What Can We Learn from the Crystal Structures of Metallacarboranes?


Abstract
:
1. Introduction
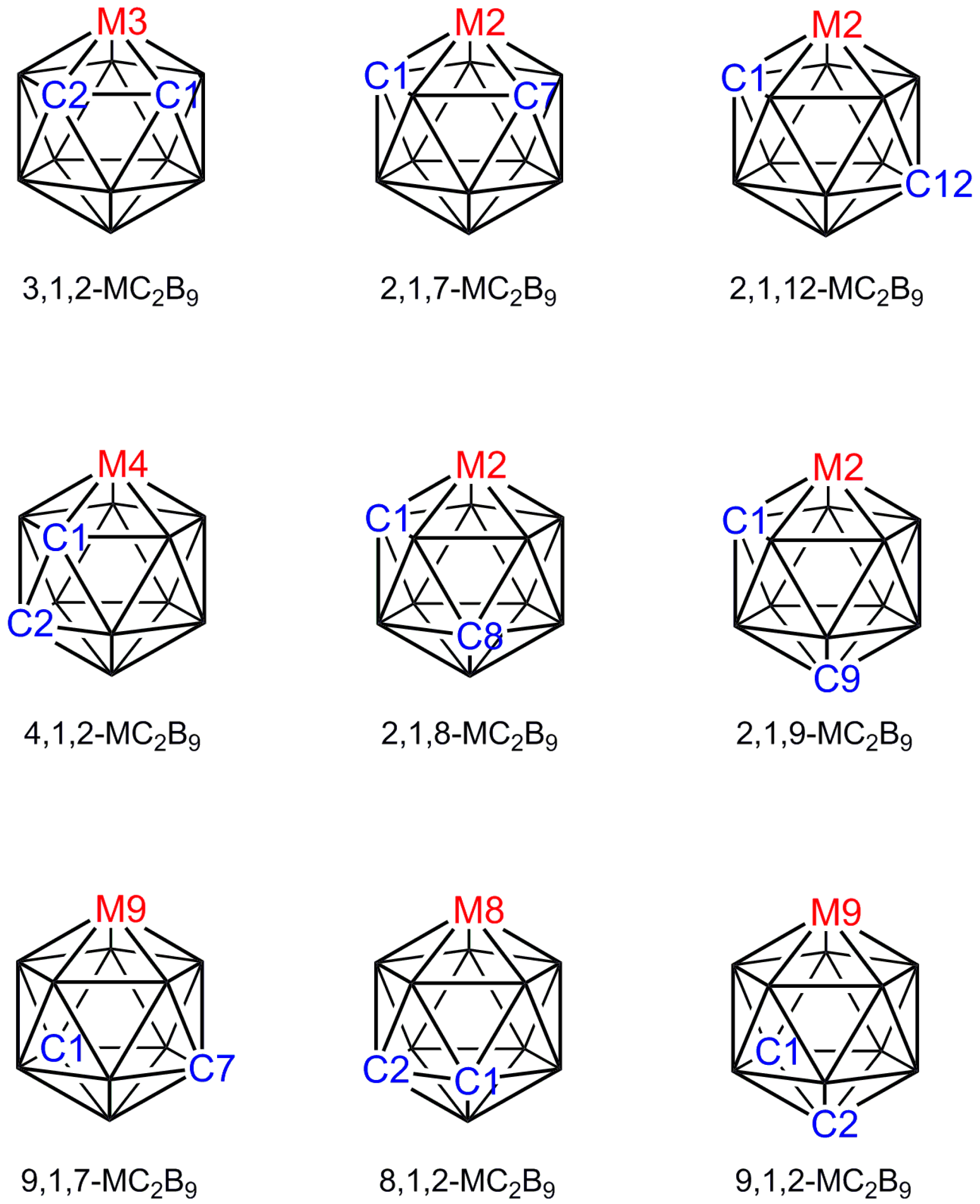
2. Distinguishing B and C Vertices
- Make use of the conventional wisdom that the lengths of cage connectivities are C–C < C–B < B–B. The basis of this is simply that C has a smaller atomic radius than B since the valence electrons experience a greater effective nuclear charge (Zeff) through imperfect shielding.
- Refine all the potential cage C and cage B atoms as B (the Prostructure) and identify the C atoms by their relatively small Ueq values, since if insufficient electron density has been assigned to an atom, the process of least-squares refinement compensates by condensing the available electron density, shrinking Ueq.
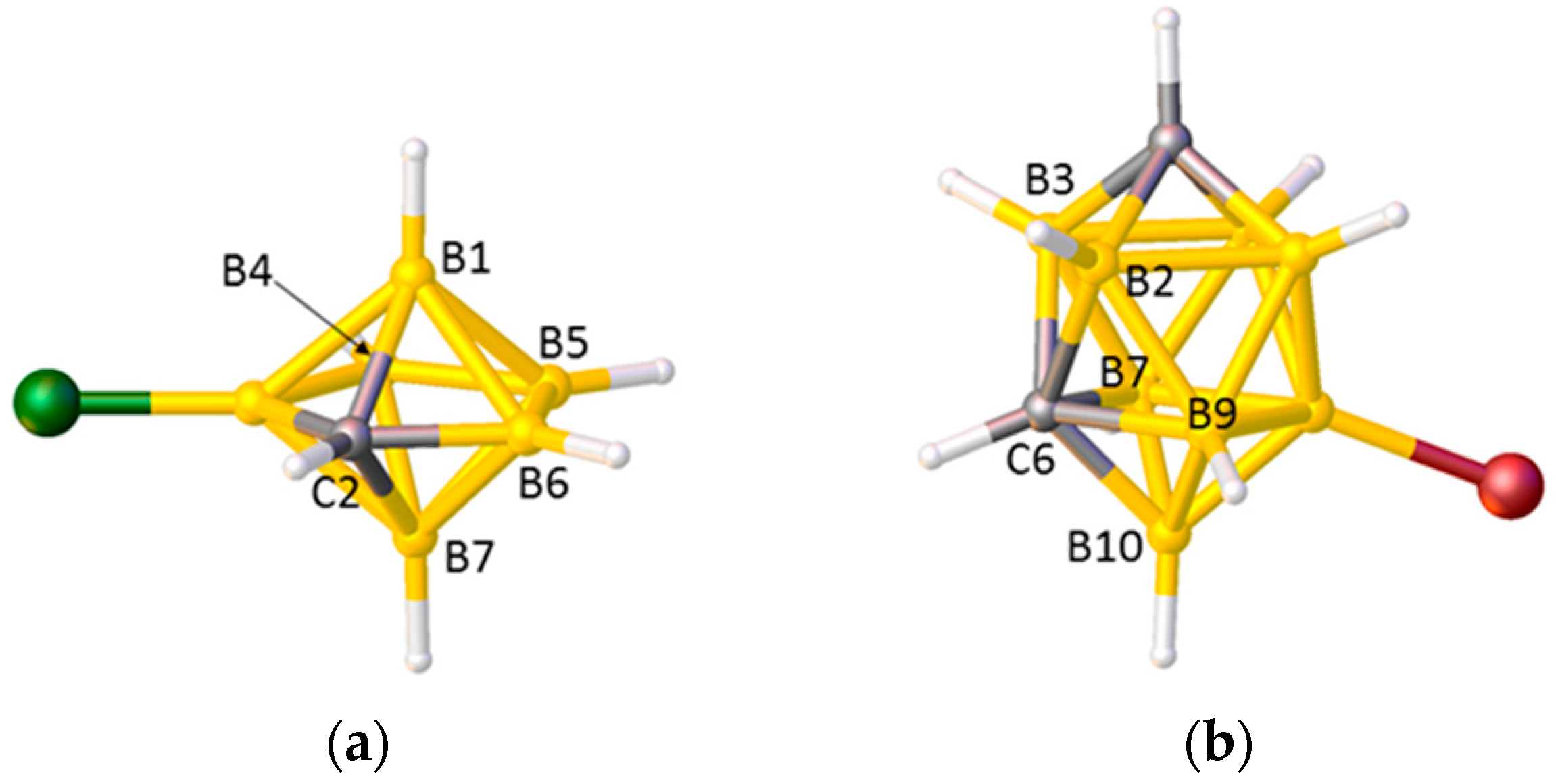
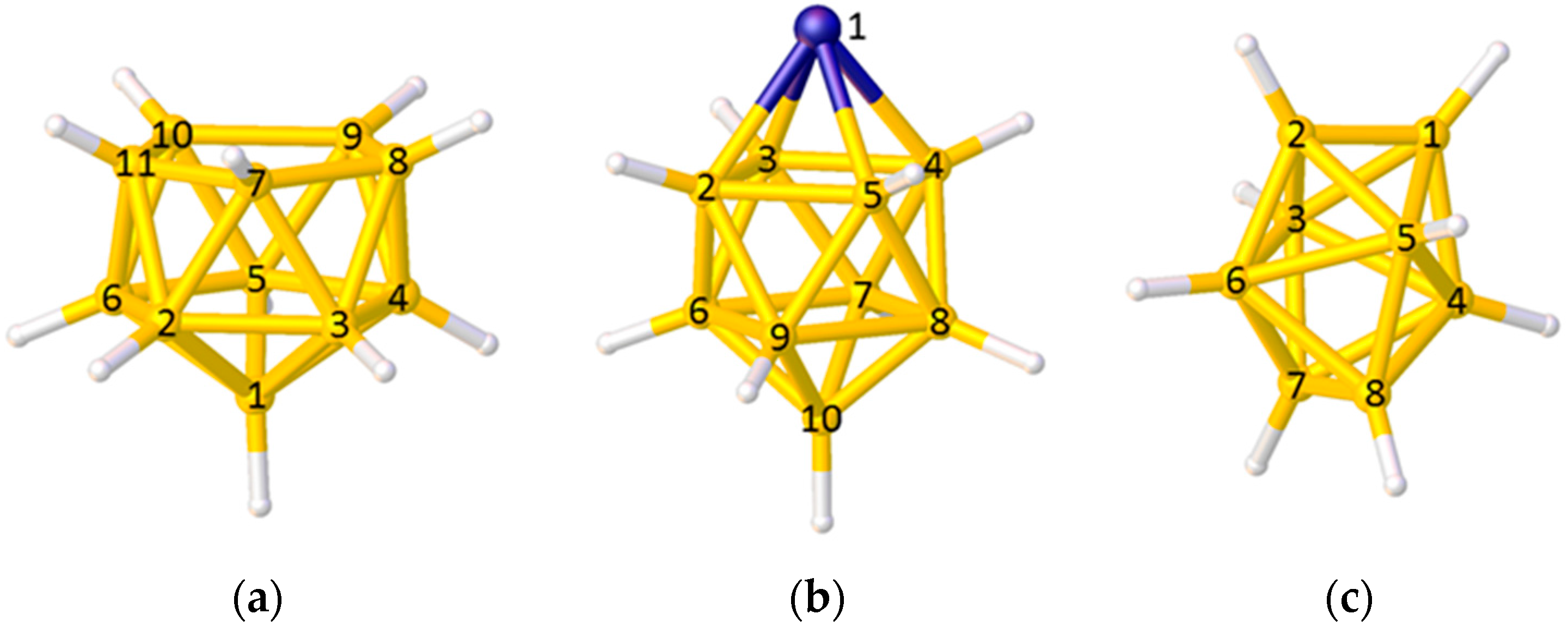
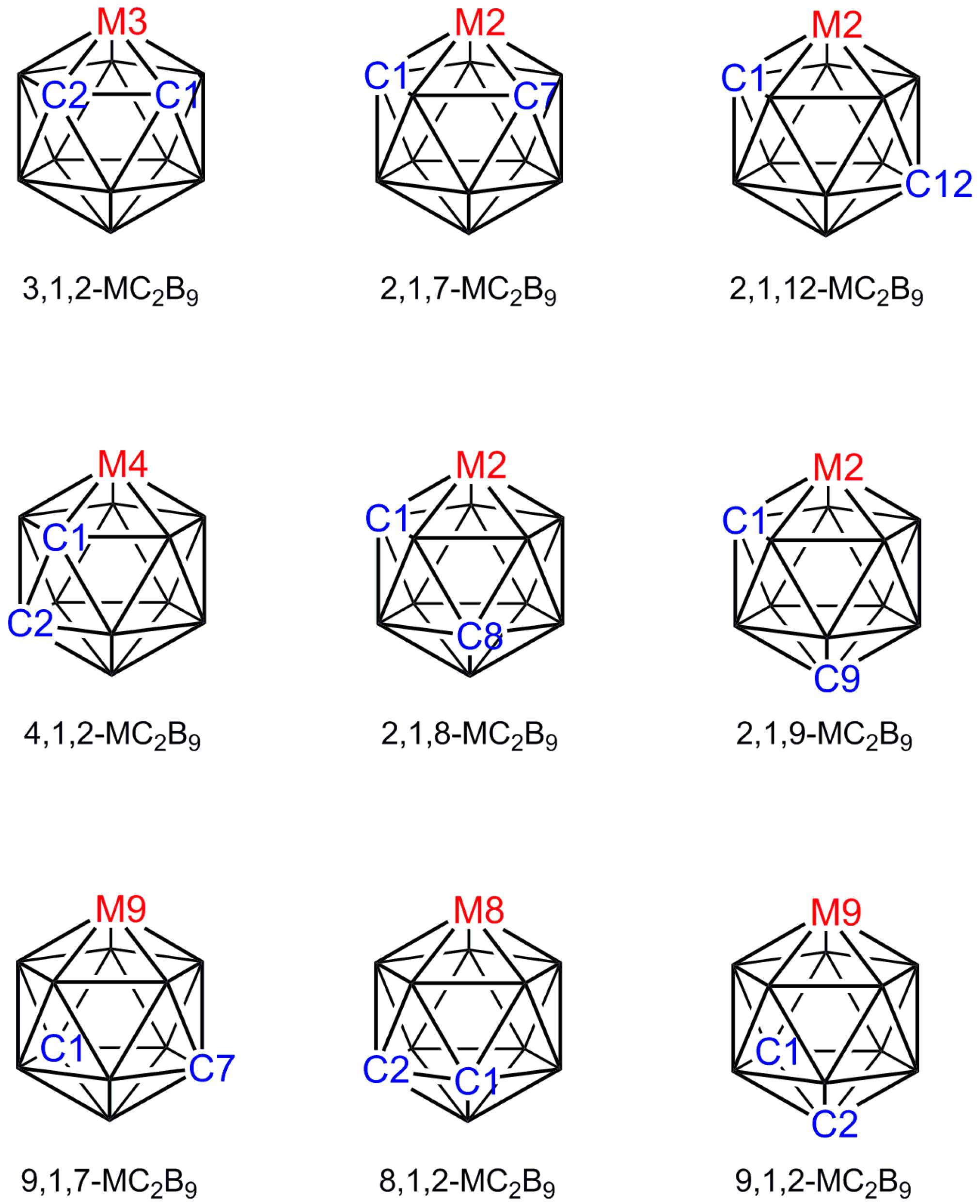
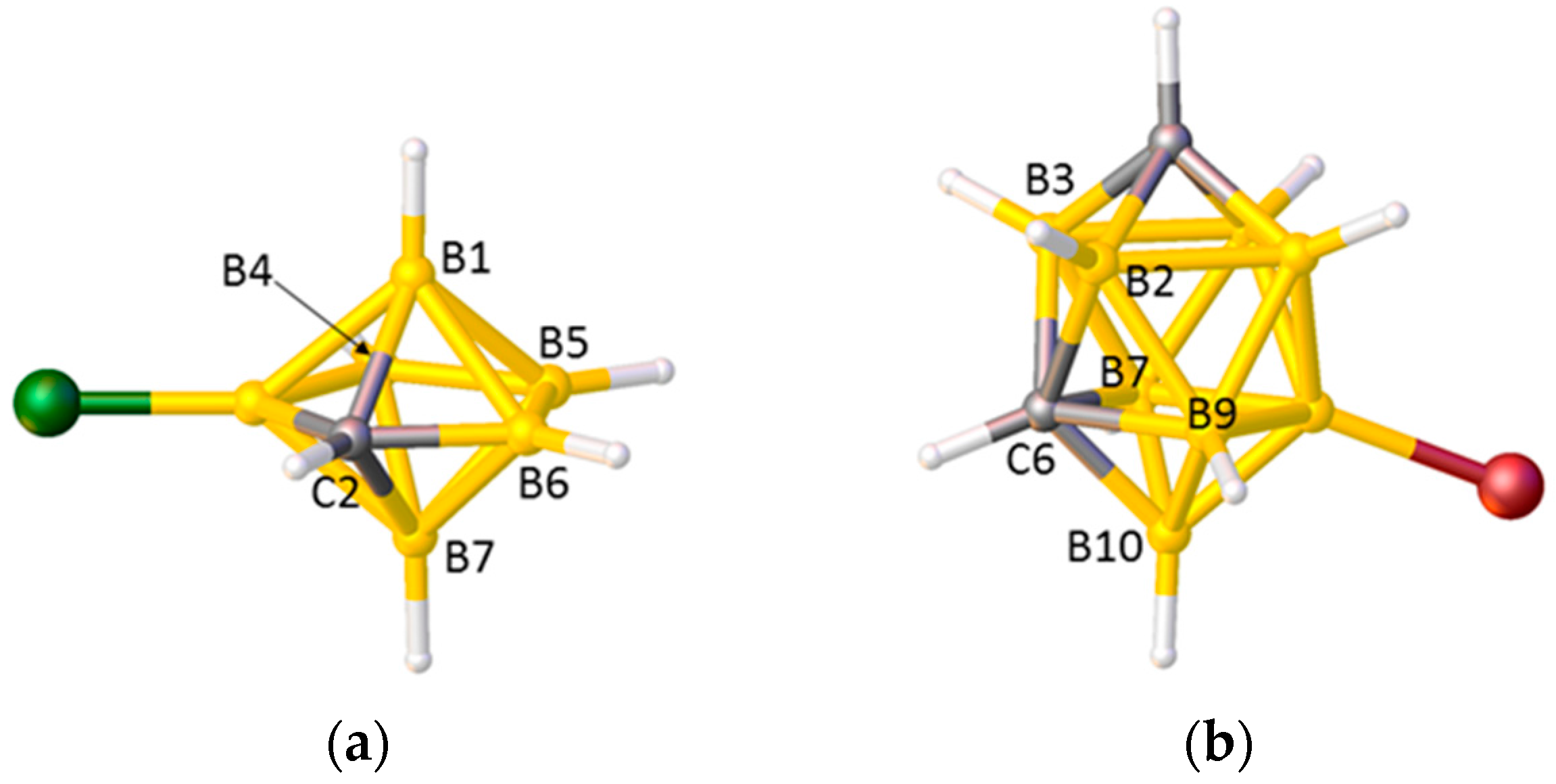
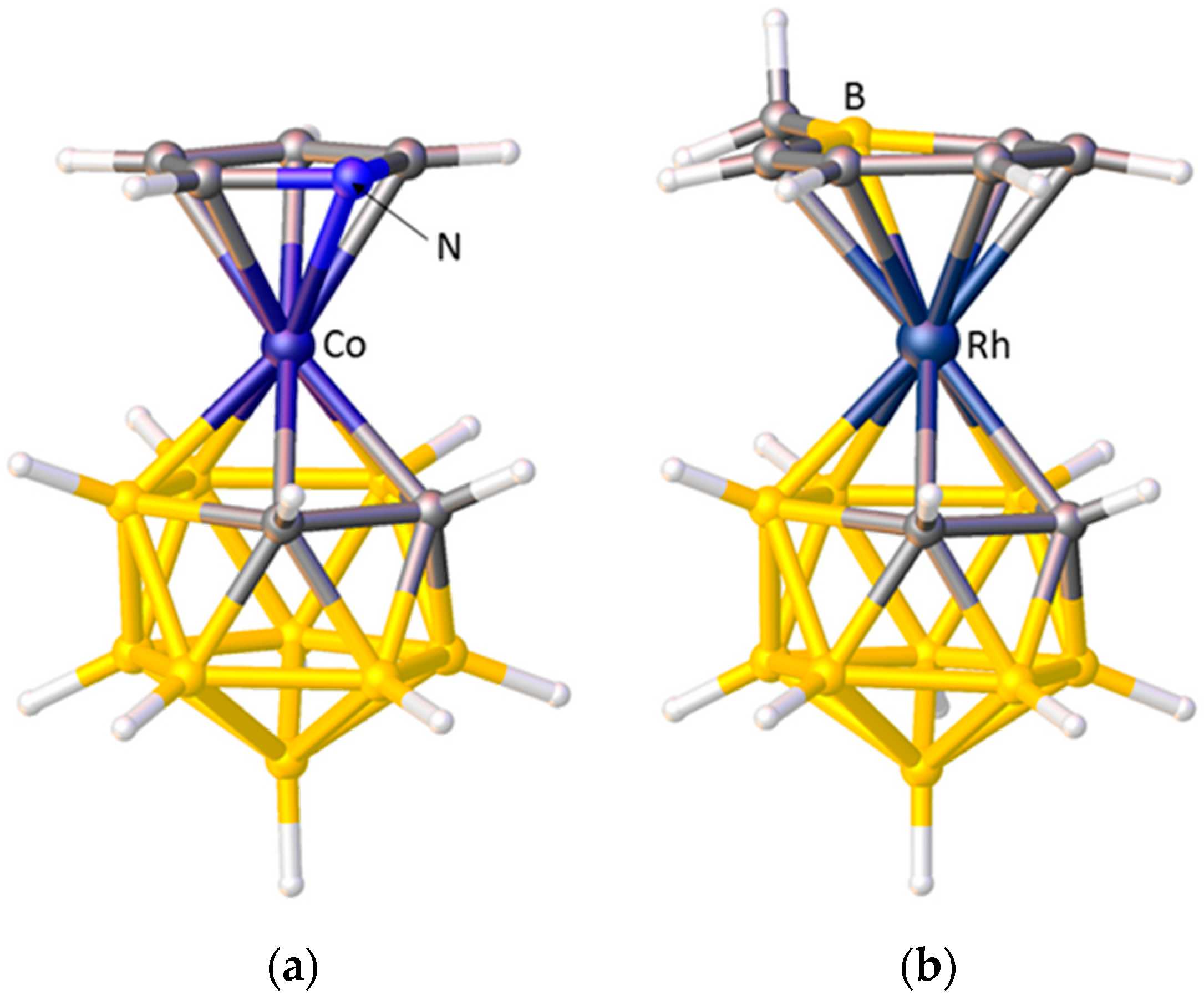
2.1. The VCD Method
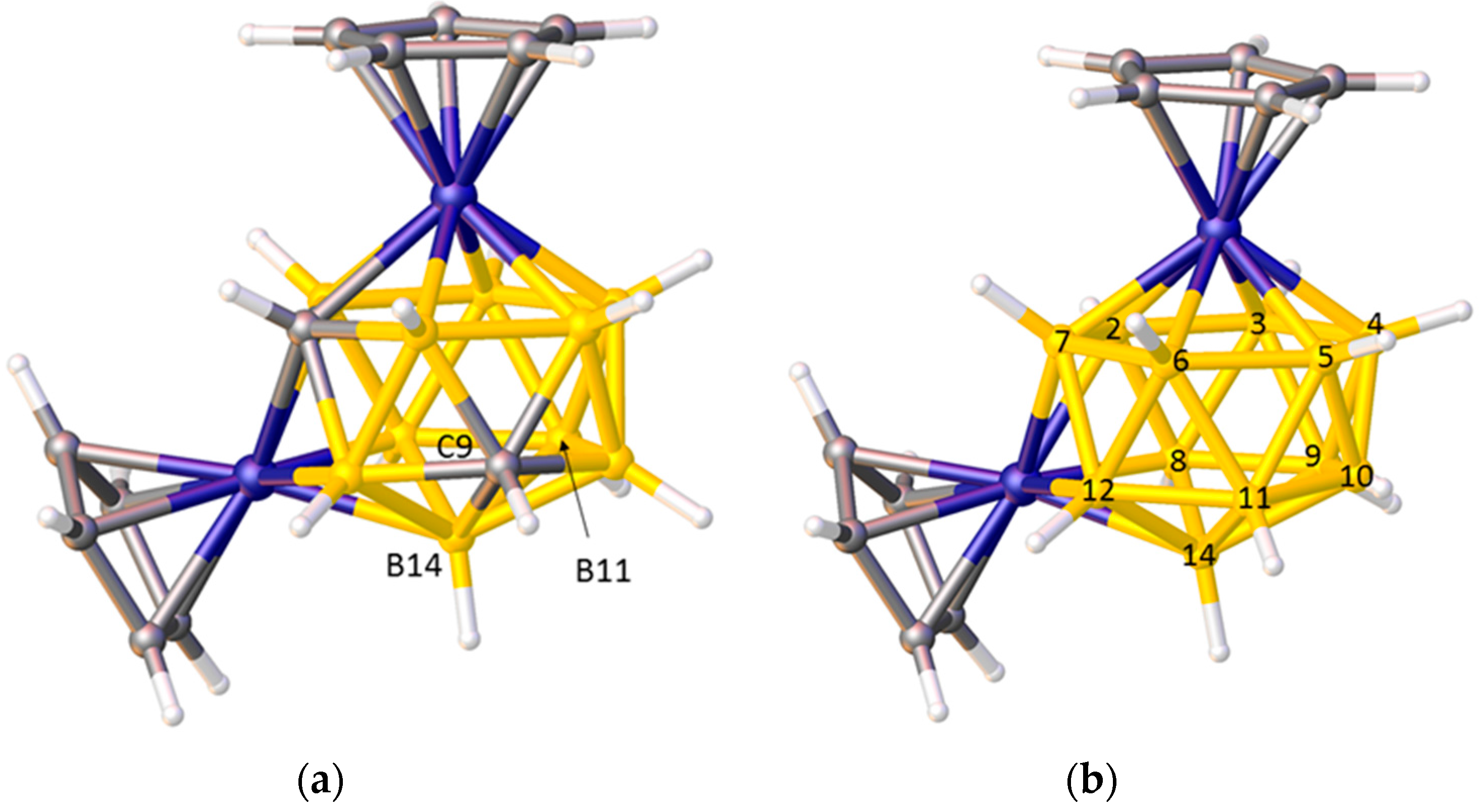
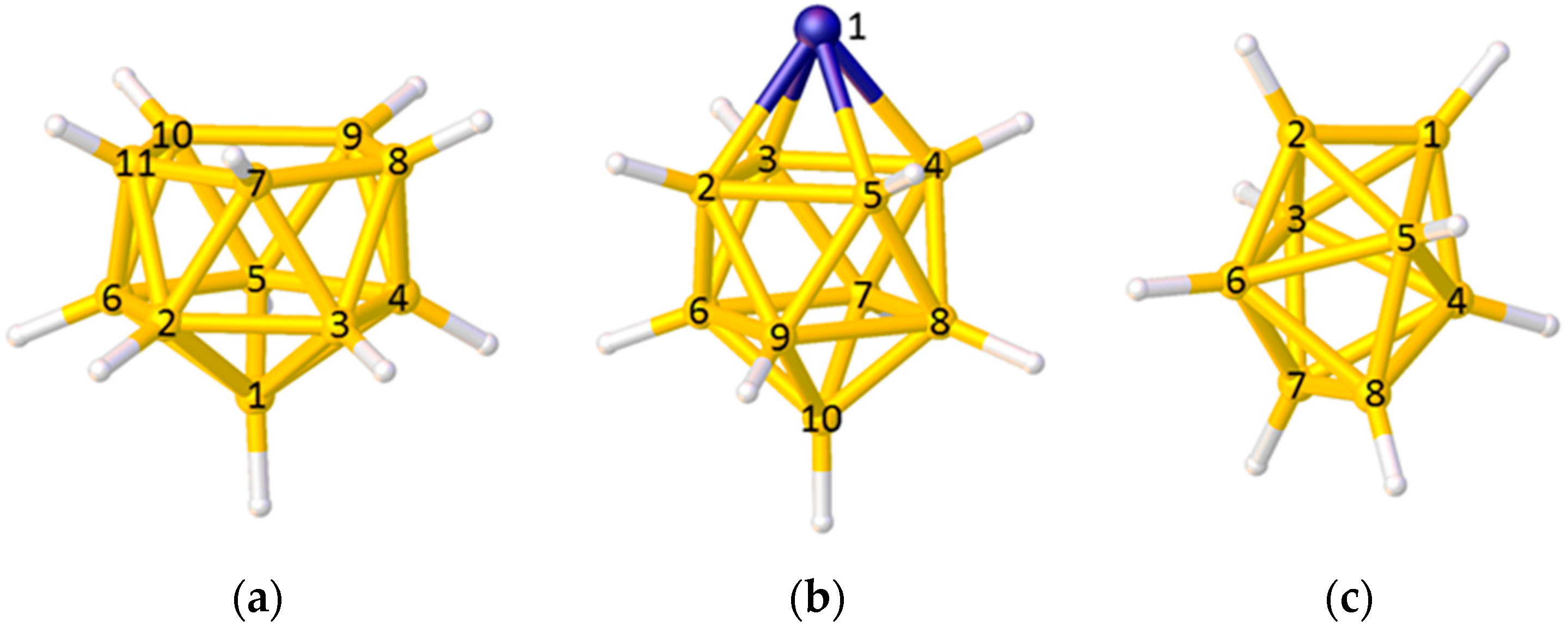
2.2. The BHD Method
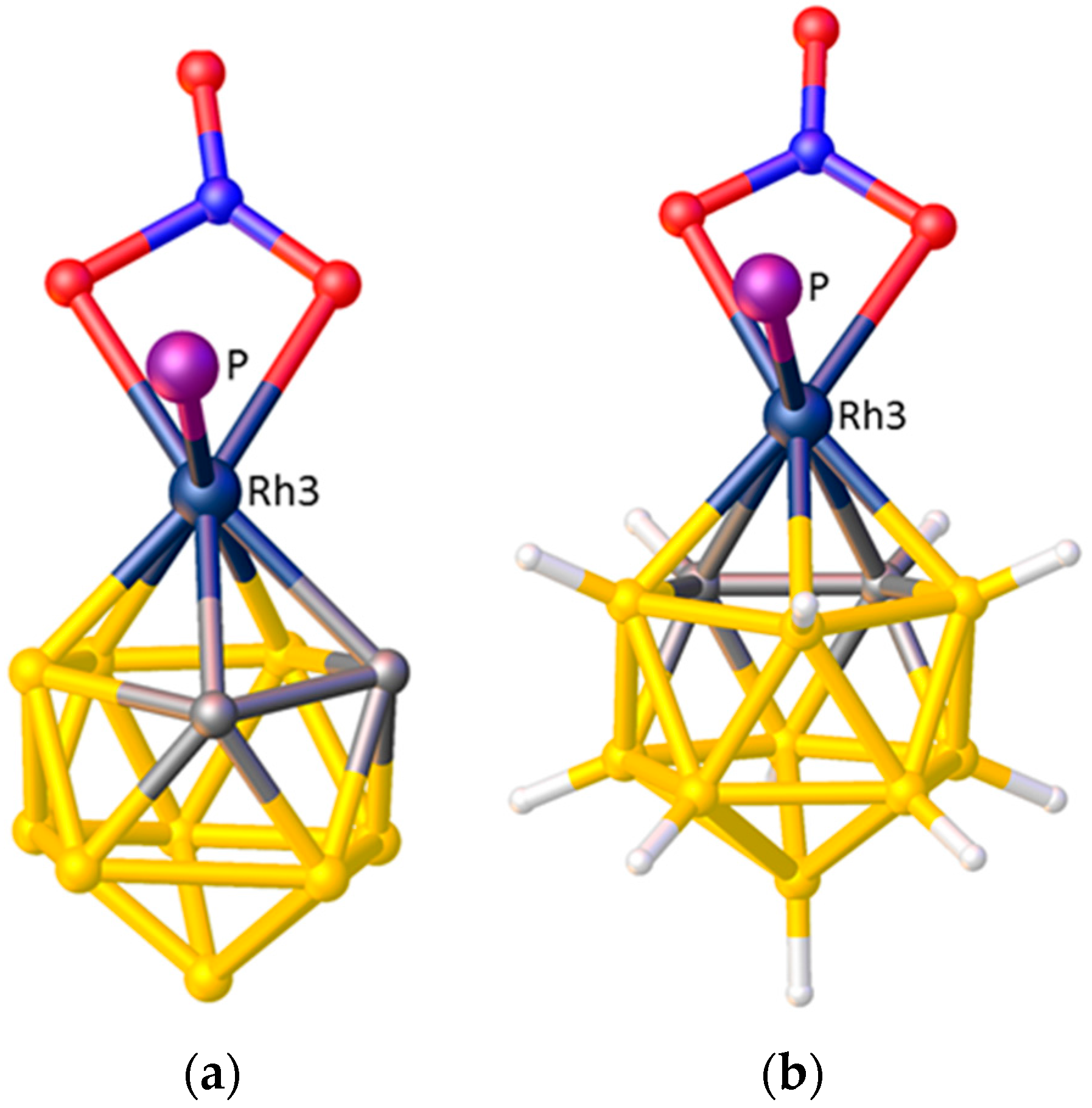
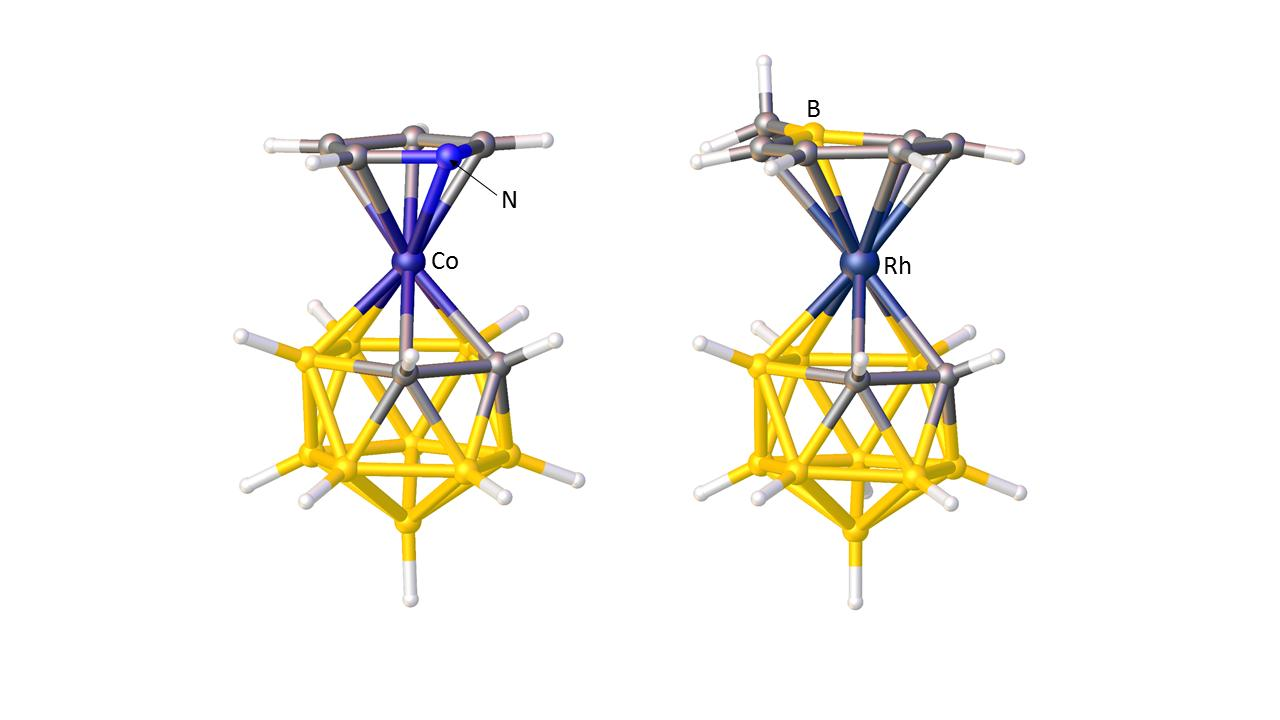
