphene. Crystals, 7(7), 219. https://doi.org/10.3390/cryst7070219