2.1. Defects, Bandgap, and Relaxation of Excited Charge Carriers in LNO
In this subsection we discuss some aspects of the material physics of LNO with big importance for the physics of polarons and polaronic luminescence. The first point are the defects and defect complexes. In LNO defects appear not only by doping but also by the slight deviations of the Li:Nb ratio from 1:1 that occur in almost all variants of LNO. The second point are the bandgap of LNO and the relaxation time of charge carriers that have been excited across this bandgap.
The defects occurring in undoped and Mg-doped LNO have a strong impact on the measured luminescence intensity, especially on the visible luminescence. At first we discuss the defects in undoped LNO. The widely used congruently-grown lithium niobate (CLN) has a
ratio of 48.45% [
1]. LNO prepared to have a
ratio closer to 50% is called stoichiometric lithium niobate (SLN). In general SLN is also only near-stoichiometric, the
ratio is not exactly at 50%. In the 70’s, the stoichiometry of such LNO samples has been still nearer to CLN than to perfect stoichiometric LNO, while in later times stoichiometry approached the perfect balance much better.
In all these samples without perfect stoichiometry (
< 50%) the lithium deficiency results in the presence of lithium vacancies
and niobium ions placed on lithium sites (so-called lithium anti-sites)
[
1]. The lithium deficiency is then related to the density of these so-called
intrinsic defects. Usually, these defects are aggregated in defect clusters where the effective charges of the constituting defects compensate each other. Several models of defect complexes formed by lithium or niobium vacancies and Nb anti-sites have been proposed [
1,
22,
29,
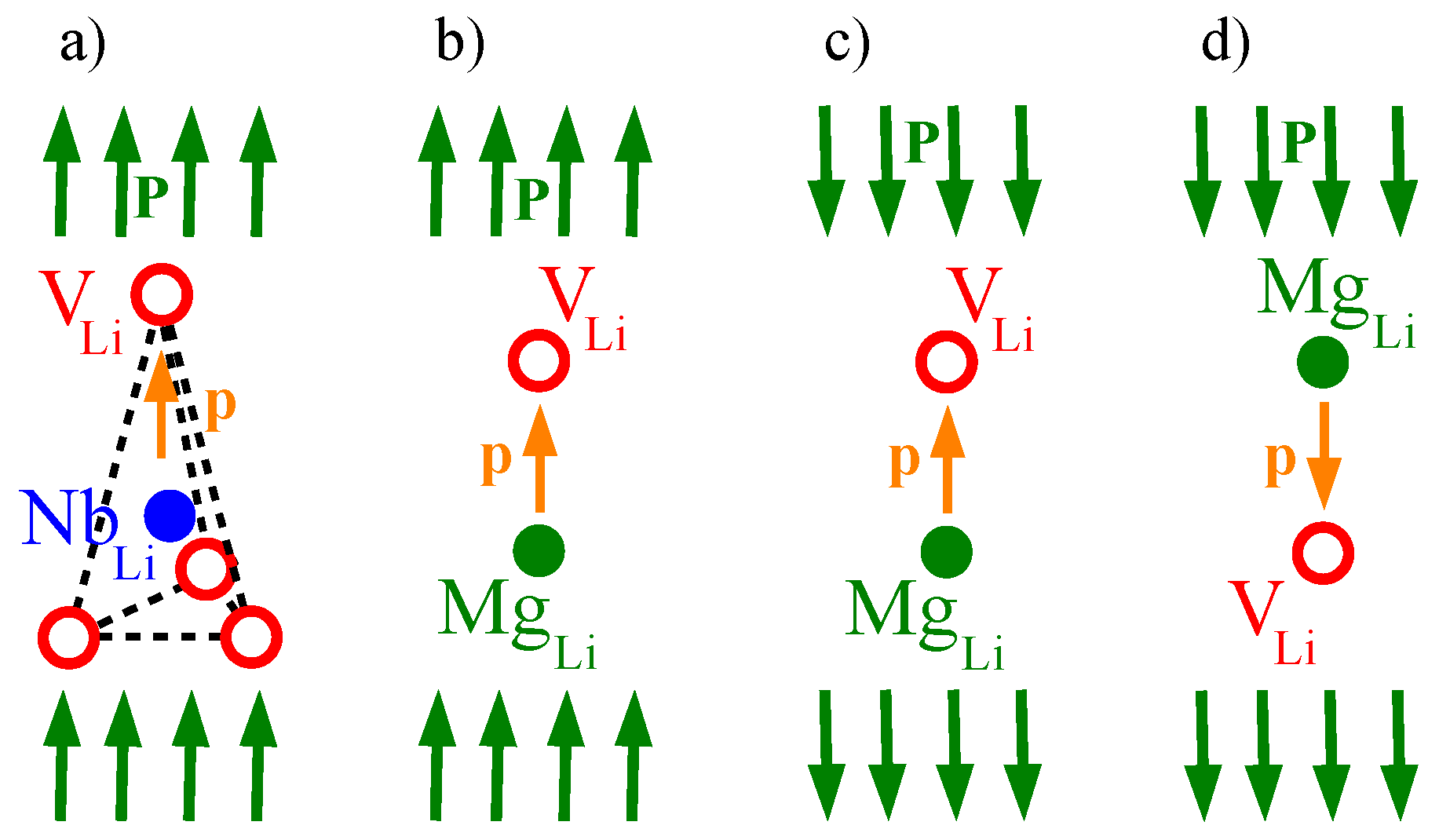
30]. The most important variant is the defect complex formed by a
anti-site, surrounded by four lithium vacancies
. In the ionic crystal the charges of the constituting ions (1 × Li
+, 1 × Nb
5+ and 3 × O
2−) compensate in each unit cell. Vacancies or defect ions cause deviations from these charges at the places of the crystal lattice, which we call here effective charges. A Nb
5+ ion on a Li site (hosting normally a Li
+ ion) yields a local charge excess of 4+:
, while a lithium vacancy (one positive charge lacking) has a negative effective charge:
. Therefore the defect cluster
+ 4
is charge-compensated. Another possible charge-compensated defect cluster model involves also niobium vacancies (with an effective charge of 5−): 5
+ 4
[
1,
2,
24,
30]. NMR measurements and subsequent simulations showed that the
+ 4
defect cluster is most probable in LNO [
28,
31,
32], although DFT energy calculations indicated that the 5
+ 4
complex is energetically similarly preferred, too [
30]. Probably both kinds of defect complexes occur in LNO with lithium deficiency. The Li:Nb ratio is determined by the density of these defect clusters.