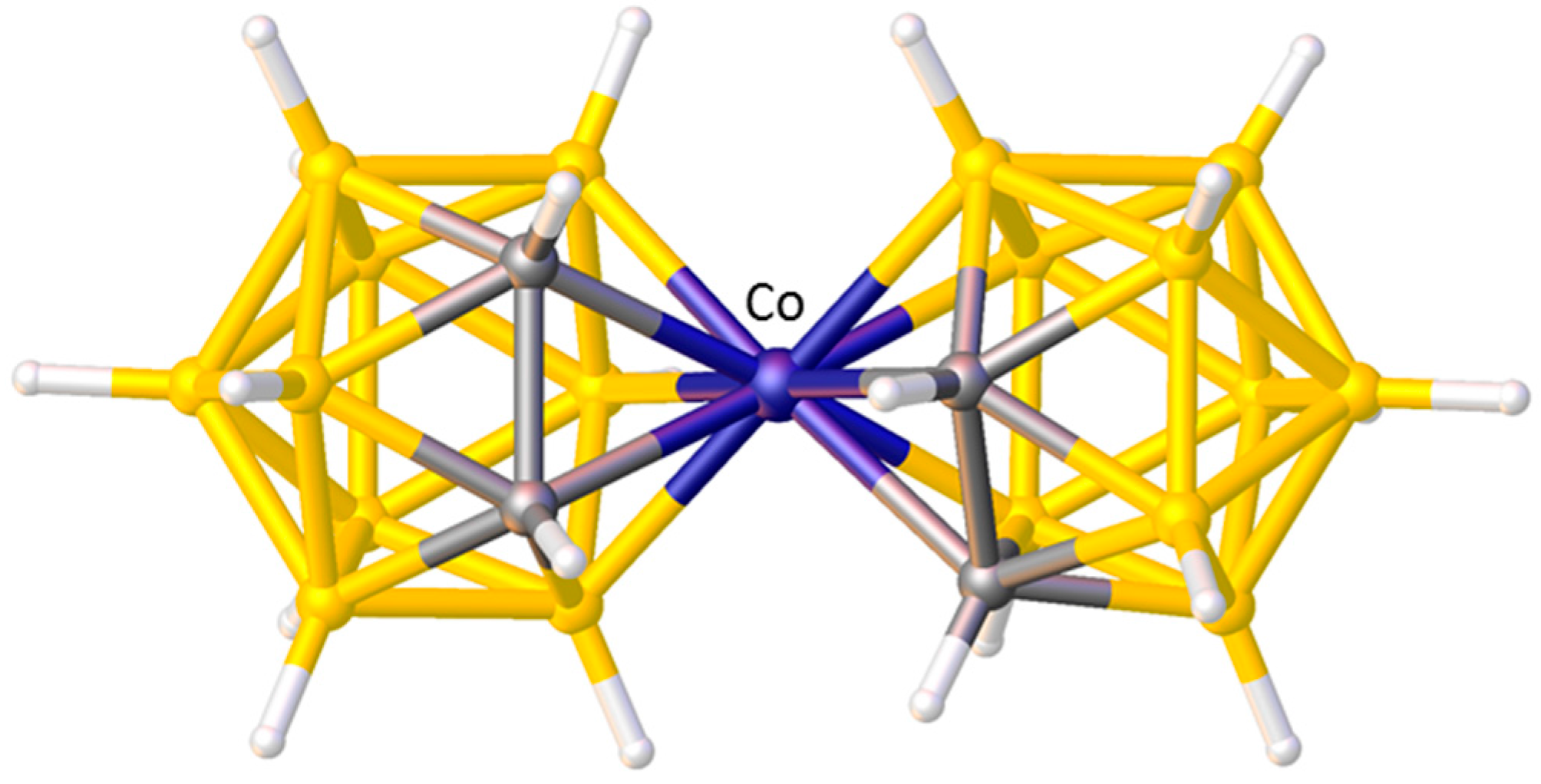
3. Structural Trans Effects in Metallacarboranes
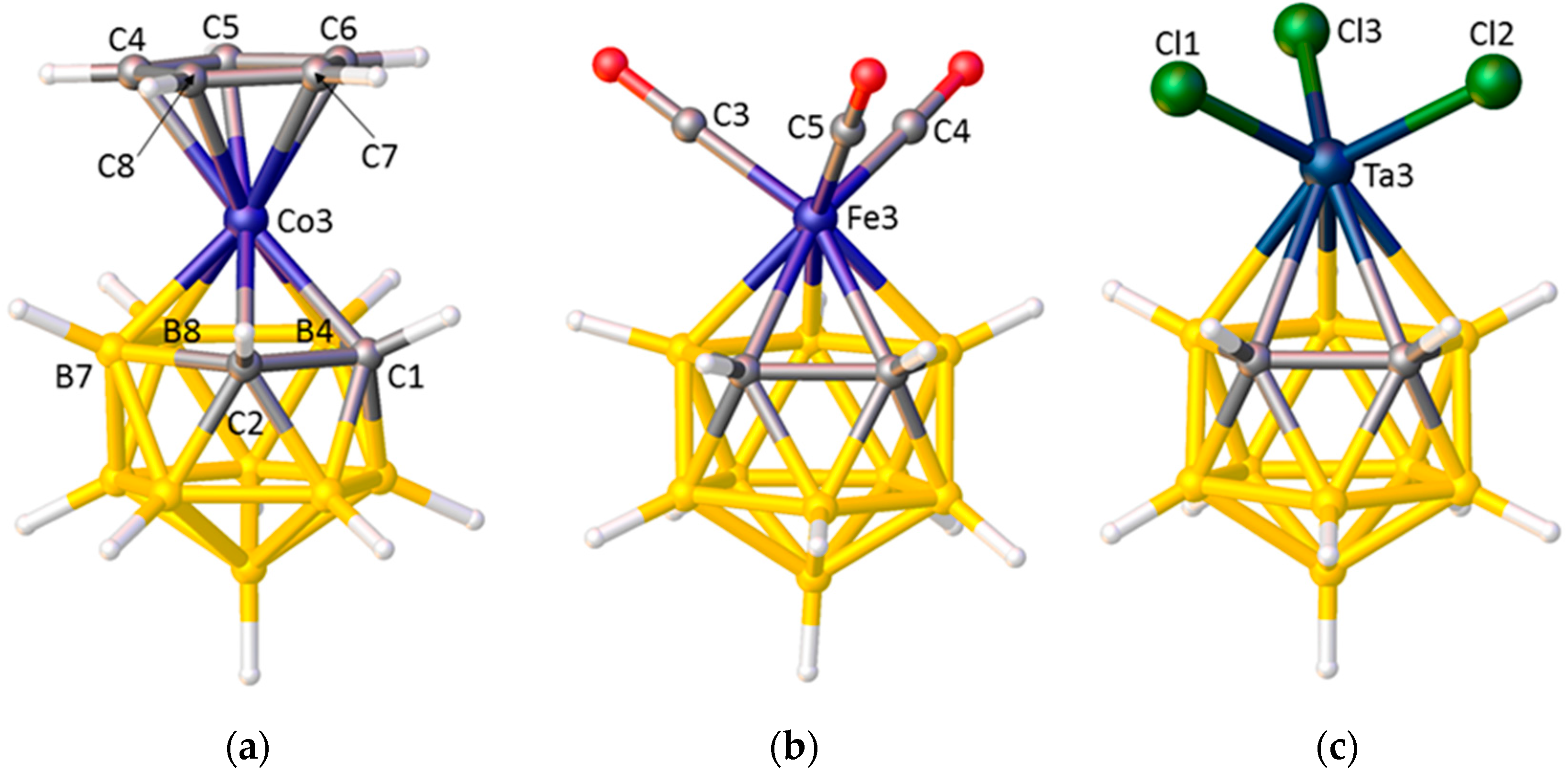
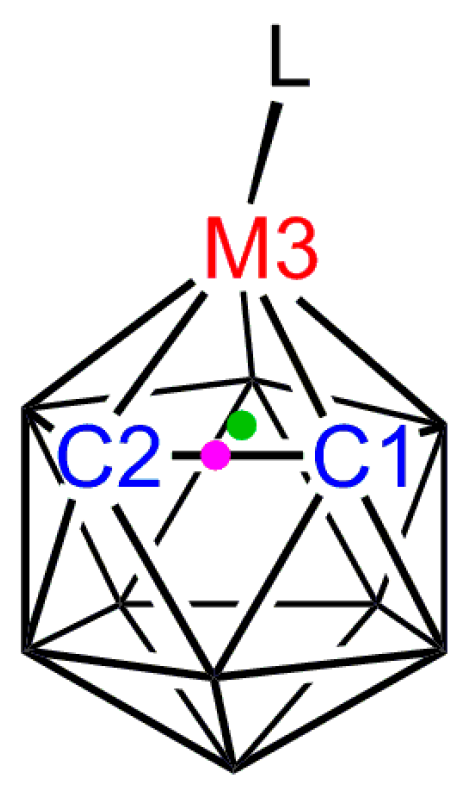
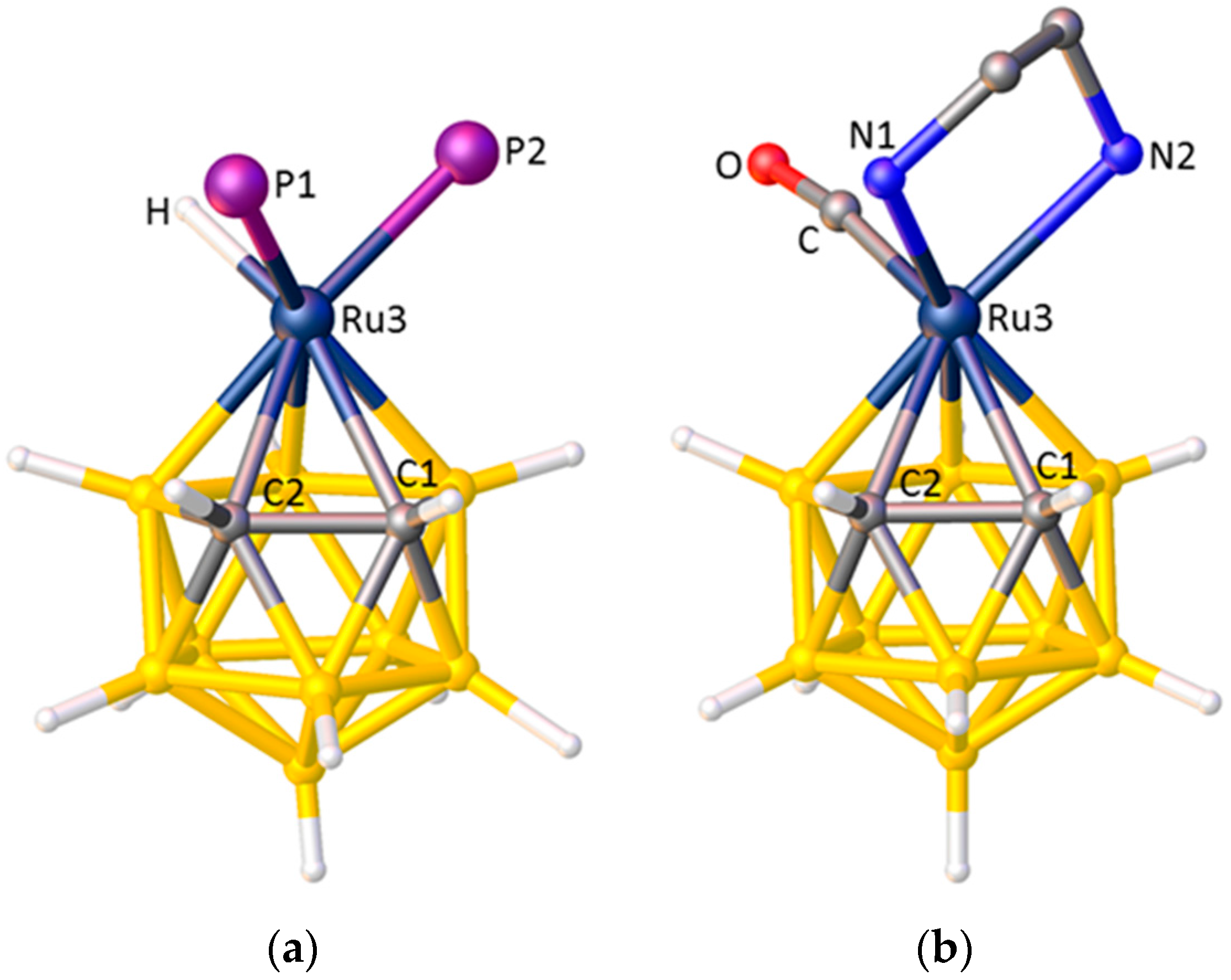
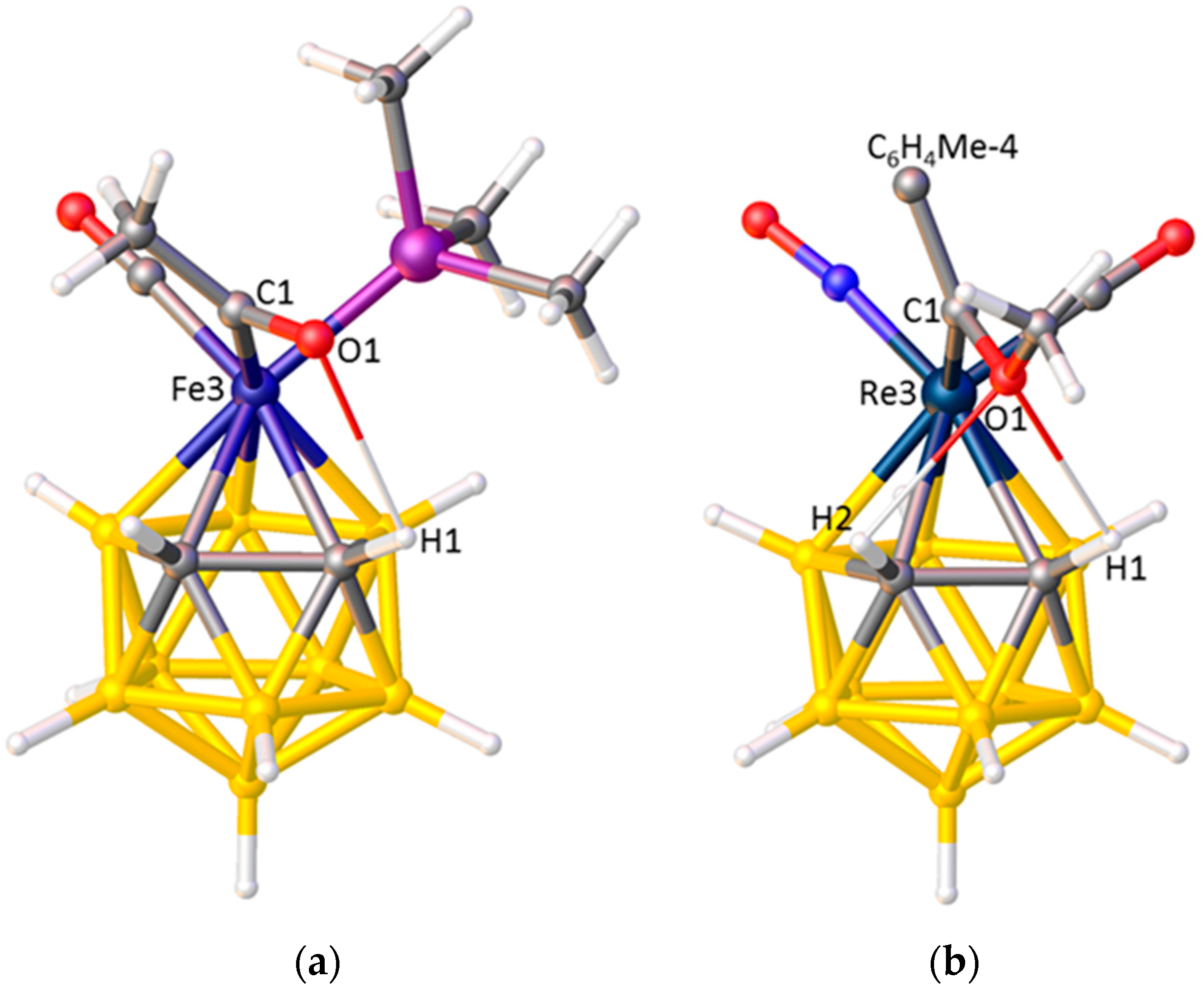
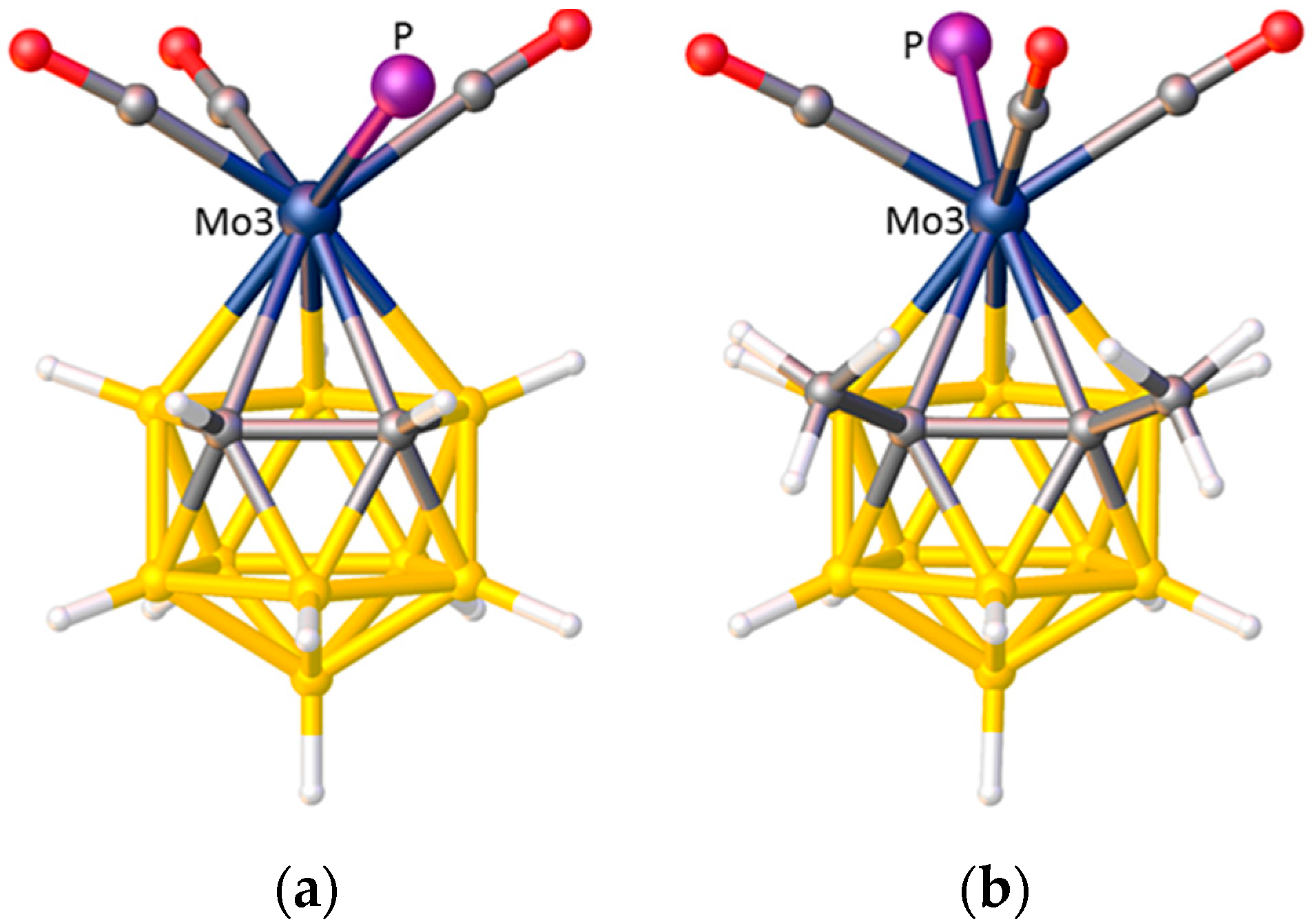
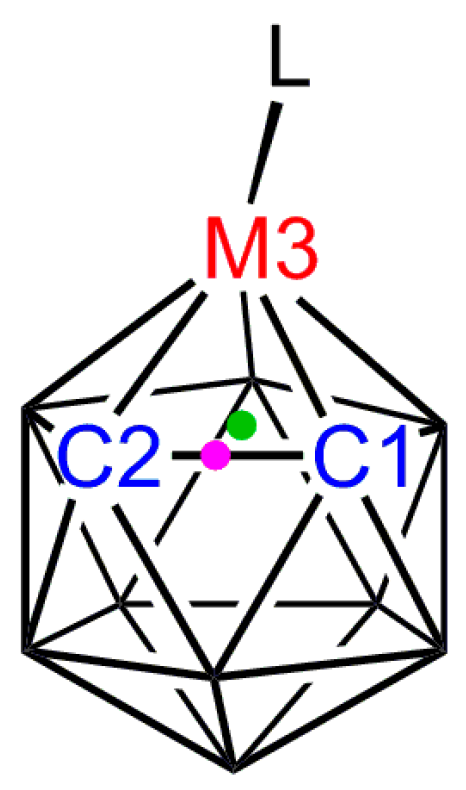
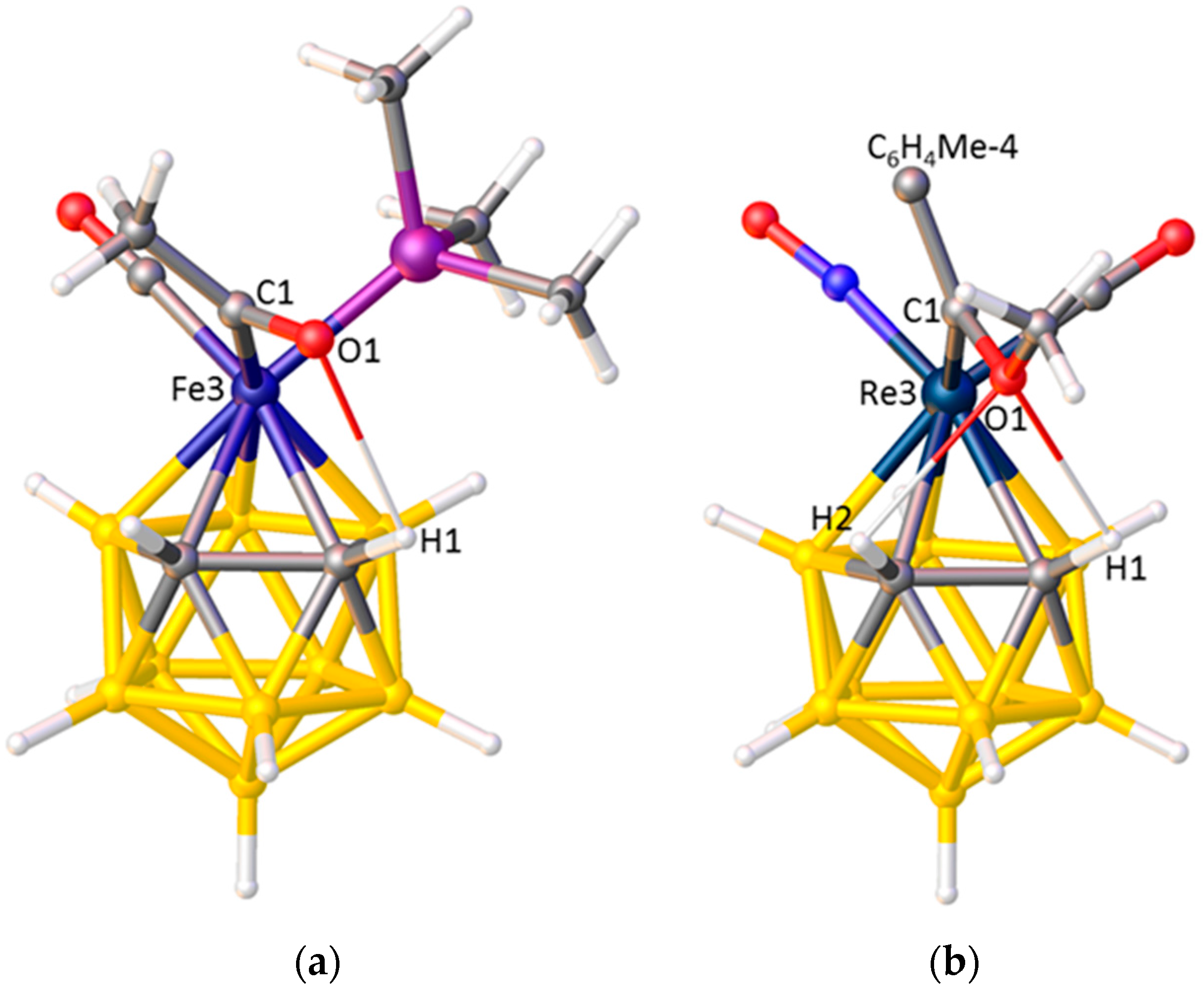
3.1. Exopolyhedral Ligand Orientation
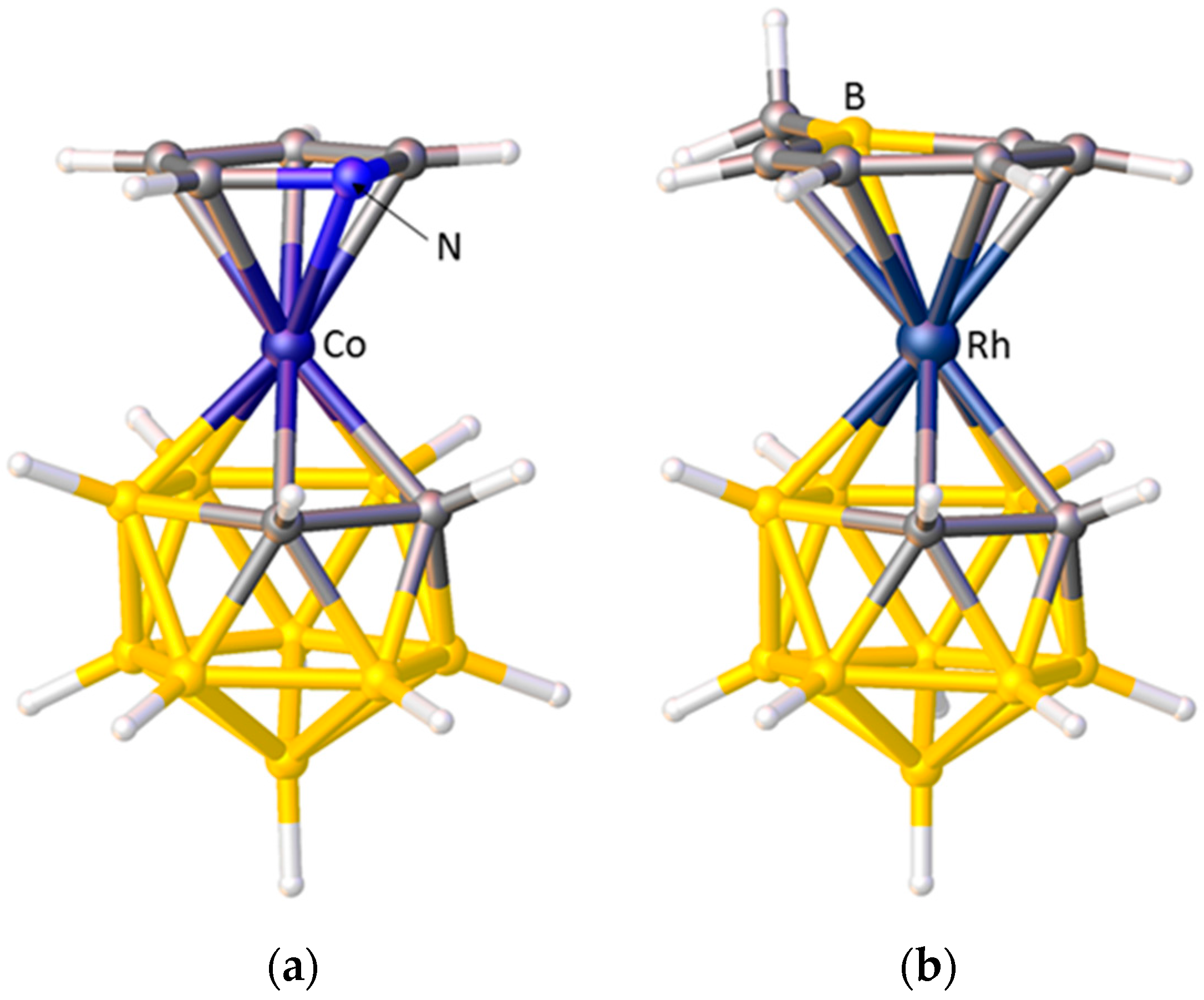
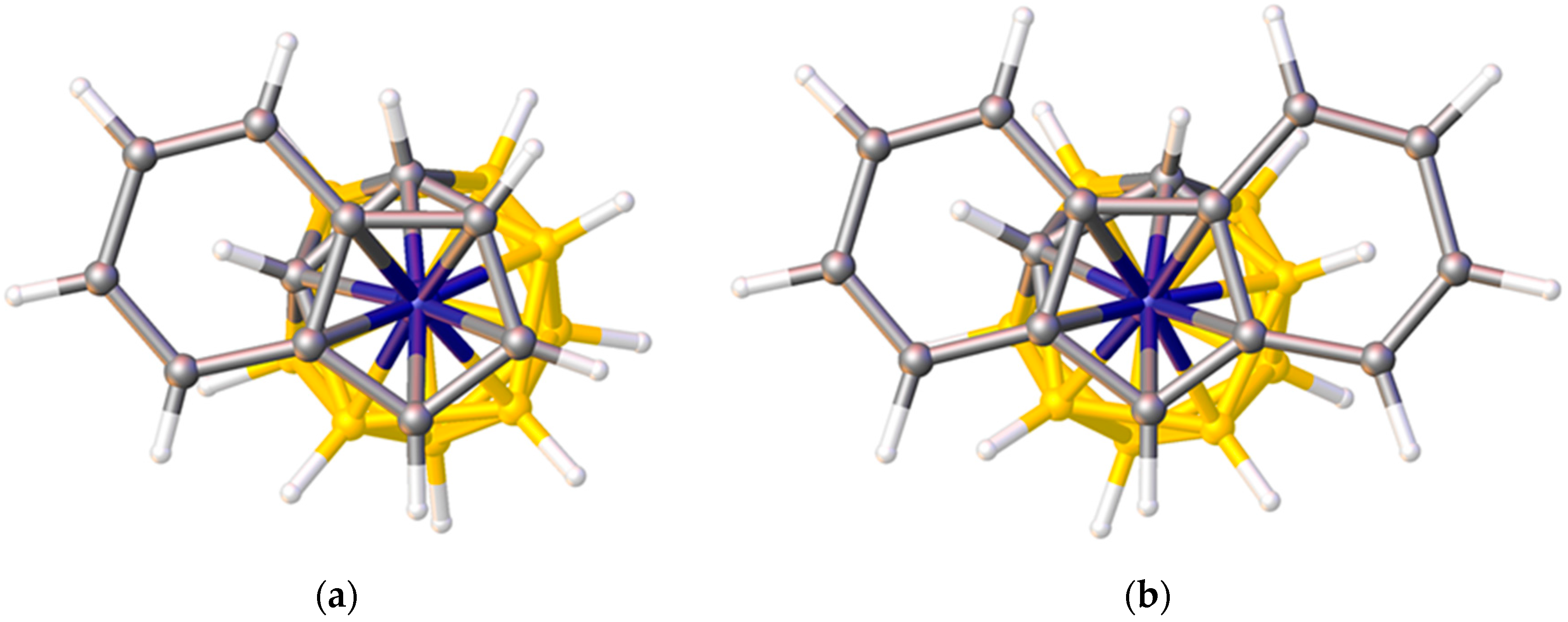
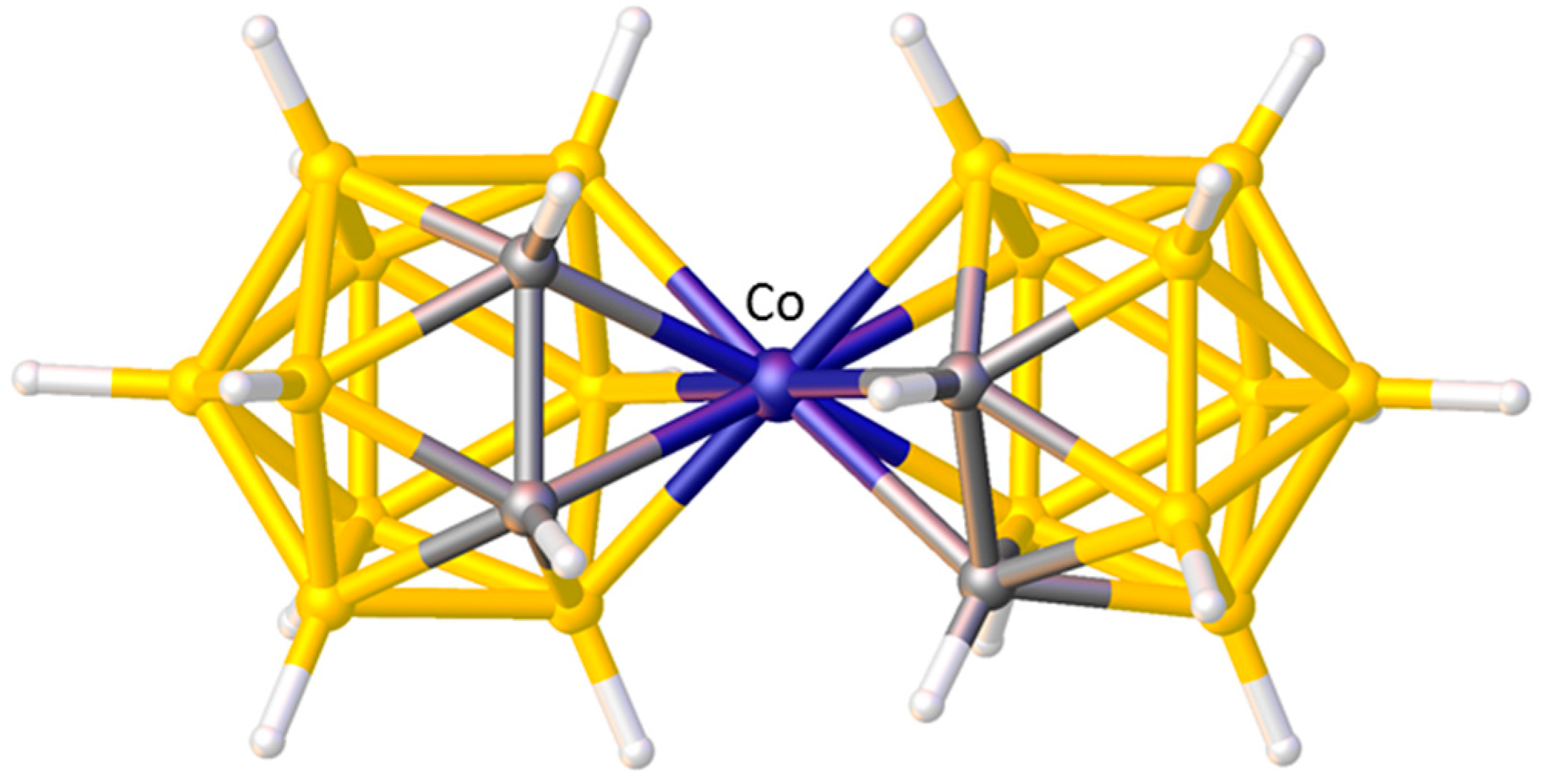
3.1.1. ELO of η-Bound Ligands
3.1.2. ELO of κ1 Ligands
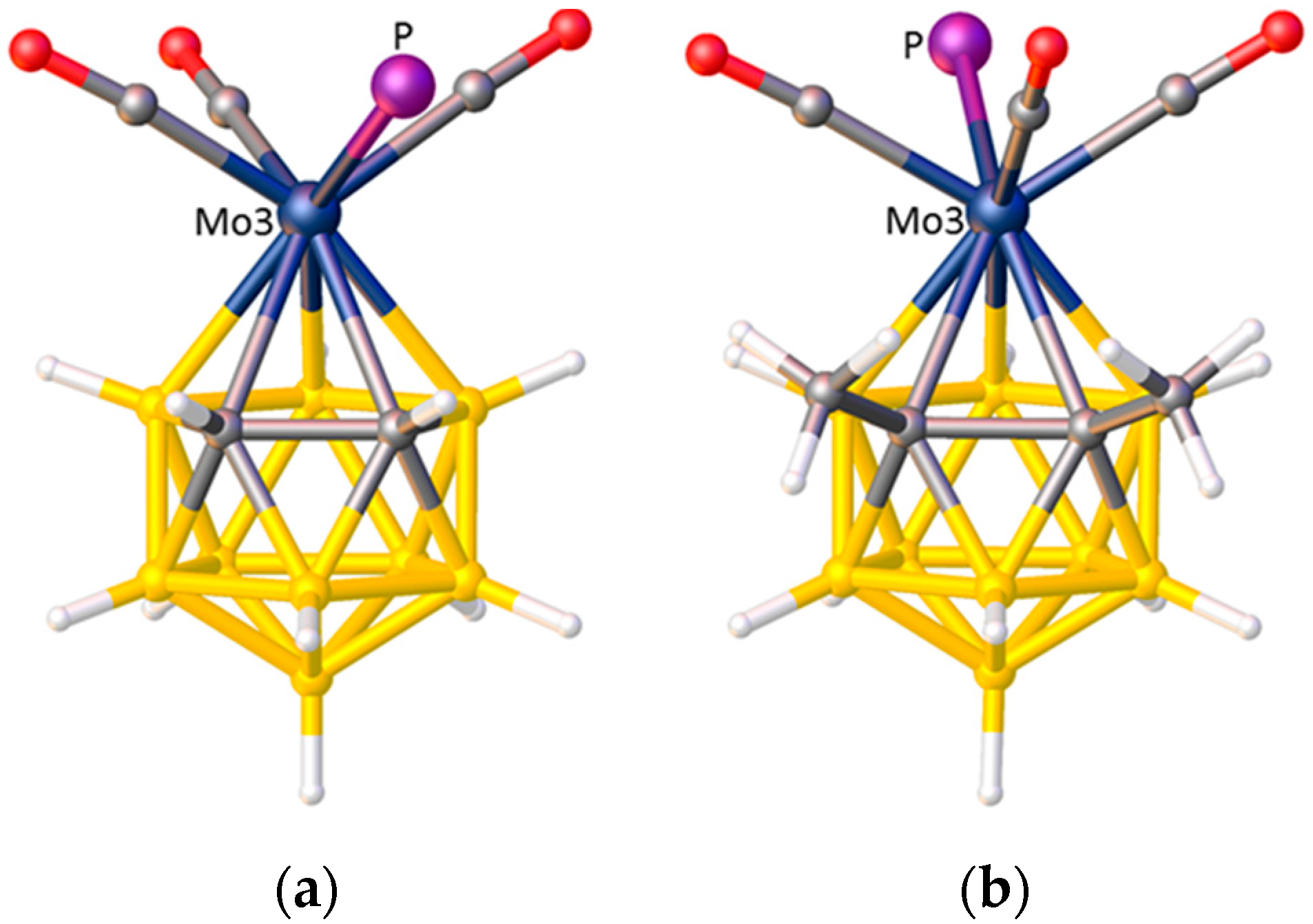
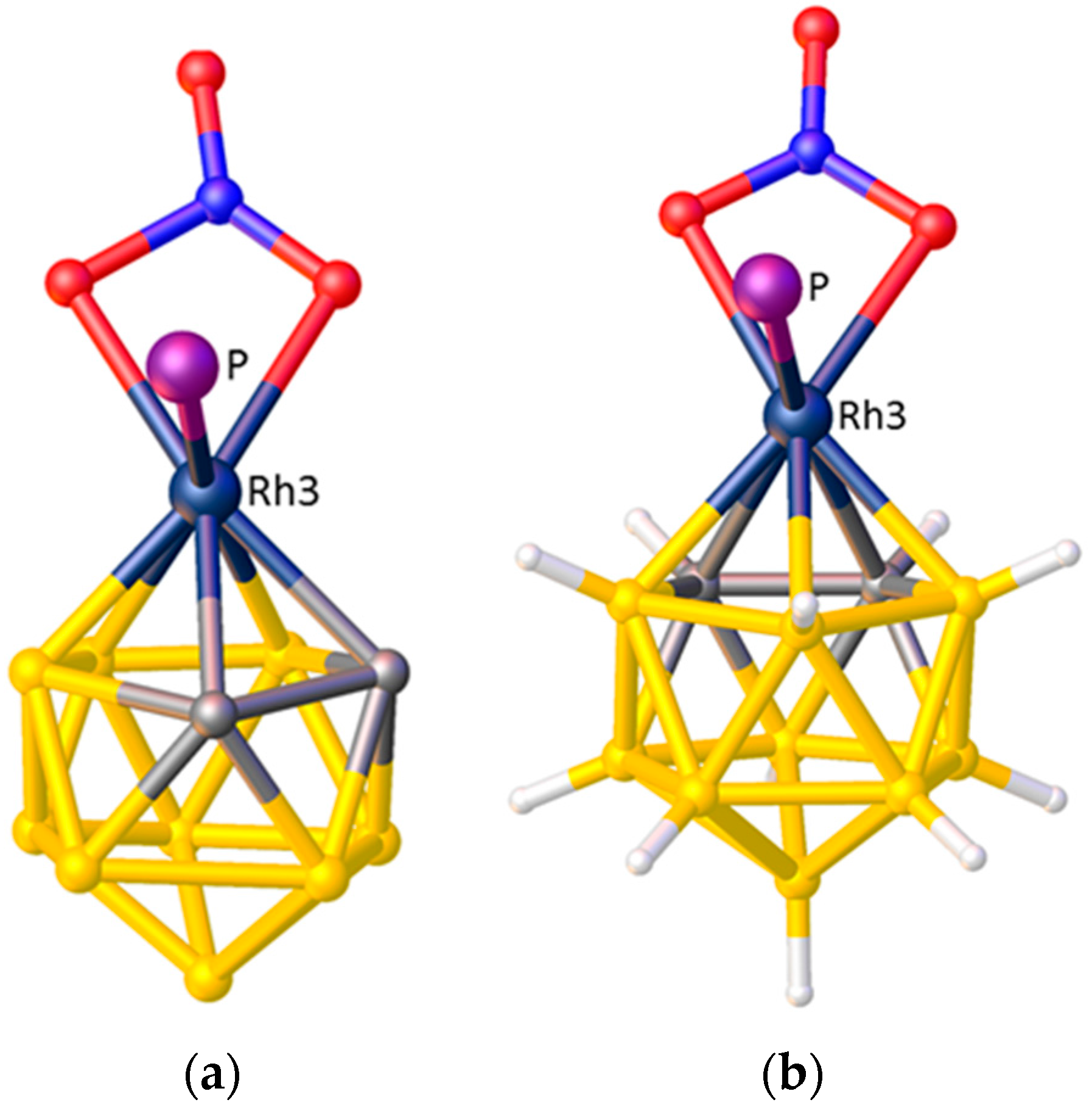
3.1.3. Applications of ELO
Rank Ordering the STEs of Exopolyhedral Ligands
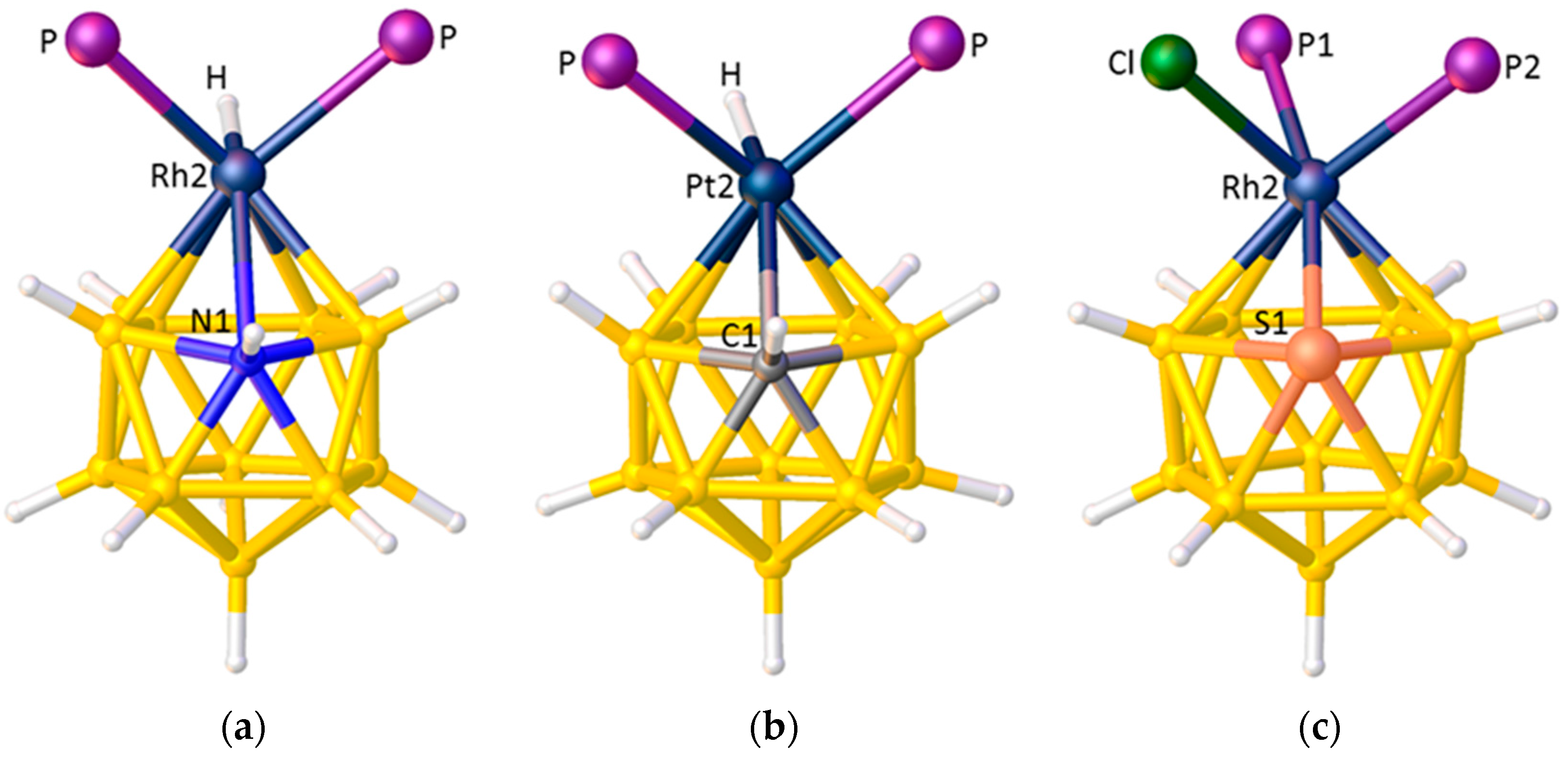
Rapid Identification of Suspect Structures
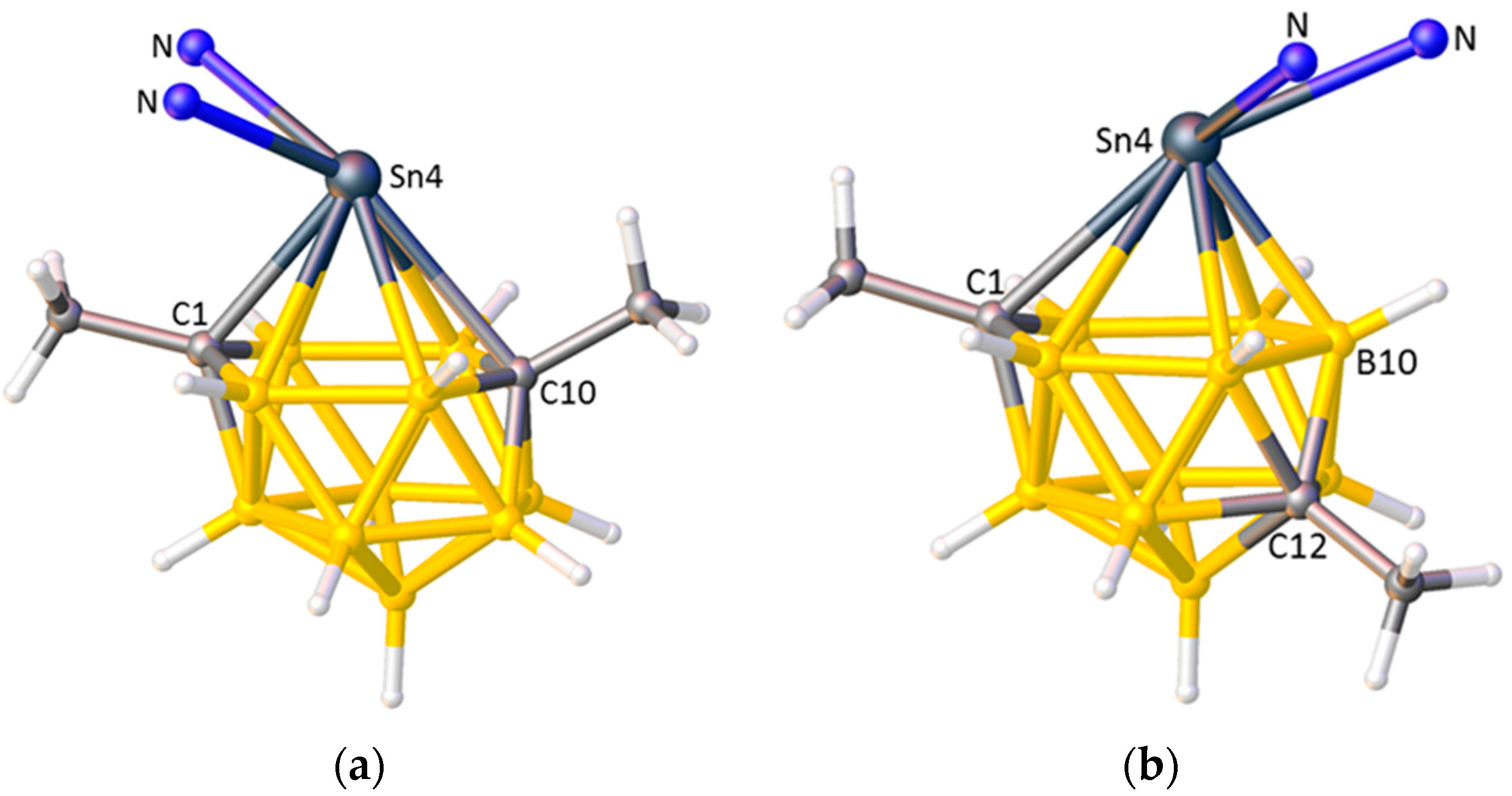
Exploring the STEs of Cage Heteroatoms
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Hawthorne, M.F.; Young, D.C.; Wegner, P.A. Carbametallic Boron Hydride Derivatives. I. Apparent Analogs of Ferrocene and Ferricinium Ion. J. Am. Chem. Soc. 1965, 87, 1818–1819. [Google Scholar] [CrossRef]