In addition to these vacancies and anti-sites, there are also niobium interstitials in LNO. In the ideal lithium niobate structure, 1/3 of the oxygen octahedra are empty [
1]. In real LNO, however, niobium interstitials
occur that occupy these ideally empty octahedra [
33,
34]. Like niobium anti-sites, these niobium interstitials are also accompanied by lithium vacancies and form defect clusters [
34].
The presence of these
anti-sites can be strongly reduced by so called
damage-resistant doping [
1]. Doping with elements like Mg, Zn, Sc—that leave the material transparent—leads to a replacement of
anti-sites by dopant ions placed on lithium sites. An investigation with varying Mg doping showed that the Mg ions do not immediately occupy lithium sites when increasing the Mg concentration from zero. At lowest dopings Mg ions occupy at first oxygen octahedra that are regularly empty; with increasing doping they are also found on lithium sites [
35]. Around a certain threshold concentration of the dopant (
doping threshold), which depends on the valence of the dopant ions and the stoichiometry of the doped LNO, the
anti-sites have been completely replaced and the dopant ions start to replace also niobium ions
on niobium sites. It is probable that the niobium intersticials
are replaced by Mg ions as well. As the removal of
anti-sites changes the optical absorption properties, especially by blueshifting the UV absorption band, this doping results in a higher resistance against high laser light densities (optical damage resistance).
In the most investigations presented in this review the used LNO samples are doped with magnesium, a widely used standard dopant for LNO. In CLN, Mg has a doping threshold in the range of 4.5 to 5 mol %, and the
anti-sites vanish completely between 3 and 5 mol % [
1]. Due to the residual Li deficiency, also near-stoichiometric SLN has still a finite doping threshold of 1 mol % for Mg, as observed by Furukawa et al. [
36]. In the group of L. Kovács, however, SLN with an even better stoichiometry was demonstrated showing a doping threshold of only 0.2 mol % [
37].
Below the doping threshold, the
defects with single effective charges (Mg
2+ on a Li
+ site) are compensated by Li vacancies, resulting in
+
clusters. Above the threshold, the additionally occurring effectively negatively charged
can be compensated in an energetically favorable manner by already existing
in
+3
defect clusters [
38].
After reporting the defects occurring in LNO we take a view on the LNO bandgap, which is also of great importance for the discussions of the polarons, their excitations and transitions. The different methods for determining the bandgap, however, yield a broader range of results ranging from about 4 to 5.4 eV. With an optical bandgap of 4 eV or larger, LNO is exceptionally transparent.
An important method for determining the bandgap was the measurement and evaluation of spectral absorption curves. This method resulted in bandgap values in a range of about 4 to 4.3 eV [
39,
40,
41,
42]. The minor absorption in the tail below 4 eV has been assigned to indirect band transitions [
40].
The bandgap, as determined by this method, decreases weakly upon elevated temperatures by about
eV/K [
39]. The bandgap of SLN showed to be about 0.1 eV larger than that of CLN, as measured by Redfield et al. [
39]. As mentioned above, at the time of these investigations perfect stoichiometry was approached worse than in later times. In more recent measurements the bandgap of SLN samples has been determined to be by 0.2–0.3 eV larger than in CLN [
43].
Reflection measurements, on the other hand, yielded strong maxima of reflectivity at energies around 5 eV [
44,
45,
46], indicating the bandgap to be at 5 eV.
Theoretical calculations based on DFT and self-consistent self-energy calculations yielded values that rather fit to the results from the reflectivity than to the absorption measurements. Thierfelder et al. calculated a value of 4.7 eV [
47], while a more recent and developed calculation resulted even in 5.4 eV [
48]. These calculation models were also able to reproduce well the dielectric function of LNO [
49].
These straying and seemingly contradictory results show the difficulties to determine the true (direct) bandgap of LNO. One may doubt the interpretation of the absorption measurements, because interactions of positive and negative charge carriers may yield absorption processes below the (real) bandgap by the excitation of certain bound states. On the other hand the calculations may be doubted because they have not been performed over more than a few unit cells, thus not considering the disorder of the real crystal. These open questions show that the exact value of the (direct) bandgap of LNO is still controversial. In the sketch in
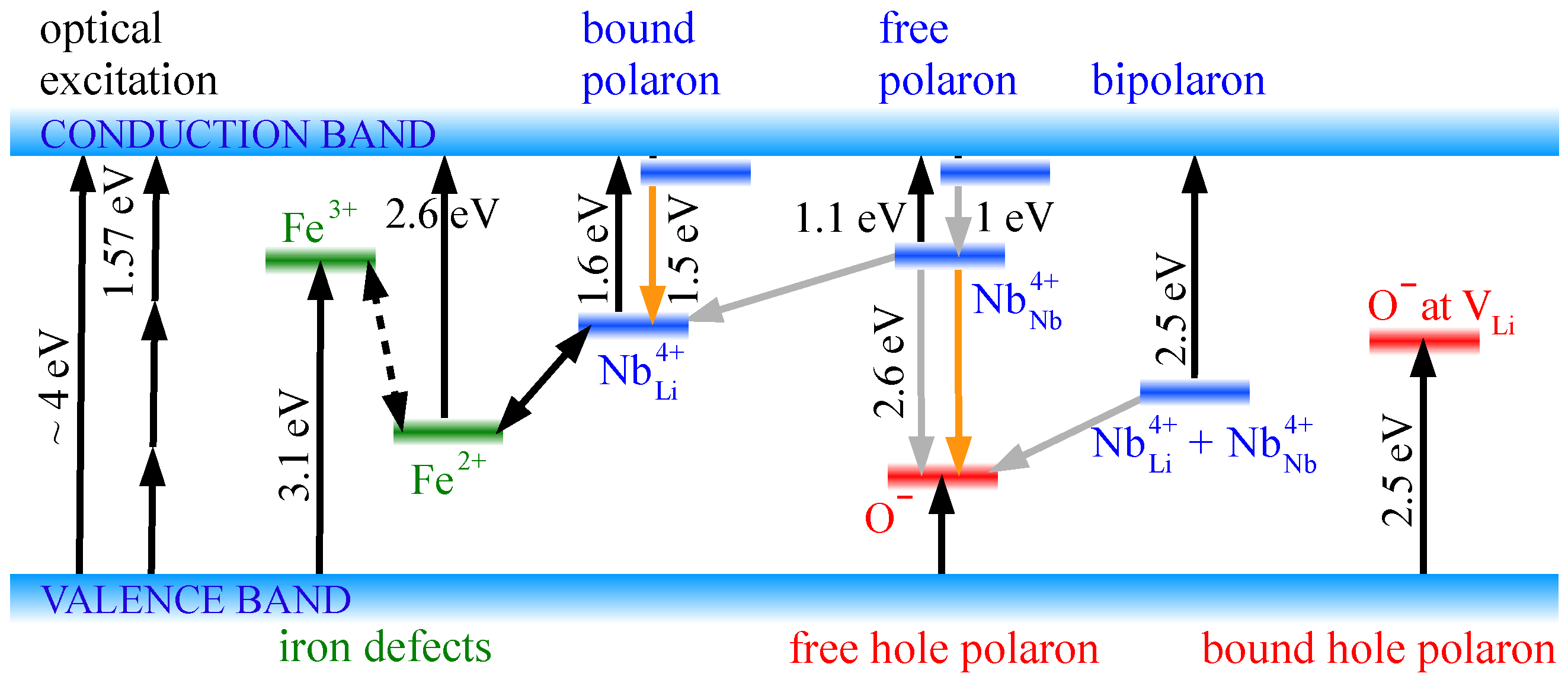
Figure 1 as well as in some discussions in this review we will refer to the widely used bandgap value of 4 eV. However, we put this value and all statements based on it under the reservation that the question of the exact bandgap value is not yet finally resolved.
After photoexcitation, electrons and holes relax into polaron states. For the discussion of polaron luminescence it is very important to consider the time scale of this relaxation as well. Pump-probe spectroscopy measurements have shown that photo-excited charge carriers relax into their respective polaron states within hundreds of femtoseconds [
50,
51,
52]. Recombination of polarons, on the other hand, occurs at time scales of 10
−7 s and longer [
15,
19]. Hence, polaron recombination and polaron formation occur at very different time scales. Thus, for the dynamics of polaronic recombination this short relaxation time can be neglected, as well as transport effects by the motion of the photo-excited charge carriers in the conduction band.
To resume the discussion of the bandgap issue, the charge carriers were excited by 4.8 eV into the conduction band via three-photon excitation [
50,
51], showing that the bandgap lies probably below 4.8 eV. In our luminescence experiments, the charge carriers were excited by about 4.7 eV—by UV as well as by multiphoton excitation. In that case the cited results on charge carrier relaxation can be well premised for our luminescence measurements.