- Zalkin, A.; Templeton, D.H.; Hopkins, T.E. The Crystal and Molecular Structure of C5H5FeB9C2H11. J. Am. Chem. Soc. 1965, 87, 3988–3990. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 831–863. [Google Scholar]
- Robertson, A.P.M.; Beattie, N.A.; Scott, G.; Man, W.Y.; Jones, J.J.; Macgregor, S.A.; Rosair, G.M.; Welch, A.J. 14-Vertex Heteroboranes with 14 Skeletal Electron Pairs: An Experimental and Computational Study. Angew. Chem. Int. Ed. 2016, 55, 8706–8710. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, M.F.; Young, D.C.; Andrews, T.D.; Howe, D.V.; Pilling, R.L.; Pitts, A.D.; Reintjes, M.; Warren, L.F.; Wegner, P.A. π-Dicarbollyl derivatives of the transition metals. Metallocene analogs. J. Am. Chem. Soc. 1968, 90, 879–896. [Google Scholar] [CrossRef]
- Kaloustian, M.K.; Wiersema, R.J.; Hawthorne, M.F. Thermal rearrangement of π-cyclopentadienyl-π-dicarbollyl derivatives of cobalt. J. Am. Chem. Soc. 1972, 94, 6679–6686. [Google Scholar] [CrossRef]
- Busby, D.C.; Hawthorne, M.F. The crown ether promoted base degradation of p-carborane. Inorg. Chem. 1982, 21, 4101–4103. [Google Scholar] [CrossRef]