2.2. Absorption in LNO by Polarons
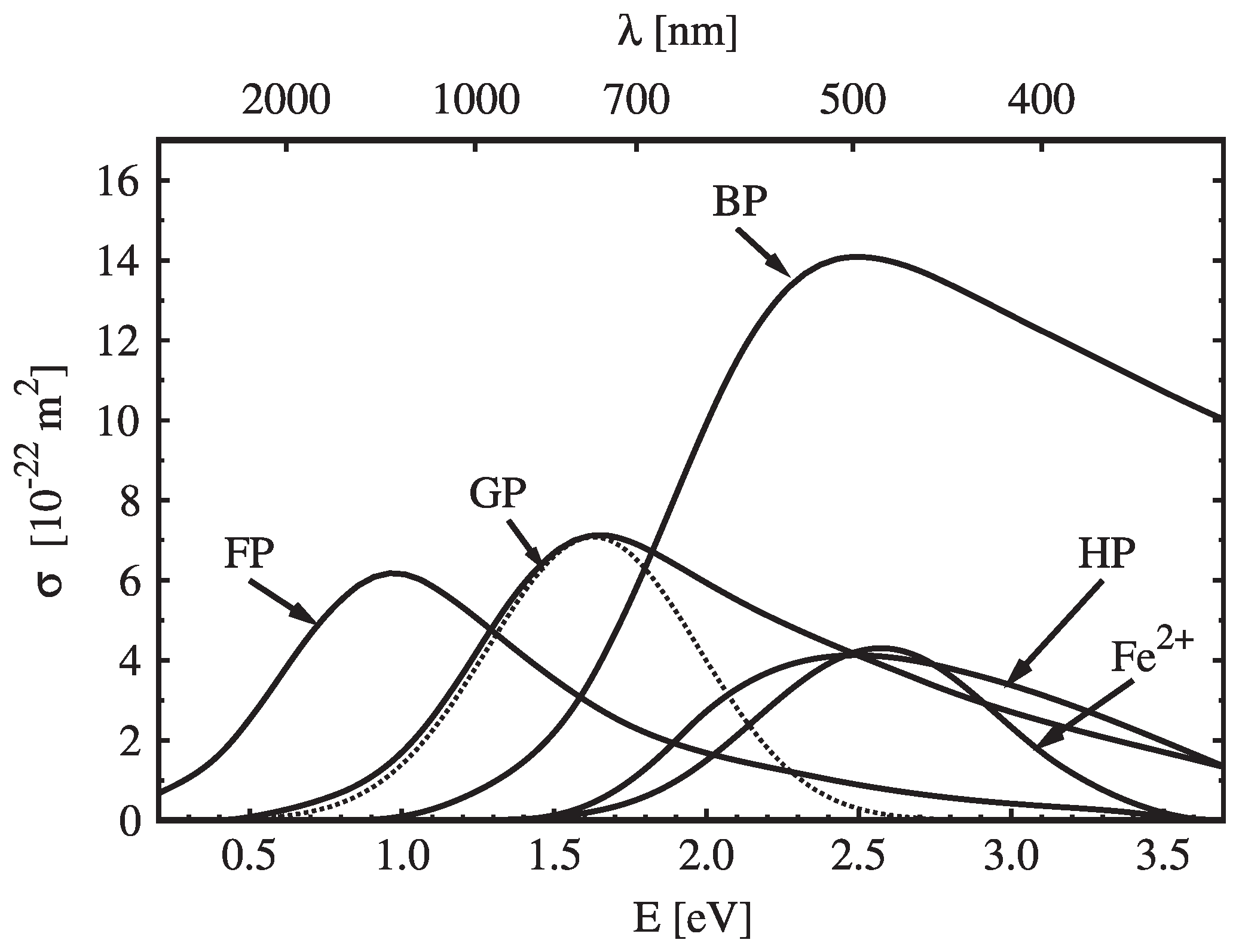
The first way to identify and characterize polaron states and their energies were measurements of their absorption bands after filling these polaron states with charge carriers by reduction. These absorption measurements allowed investigations of the influence of physical parameters on the polarons, thus clarifying their energy levels and physical nature. Doping these samples with Mg or Fe allowed further insights into the physics of these polaron states.
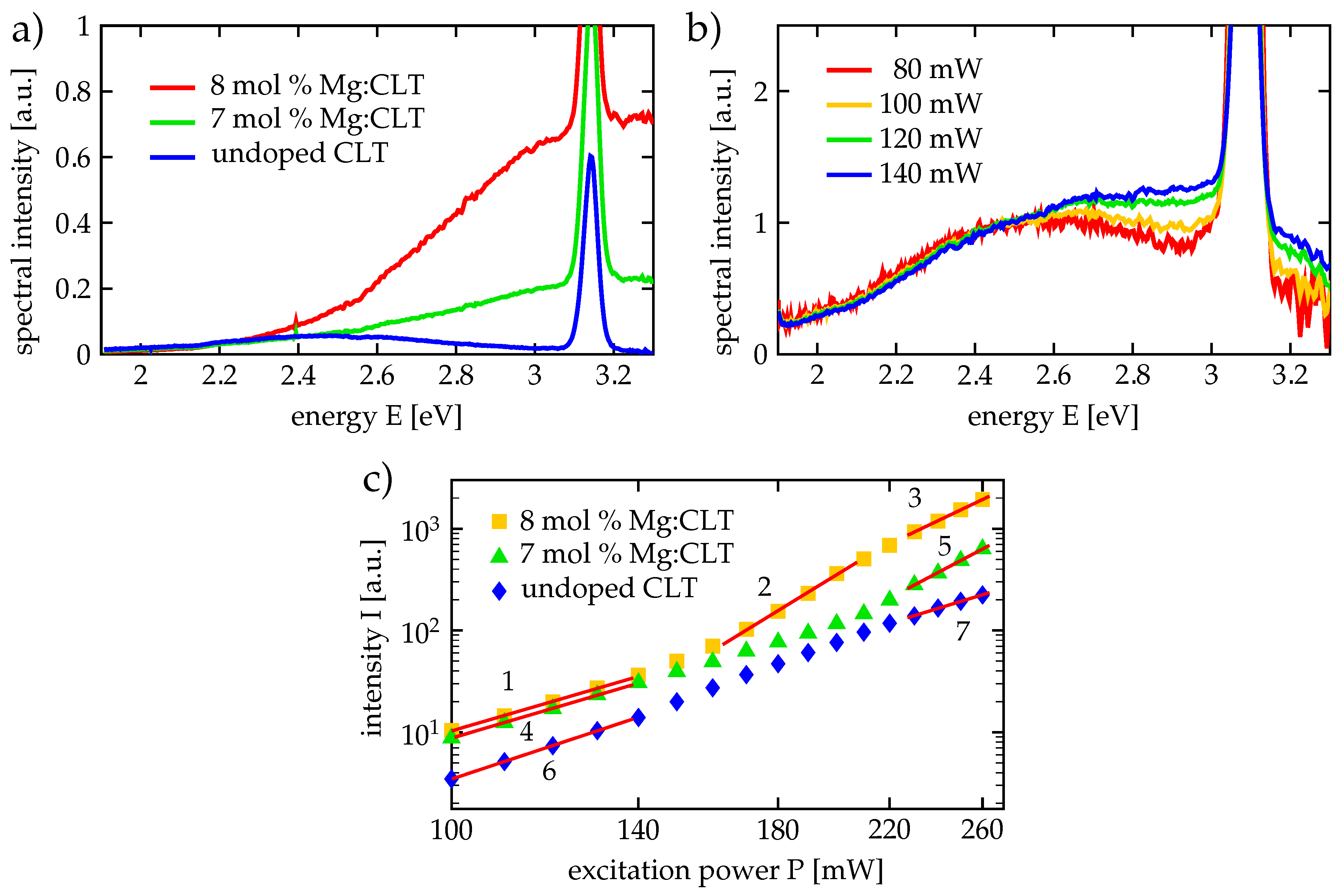
First light-induced absorption spectroscopy experiments have been carried out by Koppitz et al. [
16,
53] at congruent lithium niobate (CLN) that had been reduced for 1 h at 800 °C in vacuum. These measurements have also been extended to Mg-doped CLN. The absorption spectra of this material revealed several broad absorption bands with characteristic peak energies that could be attributed to the optical bands of electron polarons:
Peak around 1.1 eV (about 1130 nm), transition from free polaron to conduction band;
Peak around 1.6 eV (about 770 nm), transition from bound polaron to conduction band;
Peak around 2.5 eV (about 500 nm), transition from bipolaron to conduction band;
Peak around 0.6 eV, transition from polarons at Mg
Nb defects to conduction band, occurring only in CLN with more than 5 mol % Mg doping, not depicted in
Figure 1.
The absorption curves (1) to (3) are drawn in
Figure 2. The assignments of these spectral features to the according polaron types were supported by the dependence of these absorption peaks on temperature and doping with Mg. With Mg doping replacing the
defects by
defects [
1], both the bound polaron and the bipolaron peaks were reduced [
16], indicating a strong relation of these peaks to charge carriers bound to
defects. After excluding other possibilities, the thermal decay of the 2.5 eV peak (3) with an activation energy of 0.27 eV could be explained by the presence of aggregates of free and bound polarons (bipolarons) that decay thermally into single polarons [
53]. The 1.6 eV peak (2), on the other hand, has been assigned to single bound polarons [
53]. The dissociation of bipolarons has also been observed under illumination with green or blue light, leading to enhanced transparency in the blue wavelength range and increased absorption in the red and near-infrared wavelength range by the shift of the absorption spectrum from the bipolaron peak (3) to the bound polaron peak (2) [
54,
55,
56]. The attribution of the 1.6 eV absorption band to the
state has been finally confirmed by measurements of magnetic circular dichroism of this absorption center [
57].
The 1.1 eV peak (1) on the other hand remains stable for high Mg doping and under annealing, thus being independent from
and other defects. Therefore that peak has been assigned to the free polarons on
places. The 0.6 peak (4), not shown in
Figure 2, appears for Mg dopings above the doping threshold of 5 mol %, where Mg starts also to replace Nb ions from Nb places [
1]. This peak probably stems from polarons slightly trapped at the occurring
defects [
16]. The formation of this peak showed an activation energy of 40 meV [
16].
For reduced or n-doped (e.g., Fe or Mg) LNO electric transport can be assumed to be dominated by hopping of free polarons [
62]. Even for nominally undoped LNO the same can be assumed due to small amounts of Fe ions generally occurring there. In these cases investigations of the electrical transport in LNO and comparison with the theoretical polaron model have provided further support for the energy level of free electron polarons. According to the polaron model [
4,
58,
63] the electron and the surrounding lattice distortion have a total energy
, while the electron itself has an energy
, which is relevant for optical absorption. Then the activation energy
for hopping to neighboring
sites is
. Reported activation energies for electrical transport in reduced or doped LNO are between 0.1 and 0.24 eV [
58,
64,
65], which is in a fairly good agreement with the absorption band around 1 eV for free polarons, therefore resulting in a theoretical hopping energy of about 0.25 eV [
4]. However, the relation
is only an approximation and, moreover,
is often smaller than
[
66], which meets the range of measured values even better. Note that the polaronic model itself is not valid at arbitrarily low temperatures. There are models that generally predict a metal-insulator transition for polarons at low temperatures, resulting in an itinerant polaron band, while inelastic processes at higher temperatures yield the known hopping behavior [
66,
67,
68,
69].