- Hanusa, T.P.; Todd, L.J. Reductive Isomerization of Icosahedral Metallocarbaboranes. Polyhedron 1985, 4, 2063–2066. [Google Scholar] [CrossRef]
- Lopez, M.E.; Ellis, D.; Murray, P.R.; Rosair, G.M.; Welch, A.J.; Yellowlees, L.J. Synthesis and/or Molecular Structures of Some Simple 2,1,7- and 2,1,12-Ruthena and Cobaltacarboranes. Collect. Czech. Chem. Commun. 2010, 75, 853–869. [Google Scholar] [CrossRef]
- Man, W.Y.; Zlatogorsky, S.; Tricas, H.; Ellis, D.; Rosair, G.M.; Welch, A.J. How to Make 8,1,2-closo-MC2B9 Metallacarboranes. Angew. Chem. Int. Ed. 2014, 53, 12222–12225. [Google Scholar] [CrossRef] [PubMed]
- Man, W.Y.; Rosair, G.M.; Welch, A.J. Reduction-induced facile isomerisation of metallacarboranes: Synthesis and crystallographic characterisation of [4-Cp-4,1,2-closo-CoC2B9H11]. Dalton Trans. 2015, 44, 15417–15419. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Welch, A.J. Indenylmetallacarboranes. 1. The 18-Valence-Electron Complex [3-(η5-C9H7)-3,1,2-CoC2B9H11] and Comparative Molecular Structures of This Complex and [3-(η5-C5H5)-3,1,2-CoC2B9H11]. Organometallics 1986, 5, 760–766. [Google Scholar] [CrossRef]
- Planas, J.G.; Viñas, C.; Teixidor, F.; Light, M.E.; Hursthouse, M.B. Polymorphism and phase transformations in cobaltacarborane molecular crystals. CrystEngComm 2007, 9, 888–894. [Google Scholar] [CrossRef]