While these findings cover all electron polaron states, the knowledge of hole polarons and their transitions is much more fragmentary, as only the absorption band of the bound hole polaron is known and almost nothing about the free hole polaron. Under two-photon-photoexcitation of undoped CLN with 532 nm picosecond pulses at a temperature of 20 K, an absorption band around 2.5 eV (500 nm, see
Figure 2) has been measured by Schirmer et al. [
8,
9]. Comparison with broad ESR spectra at
ions (and the knowledge of similarly broad hole absorption spectra in other materials) resulted in the assignment of this peak to bound hole polarons that are probably bound to lithium vacancies [
2,
9,
11,
70]. Compared to electron polarons the absorption energy of 2.5 eV is rather high, however, this seems to be typical for O
− hole polarons in oxides [
70]. At room temperature or under additional HeNe laser light illumination this peak is still visible, but narrower and considerably weaker [
8,
9]. This influence of HeNe laser light may indicate a recombination of the bound hole polarons with electron polarons or conversions of the bound hole polarons into free hole polarons. More recent pump-probe investigations on CLN by Herth et al. [
12] measured decay times of about milliseconds for that absorption band, at room temperature and also under picosecond excitation at 532 nm. As no free polaron band was observed under these excitation conditions, the formation of bipolarons as a cause of this band has been excluded [
12,
71] and the assignment to generated bound holes has been confirmed [
12]. However, almost no data is known about the energy levels and physical properties of free hole polarons.
Ionic iron defects and their energy levels are very important for the infrared polaronic luminescence to be reviewed below. For that reason it is necessary to discuss here some additional findings on absorption measurements on iron doped CLN as well as on transitions between Fe defect ion states and polaron states. Fe defect ions are known to almost exclusively occupy Li sites [
59,
72,
73]. Note that small iron impurities are generally present in every LNO sample. Higher Fe doping, however, will strongly increase all optical effects caused by these ions.
In Fe-doped CLN an absorption band around 2.6 eV has been found (see
Figure 2) [
59,
74] that has been attributed to the
+
excitation process [
74]. Investigations of Kurz et al. included the reduction of Fe-doped CLN samples by annealing and monitoring their
and
concentrations using Mössbauer and EPR measurements. These experiments resulted in a strong linear correlation between
concentration and absorption at the 2.6 eV band, thus confirming the attribution of this band to the
+
excitation process [
59]. Another charge-transfer
+
+ Fe
2+ has been mentioned by Schirmer et al. and attributed to an absorption band around 3.1 eV [
2].
Furthermore, the generation of bound polarons by photo-excitation of electrons from Fe
2+ ions has been demonstrated by exciting Fe-doped LNO crystals with nanosecond laser light pulses at 532 nm. This excitation resulted in an increased absorption in the red wavelength range [
71,
75]. These experiments have been carried out with different concentrations of Fe
2+ and
, using CLN as well as SLN and further variations of stoichiometry. The observed absorption increased with Fe
2+ concentration, but was weaker in samples with lower
concentration. Moreover, a decrease in the concentration of Fe
2+ ions and of
increased the transition time of the growing absorption [
75,
76]. This supports that the pump light excites a charge transfer at Fe
2+, creating a polaron, leaving a Fe
3+ ion behind and yielding an increase in absorption in the red and near-infrared wavelength range. The rate of this reaction depends then on both the Fe
2+ and
anti-site concentrations. The relation of this phenomenon with the absorption peak in the near-infrared and the concentration of
anti-sites indicates that the involved polaron is indeed the bound polaron. This has been confirmed by the time-dependent investigations on the absorption at 2.5 eV by Herth et al. [
12]. Pump-probe absorption measurements of 0.1 mol % Fe:CLN under nanosecond-pulsed illumination at 532 nm showed an increase of the Fe
2+ absorption band within the first 10
−4 s after the exciting pulse. This increase showed a correlation with the decay of the bound polaron band at the same time. Thus, the increase of the Fe
2+ concentration could be directly correlated with the decrease of bound polaron concentration, as the bound polarons (created through the exciting pulse) recombine again with the Fe
3+ ions to Fe
2+ ions [
12]. A direct transition from Fe
2+ into free electron polaron states by optical absorption, however, has not been reported—probably because such a transition is forbidden.
After pumping, the relaxation of the bound polaron absorption in the red wavelength followed a stretched-exponential law. The relaxation of these bound polaron states back to Fe
2+ states means a return of the negative charge carriers from the hosting
sites to the
ions, converting them back to the
state. As the nearest neighbor distances of
and iron sites are randomly distributed, the decay of excited bound polarons follows a superposition of exponential decays with different time constants, resulting in a stretched-exponential behavior with decay times in the microsecond range [
77]. Like the transition rate mentioned above, this decay process slowed down at smaller densities of
anti-sites due to the resulting larger average distance to iron defects [
75,
76]. A deeper analysis of the physics of stretched-exponential luminescence has already been given for the example of the luminescence of Si nanocrystals [
78]. As will be discussed below, Herth et al. [
71] confirmed this behavior for the bound polarons in CLN. Furthermore, they found also reduced polaron lifetimes after introducing co-dopants such as Cu, providing more decay channels. An Arrhenius-like behavior of the polaron decay with an activation energy of 0.38 eV was observed, too [
71]. If we would use the polaron hopping model for the escape of the charge carrier from the
state back to the iron ion, this activation energy would well agree with the optical absorption energy of 1.6 eV.
2.3. Near-Infrared Photoluminescence Band in LNO
Up to now we have discussed the different polaron states occurring in LNO and their physical behavior. This is a necessary basis to understand the photoluminescence phenomena that are mediated by these polaron states. In this section, we will discuss the near-infrared luminescence observed around 1.3–1.5 eV. It has been shown that this band stems from the relaxation of excited bound polarons and that its intensity depends also strongly on the presence of ionic iron defects. The latter is the case due to the role of iron ions for the generation of bound polarons by photoexcitation.
First measurements of a luminescence band around 1.5 eV have been carried out in slightly Fe-doped CLN (0.015 mol % Fe) upon UV laser light illumination at 325 nm [
79]. In untreated Fe-doped samples, the luminescence is weak, while it is strongly enhanced after reduction (by annealing for 20 h at 520 °C in LiCO
3). Due to the donated electrons, the charge transfer
resulting from the reduction process [
80,
81] indicates a strong relation between that luminescence and the presence of polarons.
Further investigations [
13,
82] on this luminescence band of undoped LNO yielded deeper insights into the physical origin of this luminescence—especially confirming its mediation by bound polarons. Exciting undoped LNO with ns pulses at either 355 nm [
13] or 514 nm [
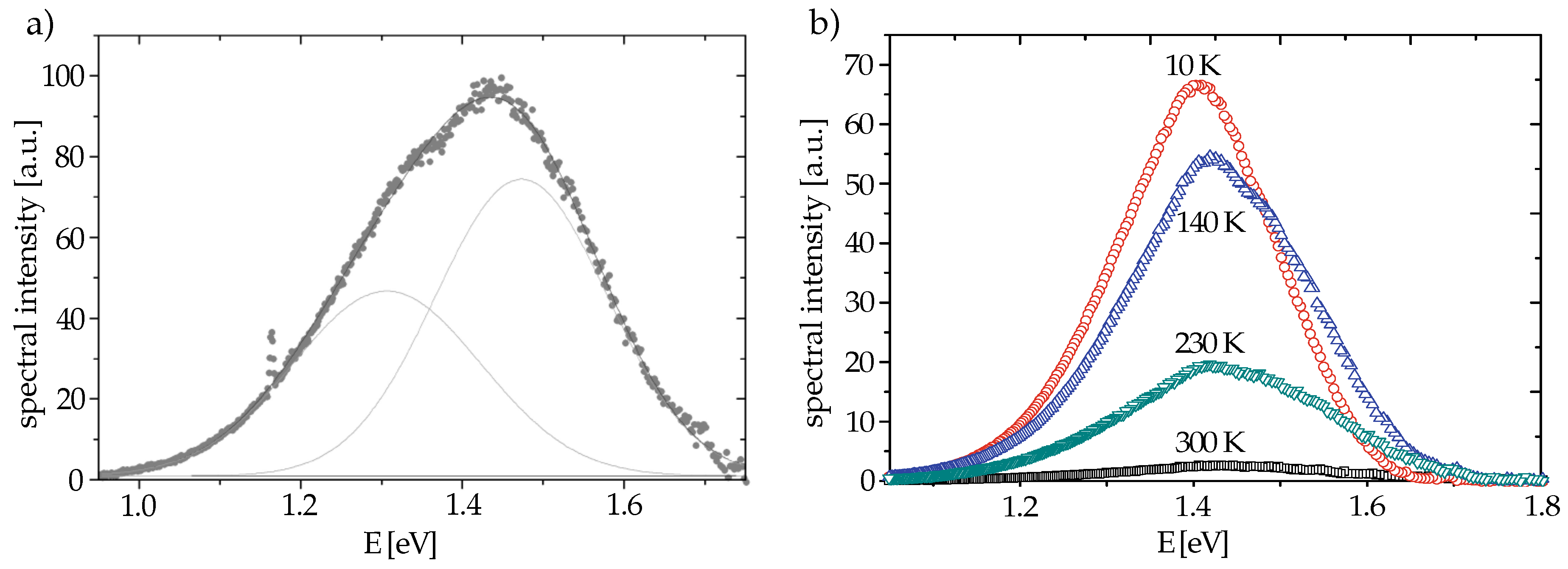
82] resulted in two infrared luminescence peaks at 1.5 eV (835 nm) and 1.3 eV (950 nm), as shown in
Figure 3a [
13,
83].
These two peaks are no fitting artifacts, as the following results show. Kostritskii et al. showed that the intensity of the 1.5 eV peak depends strongly on the intrinsic defect concentration, which has been revealed by comparison of congruent lithium niobate (CLN) and stoichiometric lithium niobate (SLN) [
82,
84]. In CLN the 1.5 eV and the 1.3 eV peaks amounted to the same order of magnitude; in SLN, however, the 1.5 eV peak was significantly weaker while the 1.3 eV peak did not change drastically. Assuming a connection of the 1.5 eV luminescence peak to the bound polaron state, this behavior fits again well with the reduced presence of lithium vacancies and therefore also of
defects that can provide bound polarons in SLN. This connection to the bound polaron state is also confirmed by a strong increase of the 1.5 eV peak in CLN upon prolonged reduction [
82,
84].