- Man, W.Y.; Rosair, G.M.; Welch, A.J. Crystal structure of a second polymorph of 2-cyclopentadienyl-1,7-dicarba-2-cobalta-closo-dodecaborane(11). Acta Crystallogr. Sect. E Struct. Rep. Online 2015, 71, m141. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.G.; Hibbert, T.G.; Howard, J.A.K.; Mackinnon, A.; Wade, K. Definitive crystal structures of ortho, meta- and para-carboranes: Supramolecular structures directed solely by C–H···O hydrogen bonding to hmpa (hmpa = hexamethylphosphoramide). Chem. Commun. 1996, 2285–2286. [Google Scholar] [CrossRef]
- Disorder between {BH} and {CH} vertices is relatively commonplace in crystallographic studies of carboranes and heterocarboranes, complicating all approaches to discriminating between B and C vertices. Although in principle such disorder could be extensive, in our experience most examples appear to involve 50:50 disorder over two vertices which is fairly easily identified and simply modelled.
- Schlüter, F.; Bernhardt, E. Synthesis of 3-DBU-closo-2-C2B6H6 and [3-Cl-closo-2-CB6H6]−—A First Straightforward Synthesis of [CB6]-closo-Clusters. Z. Anorg. Allg. Chem. 2010, 636, 2462–2466. [Google Scholar] Note that some of the distances given in the legend to Figure 5 in this paper do not agree with those calculated from the deposited CIF.
- Bakardjiev, M.; Štíbr, B.; Holub, J.; Padělková, Z.; Růžička, A. Simple Synthesis, Halogenation and Rearrangement of closo-1,6-C2B8H10. Organometallics 2015, 34, 450–454. [Google Scholar] [CrossRef]
- McAnaw, A.; Scott, G.; Elrick, L.; Rosair, G.M.; Welch, A.J. The VCD method—A simple and reliable way to distinguish cage C and B atoms in (hetero)carborane structures determined crystallographically. Dalton Trans. 2013, 42, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Basato, M.; Biffis, A.; Buscemi, G.; Callegaro, E.; Polo, M.; Tubaro, C.; Venzo, A.; Vianini, C.; Graiff, C.; Tiripicchio, A.; Benetollo, F. Reaction of Cyclopentadienyl Ruthenium Complexes with a Carborane Anion: Effect of the Spectator Ligands on the Substitution Site. Organometallics 2007, 26, 4265–4270. [Google Scholar] [CrossRef]
- Note that in all cases in this review where we have analysed literature structures we have confirmed the cage C atom positions ex-post by the VCD method.
- Burke, A.; McIntosh, R.; Ellis, D.; Rosair, G.M.; Welch, A.J. 13-Vertex carbacobaltaboranes: Synthesis and molecular structures of the 4,1,6-, 4,1,8- and 4,1,12-isomers of Cp*CoC2B10H12. Collect. Czech. Chem. Commun. 2002, 67, 991–1006. [Google Scholar] [CrossRef]
- McAnaw, A.; Lopez, M.E.; Ellis, D.; Rosair, G.M.; Welch, A.J. Asymmetric 1,8/13,2,x-M2C2B10 14-vertex metallacarboranes by direct electrophilic insertion reactions; the VCD and BHD methods in critical analysis of cage C positions. Dalton Trans. 2014, 43, 5095–5105. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Allen, F.H. The Cambridge Structural Database in Retrospect and Prospect. Angew. Chem. Int. Ed. 2015, 53, 662–671, For this search we used ConQuest version 1.19, 2016. [Google Scholar] [CrossRef] [PubMed]
- Mingos, D.M.P.; Forsyth, M.I.; Welch, A.J. Molecular and Crystal Structure of 3,3-Bis(triethylphosphine)-1,2-dicarba-3-platinadodecaborane(11) and Molecular-orbital Analysis of the “Slip” Distortion in Carbametallaboranes. J. Chem. Soc. Dalton Trans. 1978, 1363–1374. [Google Scholar] [CrossRef]
- Garcia, M.P.; Green, M.; Stone, F.G.A.; Somerville, R.G.; Welch, A.J.; Briant, C.E.; Cox, D.N.; Mingos, D.M.P. Metallaborane Chemistry. Part 14. Icosahedral η6-Arene Carbametallaboranes of Iron and Ruthenium; Molecular Structures of closo-[1-(η6-C6H5Me)-2,4-Me2-1,2,4-FeC2B9H9] and closo-[3-(η6-C6H6)-3,1,2-RuC2B9H11]. J. Chem. Soc. Dalton Trans. 1985, 2343–2348. [Google Scholar] [CrossRef]
- Lee, S.S.; Knobler, C.B.; Hawthorne, M.F. Synthesis and structural characterization of mononuclear iron(II) ferracarboranes. Organometallics 1991, 10, 670–677. [Google Scholar] [CrossRef]
- Uhrhammer, R.; Crowther, D.J.; Olson, J.D.; Swenson, D.C.; Jordan, R.F. Synthesis and characterization of tantalum(V) dicarbollide complexes. Organometallics 1992, 11, 3098–3104. [Google Scholar] [CrossRef]
- Powley, S.L.; Rosair, G.M.; Welch, A.J. Further studies of the Enhanced Structural Carborane Effect: Tricarbonylruthenium and related derivatives of benzocarborane, dihydrobenzocarborane and biphenylcarborane. Dalton Trans. 2016, 45, 11742–11752. [Google Scholar] [CrossRef] [PubMed]
- Lamrani, M.; Gómez, S.; Viñas, C.; Teixidor, F.; Sillanpää, R.; Kivekäs, R. First examples of mixed (h5-pyrrolyl)(dicarbollide) sandwich cobalt-carboranes. New J. Chem. 1996, 20, 909–912. [Google Scholar]
- Loginov, D.A.; Starikova, Z.A.; Petrovskii, P.V.; Kudinov, A.R. Synthesis and structure of (boratabenzene)rhodacarborane (η-7,8-C2B9H11)Rh(η-C5H5BMe). Russ. Chem. Bull. Int. Ed. 2010, 59, 654–656. [Google Scholar] [CrossRef]
- Shirokii, V.L.; Knizhnikov, V.A.; Konovalov, T.P.; Zubreichuk, Z.P.; Erdman, A.A.; Nefedov, S.E.; Eremenko, I.L.; Yanovskii, A.I.; Struchkov, Y.T.; Mayer, N.A. Electrochemical synthesis of π-indenyl-π-(3)-1,2-dicarbollyliron(III). Russ. Chem. Bull. Int. Ed. 1993, 42, 732–733. [Google Scholar] [CrossRef]
- Lewis, Z.G.; Welch, A.J. Synthesis and Structure of 3-Fluorenyl-3-cobalta-1,2-dicarba-closo-dodecaborane(11), 3-(η5-C13H9)-3,1,2-closo-CoC2B9H11. Acta Cryst. 1992, C48, 53–57. [Google Scholar]
- Scott, G.; Ellis, D.; Rosair, G.M.; Welch, A.J. Icosahedral and supraicosahedral naphthalene ruthenacarboranes. J. Organomet. Chem. 2012, 721–722, 78–84. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Zink, J.I.; Skelton, J.M.; Bayer, M.J.; Liu, C.; Livshits, E.; Baer, R.; Neuhauser, D. Electrical or Photocontrol of the Rotary Motion of a Metallacarborane. Science 2004, 303, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Hendershot, S.L.; Jeffrey, J.C.; Jelliss, P.A.; Mullica, D.F.; Sappenfield, E.L.; Stone, F.G.A. Reaction of nido-7,8-C2B9H13 with Dicobalt Octacarbonyl: Crystal Structures of the Complexes [Co2(CO)2(η5-7,8-C2B9H11)2], [Co2(CO)(PMe2Ph)(η5-7,8-C2B9H11)2], and [CoCl(PMe2Ph)2(η5-7,8-C2B9H11)]. Inorg. Chem. 1996, 35, 6561–6570. [Google Scholar] [CrossRef] [PubMed]
- Tyurin, A.P.; Smol’yakov, A.F.; Dolgushin, F.M.; Godovikov, A.A.; Chizhevsky, I.T. Synthesis of 12-vertex mixed ligand closo-cobaltcarborane complexes and molecular structure of [3,3-(Ph2P(CH2)2PPh2)-3-Cl-closo-3,1,2-CoC2B9H11]. Russ. Chem. Bull. Int. Ed. 2013, 62, 1938–1940. [Google Scholar] [CrossRef]
- Ferguson, G.; McEneaney, P.A.; Spalding, T.R. 3-Chloro-3,3-bis(triphenylphosphine-P)1,2-dicarba-3-rhoda-closo-dodecaborane-Dichloromethane (1/1.1). Acta Cryst. 1996, C52, 2710–2713. [Google Scholar] [CrossRef]
- Chizhevsky, I.T.; Pisareva, I.V.; Vorontzov, E.V.; Bregadze, V.I.; Dolgushin, F.M.; Yanovsky, A.I.; Struchkov, Y.T.; Knobler, C.B.; Hawthorne, M.F. Synthesis, structure and reactivity of a novel monocarbon hydridorhodacarborane closo-2,2-(Ph3P)2-2-H-1-(Me3N)-2,1-RhCB10H10 molecular structure of 16-electron closo-2-(Ph3P)-2-Cl-1-(Me3N)-2,1-RhCB10H10 and closely related 18-electron closo-3,3-(Ph3P)2-3-Cl-3,1,2-RhC2B9H11. J. Organomet. Chem. 1997, 536–537, 223–231. [Google Scholar]
- Ferguson, G.; Pollock, J.; McEneaney, P.A.; O’Connell, D.P.; Spalding, T.R.; Gallagher, J.F.; Maciás, R.; Kennedy, J.D. An air-stable, cationic metallacarborane without a charge-compensated carborane ligand. Chem. Commun. 1996, 679–681. [Google Scholar] [CrossRef]
- Tyurin, A.P.; Dolgushin, F.M.; Smol’yakov, A.F.; Grishin, I.D.; D’yachihin, D.I.; Turmina, E.S.; Grishin, D.F.; Chizhevsky, I.T. Synthesis and characterization of mixed-ligand ferracarboranes. Direct metalation of the nido-carborane [nido-7,8-C2B9H12]− mono-anion with 14-e [Ph2P(CH2)nPPh2]FeCl2 (n = 2, 3). J. Organomet. Chem. 2013, 747, 148–154. [Google Scholar] [CrossRef]
- Chizhevsky, I.T.; Lobanova, I.A.; Petrovskii, P.V.; Bregadze, V.I.; Dolgushin, F.M.; Yanovsky, A.I.; Struchkov, Y.T.; Chistyakov, A.L.; Stankevich, I.V.; Knobler, C.B.; Hawthorne, M.F. Synthesis of Mixed-Metal (Ru–Rh) Bimetallacarboranes via exo-nido- and closo-Ruthenacarboranes. Molecular Structures of (η4-C8H12)Rh(μ-H)Ru(PPh3)2(η5-C2B9H11) and (CO)(PPh3)Rh(μ-H)Ru(PPh3)2(η5-C2B9H11) and Their Anionic closo-Ruthenacarborane Precursors. Organometallics 1999, 18, 726–735. [Google Scholar] [CrossRef]
- Cheredilin, D.N.; Kadyrov, R.; Dolgushin, F.M.; Balagurova, E.V.; Godovikov, I.A.; Solodovnikov, S.P.; Chizhevsky, I.T. Chiral paramagnetic closo-ruthenacarboranes via phosphine–diphosphine displacement reaction of “three-bridge” exo-nido-ruthenacarboranes: Molecular Structure of (−)-[closo-3-Cl-3,3-{(Ph2PCHCH3)2CH2}-3,1,2-RuC2B9H11] and its ortho-cycloboronated derivative. Inorg. Chem. Commun. 2005, 8, 614–618. [Google Scholar] [CrossRef]
- Cheredilin, D.N.; Dolgushin, F.M.; Grishin, I.D.; Kolyakina, E.V.; Nikiforov, A.S.; Solodovnikov, S.P.; Il’in, M.M.; Davankov, V.A.; Chizhevsky, I.T.; Grishin, D.F. Facile method for the synthesis of ruthenacarboranes, diamagnetic 3,3-[Ph2P(CH2)nPPh2]-3-H-3-Cl-closo-3,1,2-RuC2B9H11 (n = 3 or 4) and paramagnetic 3,3-[Ph2P(CH2)nPPh2]-3-Cl-closo-3,1,2-RuC2B9H11 (n = 2 or 3), as efficient initiators of controlled radical polymerization of vinyl monomers. Russ Chem. Bull. 2006, 55, 1163–1170. [Google Scholar]
- Grishin, I.D.; D’yachihin, D.I.; Piskunov, A.V.; Dolgushin, F.M.; Smol’yakov, A.F.; Il’in, M.M.; Davankov, V.A.; Chizhevsky, I.T.; Grishin, D.F. Carborane Complexes of Ruthenium(III): Studies on Thermal Reaction Chemistry and the Catalyst Design for Atom Transfer Radical Polymerization of Methyl Methacrylate. Inorg. Chem. 2011, 50, 7574–7585. [Google Scholar] [CrossRef] [PubMed]
- Grishin, I.D.; D’yachihin, D.I.; Turmina, E.S.; Dolgushin, F.M.; Smol’yakov, A.F.; Piskunov, A.V.; Chizhevsky, I.T.; Grishin, D.F. Mononuclear closo-ruthenacarborane complexes containing a rare eight-membered metal-diphosphine ring. J. Organomet. Chem. 2012, 721–722, 113–118. [Google Scholar] [CrossRef]
- McKay, D.; Welch, A.J. Unpublished results.
- Anderson, S.; Mullica, D.F.; Sappenfield, E.L.; Stone, F.G.A. Carborane Complexes of Ruthenium: A Convenient Synthesis of [Ru(CO)3(η5-7,8-C2B9H11)] and a Study of Reactions of This Complex. Organometallics 1995, 14, 3516–3526. [Google Scholar] [CrossRef]
- Jeffery, J.C.; Jelliss, P.A.; Psillakis, E.; Rudd, G.E.A.; Stone, F.G.A. Synthesis, crystal structure and some reactions of the ruthenacarborane complex [Ru(CO)2(MeC≡CPh)(η5-7,8-C2B9H11)]. J. Organomet. Chem. 1998, 562, 17–27. [Google Scholar] [CrossRef]
- Jelliss, P.A.; Mason, J.; Nazzoli, J.M.; Orlando, J.H.; Vinson, A.; Rath, N.P.; Shaw, M.J. Synthesis and Characterisation of Ruthenacarborane Complexes Incorporating Chelating N-Donor Ligands: Unexpected Luminescence from the Complex [3-CO-3,3-{κ2-Me2N(CH2)2NMe2}-closo-3,1,2-RuC2B9H11]. Inorg. Chem. 2006, 45, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.J.; Glenwright, S.J. Trans-effects in octahedral transition metal complexes. Coord. Chem. Rev. 2000, 203, 5–80. [Google Scholar] [CrossRef]
- See, R.F.; Kozina, D. Quantification of the trans influence in d8 square planar and d6 octahedral complexes: A database study. J. Coord. Chem. 2013, 66, 490–500. [Google Scholar] [CrossRef]
- Robertson, A.P.M.; Reckziegel, A.; Jones, J.J.; Rosair, G.M.; Welch, A.J. Balancing Steric and Electronic Effects in Carbonyl-Phosphine Molybdacarboranes. Eur. J. Inorg. Chem. 2017. [Google Scholar] [CrossRef]
- Vickers, P.W.; Pearson, J.M.; Ghaffar, T.; Adams, H.; Haynes, A. Kinetics and thermodynamics of C–Cl bond activation by [Ir(CO)2Cl2]−. J. Phys. Org. Chem. 2004, 17, 1007–1016. [Google Scholar] [CrossRef]
- Gunnoe, T.B.; White, P.S.; Templeton, J.L. Synthesis and Deprotonation/Alkylation Reactions of the Chiral Carben Complex Tp′(CO)(NO)Mo=C(OMe)(Me). Organometallics 1997, 16, 370–377. [Google Scholar] [CrossRef]
- Du, S.; Ellis, D.D.; Jelliss, P.A.; Kautz, J.A.; Malget, J.M.; Stone, F.G.A. Studies with the Ruthenacarborane Complex [Ru(CO)(PPh3)(THF)(η5-7,8-C2B9H11)]: Reactions with Terminal Alkynes. Organometallics 2000, 19, 1983–1992. [Google Scholar] [CrossRef]
- Lee, S.S.; Knobler, C.B.; Hawthorne, M.F. A Ferracarborane Analogue to [Fp]−. Synthesis and Reactions of [closo-3,3-(CO)2-3,1,2-FeC2B9H11]2−. Organometallics 1991, 10, 1054–1063. [Google Scholar] [CrossRef]
- Ellis, D.D.; Jelliss, P.A.; Stone, F.G.A. Rhenacarborane complexes with nitrosyl and alkylidene ligands. Structures of the complexes [[Re{=C(OMe)C6H4Me-4}(NO)(η5-7,8-C2B9H11)] and [Re(NO)(CNBut){η5,σ-7-C=N(H)But-7,8-C2B9H10}]. Chem. Commun. 1999, 2385–2386. [Google Scholar] [CrossRef]
- Demidowicz, Z.; Teller, R.G.; Hawthorne, M.F. Synthesis and Molecular Structure of [3-(PPh3)-3,3-(NO3)-3,1,2-RhC2B9H11], a Versatile Metallocarbaborane Reagent. J. Chem. Soc. Chem. Comm. 1979, 831–832. [Google Scholar] [CrossRef]
- Hansen, H.-P.; Müller, J.; Englert, U.; Paetzold, P. Azarhoda-closo-dodecaborane. Angew. Chem. Int. Ed. 1991, 30, 1377–1379. [Google Scholar] [CrossRef]
- Batten, S.A.; Jeffery, J.C.; Jones, P.L.; Mullica, D.F.; Rudd, M.D.; Sappenfield, E.L.; Stone, F.G.A.; Wolf, A. Synthesis of the Reagent [Na][Pt(PEt3)2(η5-7-CB10H11)] and Preparation of the Platinacarborane Complexes [PtX(PEt3)2(η5-7-CB10H11)] (X = H, Au(PPh3), Cu(PPh3), HgPh). Inorg. Chem. 1997, 36, 2570–2577. [Google Scholar] [CrossRef]
- Macías, R.; Thornton-Pett, M.; Holub, J.; Spalding, T.R.; Faridoon, Y.; Štíbr, B.; Kennedy, J.D. Polyhedral metallathiaborane chemistry: Synthesis and characterisation of metallathiaboranes based on the twelve-vertex icosahedral closo-{MSB10H10} unit, where M is Rh or Ir. J. Organomet. Chem. 2008, 693, 435–445. [Google Scholar] Note that the Rh–P distances given in Table 4 in this paper do not correspond to the atom labelling in Figure 2.
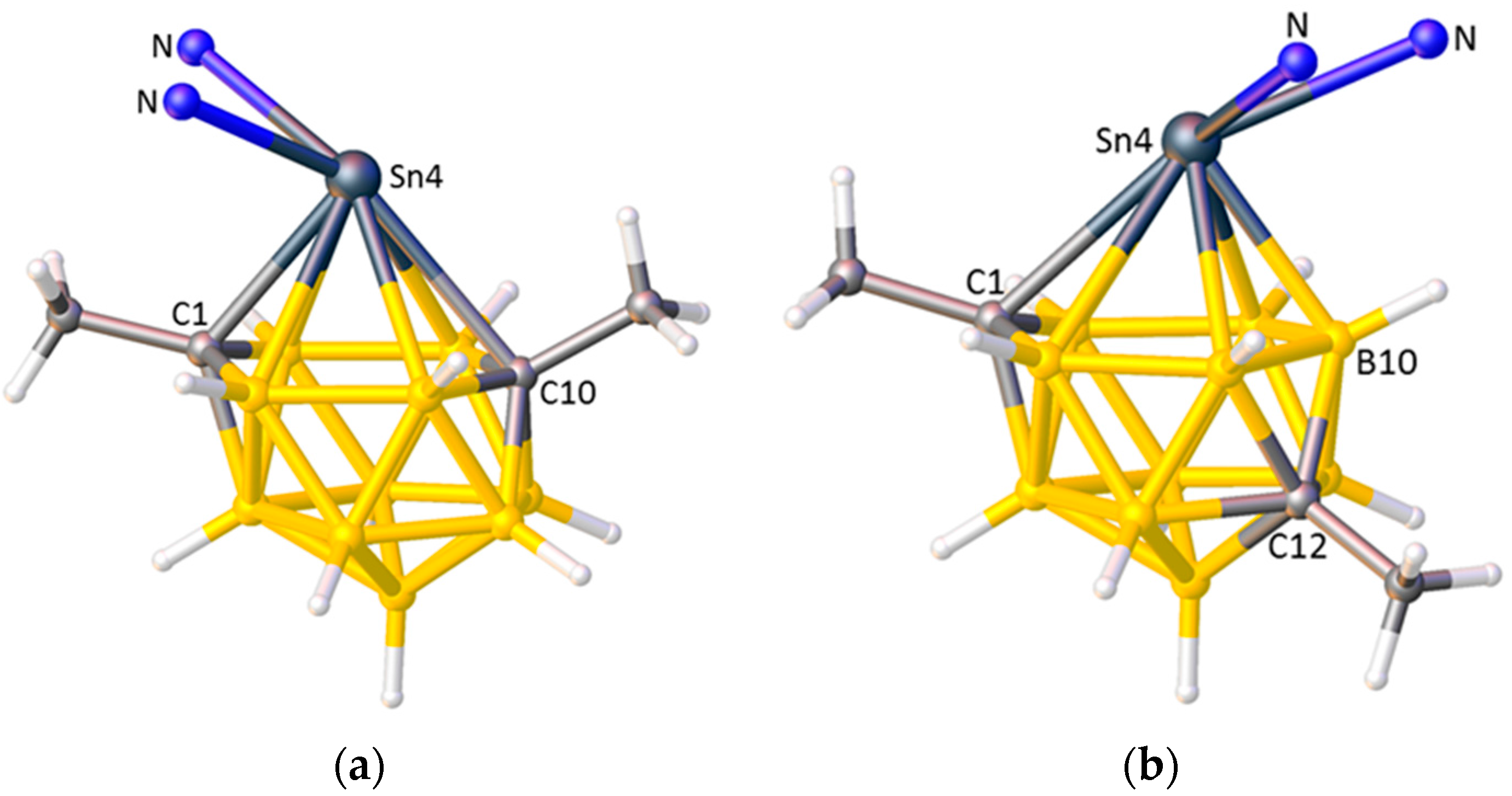
- Abram, P.D.; Ellis, D.; Rosair, G.M.; Welch, A.J. Exopolyhedral ligand flipping on isomerisation of novel supraicosahedral stannacarboranes. Chem. Commun. 2009, 5403–5405. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vertex | Ueq | Vertex | Ueq |
|---|---|---|---|
| 2 | 0.0089(9) | 8 | 0.0163(10) |
| 3 | 0.0209(12) | 9 | 0.0269(14) |
| 4 | 0.0311(16) | 10 | 0.0308(18) |
| 5 | 0.0308(15) | 11 | 0.0340(17) |
| 6 | 0.0237(13) | 12 | 0.0231(12) |
| 7 | 0.0178(11) | 14 | 0.0210(12) |
| Vertex | VCD | Vertex | VCD | Vertex | VCD |
|---|---|---|---|---|---|
| 1 | 1.546(5) | 5 | 1.668(6) | 9 | 1.672(7) |
| 2 | 1.696(7) | 6 | 1.668(7) | 10 | 1.680(9) |
| 3 | 1.698(7) | 7 | 1.546(4) | 11 | 1.690(6) |
| 4 | 1.694(6) | 8 | 1.679(7) | 12 | 1.656(6) |
| Vertex | VCD | Vertex | VCD | Vertex | VCD |
|---|---|---|---|---|---|
| 1 | 1.702(2) | 5 | 1.668(3) | 9 | 1.678(3) |
| 2 | 1.577(2) | 6 | 1.672(3) | 10 | 1.680(3) |
| 3 | 1.671(3) | 7 | 1.690(3) | 11 | 1.691(3) |
| 4 | 1.674(2) | 8 | 1.687(3) | 12 | 1.697(3) |
| Vertex | Vertex–H 1 | Vertex | Vertex–H 1 | ||
|---|---|---|---|---|---|
| 2 | 0.46(7) | 0.99(6) | 8 | 1.08(6) | 1.08(6) |
| 3 | 1.06(7) | 1.05(5) | 9 | 1.07(7) | 1.04(7) |
| 4 | 1.06(7) | 1.05(7) | 10 | 0.34(9) | 0.86(8) |
| 5 | 1.07(7) | 1.08(6) | 11 | 1.05(7) | 1.05(7) |
| 6 | 1.18(6) | 1.19(6) | 12 | 1.09(7) | 1.09(7) |
| 7 | 1.14(6) | 1.13(5) | 14 | 1.13(7) | 1.13(7) |
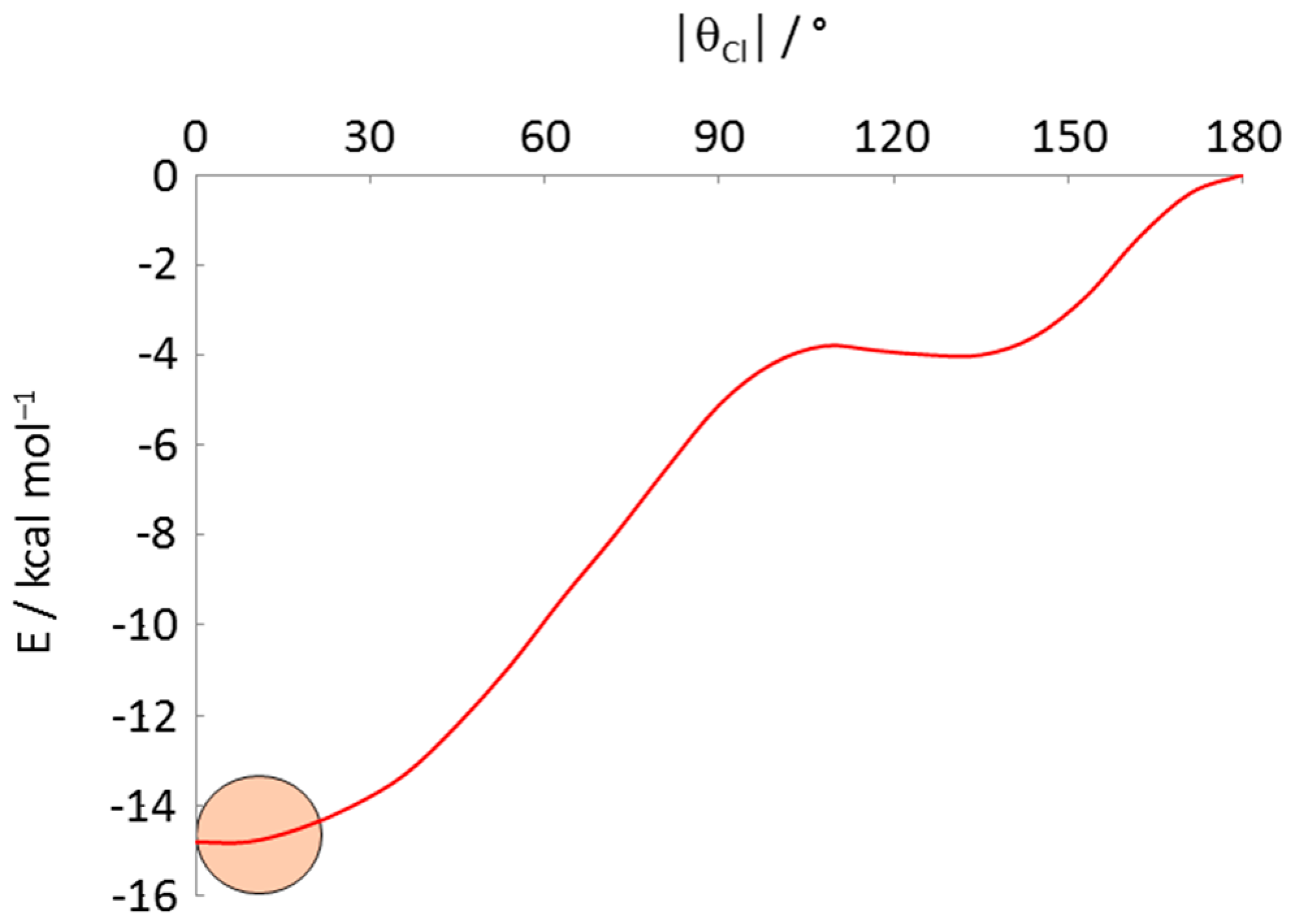
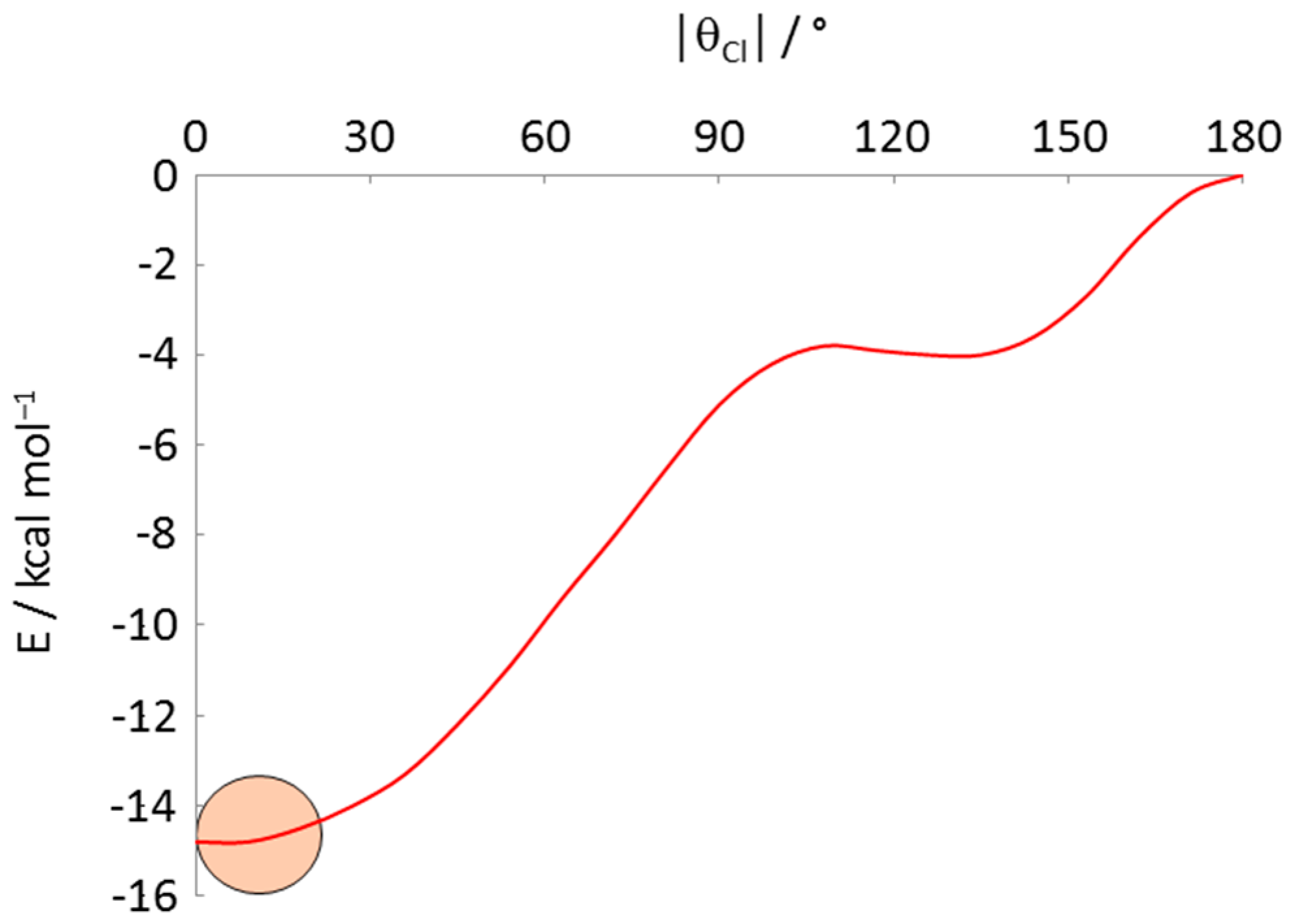
| Refcode | M | Phosphine | θCl | θP1 | θP2 | Ref. |
|---|---|---|---|---|---|---|
| TUBLUX | Co | PMe2Ph | −15.5 | 99.3 | −128.9 | [38] |
| HIPQII | Co | dppe/2 | 11.4 | −102.9 | 131.6 | [39] |
| TELCIW 1 | Rh | PPh3 | 12.6 | −98.3 | 127.6 | [40] |
| NITWOC 1 | Rh | PPh3 | −12.4 | 98.5 | −127.2 | [41] |
| ZOTVOT | Rh | PPh2Me | 9.1 | −107.5 | 127.9 | [42] |
| HIMRUS | Fe | dppe/2 | 9.4 | −113.8 | 131.6 | [43] |
| HIMROM | Fe | dppp3/2 2 | −10.7 | 102.3 | −134.1 | [43] |
| CEHCEX | Ru− | PPh3 | −12.9 | 99.5 | −127.2 | [44] |
| QAQQAB | Ru | 2,4-dppp5/2 3 | −15.8 | 99.6 | −137.5 | [45] |
| QEXWEW | Ru | dppe/2 | 15.6 | −114.4 | 132.9 | [46] |
| UZUYEU | Ru | dppb/2 | −14.6 | 101.0 | −138.5 | [47] |
| SEMZIV | Ru | 1,5-dppp5/2 4 | 18.7 | −95.2 | 143.2 | [48] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welch, A.J. What Can We Learn from the Crystal Structures of Metallacarboranes? Crystals 2017, 7, 234. https://doi.org/10.3390/cryst7080234
Welch AJ. What Can We Learn from the Crystal Structures of Metallacarboranes? Crystals. 2017; 7(8):234. https://doi.org/10.3390/cryst7080234
Chicago/Turabian StyleWelch, Alan J. 2017. "What Can We Learn from the Crystal Structures of Metallacarboranes?" Crystals 7, no. 8: 234. https://doi.org/10.3390/cryst7080234
APA StyleWelch, A. J. (2017). What Can We Learn from the Crystal Structures of Metallacarboranes? Crystals, 7(8), 234. https://doi.org/10.3390/cryst7080234