The 1.3 eV peak, however, vanishes under that reduction. This may indicate a relation of that peak to hole polarons, as the reduction floods the substrate with negative charge carriers not only appearing as electron polarons but also filling the remaining holes. Maybe it is even a counterpart of the bound polaron luminescence for free or bound holes—a transition from free hole states or from excited bound hole states back to the bound hole ground state. Unfortunately, no further investigations have been reported for that peak so far. Therefore no data are available for the determination of the deeper physical nature of that peak—probably because of problems to separate both peaks spectrally. However, we must reject the claim of Kostritskii et al. [
82,
84] to assign the 1.3 eV peak to the recombination of free electron polarons with hole polarons. The recombination of free electron polarons with free hole polarons has already been assigned to the luminescence in the visible wavelength range to be discussed below. Moreover, the time dependence of such polaronic recombinations would result in a stretched-exponential behavior [
19,
76], which has not been observed in the time-dependent measurements of the NIR luminescence band to be discussed below.
Further measurements of temporal, thermal, and doping behavior confirmed and clarified the role of the bound polaron state in the 1.5 eV luminescence peak. Unfortunately, in all these measurements to be discussed in the following, no differentiation has been made between the 1.5 and 1.3 eV peaks, possibly due to difficulties to separate these two overlapping peaks. Conclusions have therefore been drawn only for the 1.5 eV peak, and not for the 1.3 eV peak.
Measurements of the 1.5 eV photoluminescence in Fe-doped CLN showed a clear increase of intensity with the doping level, while lifetime and temperature dependence were not influenced [
14]. This supports the role of Fe not as a luminescence center itself but as a donor providing bound electron polarons as luminescence centers. The luminescence of the 1.5 eV peak has been interpreted as the result of a two-step excitation process: A charge transfer
+
+
is followed by a transition into an excited state (
)* slightly below the conduction band edge (
Figure 1). The luminescence energy of 1.5 eV agrees well with the bound polaron energy of 1.6 eV, when (
)* is identified with a shallow trap around 0.1 eV below the conduction band edge. The mono-exponential behavior of the luminescence decay (with a unique decay time) supports the interpretation that the bound polaron and excited state (
)* are on the same site, since a random spatial separation would result in a stretched exponential decay as a superposition of exponentials with different decay times [
19,
76,
77].
Moreover, strongly Fe-doped CLN (1 mol % Fe) showed a quadratic increase of the the luminescence intensity with the illumination intensity, according to a two-step excitation [
14]. For lower doping concentrations than 0.05 mol % Fe, the dependence of the luminescence on the illumination intensity reached a linear regime, because the smaller number of Fe ions was already completely excited at the used illumination intensities (saturation regime). In this regime, the found direct proportionality between the luminescence intensity and doping concentration supports the model of polaron excitation by charge transfer. The observation of luminescence in nominally undoped LNO may be related to the Fe impurities that are generally present in this material.
In addition to these doping dependencies, Harhira et al. found also a strong temperature dependence of the luminescence intensity, which is typical for polaron-mediated luminescence, as shown in
Figure 3b. Due to non-radiative transitions the 1.5 eV luminescence peak decreases strongly with temperature and is at room temperature about 2 orders of magnitude weaker than at 10 K. This dependence on temperature yields an activation energy of 0.22 eV. However, the radiative decay time of 9 µs was observed to be independent on temperature [
13]. As the extracted activation energy is near the free polaron hopping energy, the authors interpreted the results in the following way: the temperature dependence is caused by a concurring non-radiative decay being mediated by hopping from the excited state (
)* to the free polaron and further down to the bound polaron (
Figure 1) [
14]. Note that no analogous luminescence had been observed for free polarons, which supports that the relaxation via the free polaron state is indeed completely non-radiative.
This interpretation, however, is still contradictory. When a certain number of bound polarons is excited and an additional temperature-dependent nonradiative decay would occur, the total measured luminescence lifetime would also be influenced by that nonradiative channel and could not be temperature-independent. We suggest here an alternative interpretation: Between the excitation of the charge carrier from the Fe2+ ion into the bound polaron state and the excitation of that polaron, the possibility exists that the charge carrier tunnels to a neighbored site. The probability not to return but to escape as a free polaron is determined by the hopping probability being governed by the hopping activation energy. As a result, the number of bound polarons being excited to the ()* state is drained by that escape process, yielding the polaron hopping activation energy for the temperature dependence of the luminescence. The luminescence process itself, however, would not be accompanied by a nonradiative decay modifying the luminescence lifetime.
2.4. Visible Luminescence Band in LNO
The second important polaron-mediated photoluminescence band found in LNO lies in the visible wavelength range. The luminescence maximum was observed around 2.6 eV (ca. 470 nm), as shown in
Figure 4a. The luminescence showed a strong dependence on Mg doping and has been associated with the recombination of free electron with free hole polarons [
15,
16,
17,
18,
19]. This interpretation has been supported by a stretched-exponential decay of this luminescence with lifetimes in the range of µs [
19,
71]. Similar to the transfer of bound polarons into iron ions described above, this recombination involves randomly distributed recombination partners being separated by random distances—resulting in a stretched-exponential instead of a mono-exponential decay [
19,
71,
76,
78].
As there is little data about the properties of free hole polarons, the measured luminescence energy can, in principle, be used to determine the hole polaron energy level. Assuming a LNO bandgap of 4–4.3 eV following to the absorption measurements [
39,
40,
41], a free polaron energy of 1.1 eV and the 2.6 eV luminescence energy leave a free hole polaron absorption energy of about 0.3–0.6 eV (
Figure 1). The calculated bandgap values of 4.7–5.4 eV [
47,
48], on the other hand (supported also by the reflection measurements in Refs. [
44,
45]), result in higher free hole polaron energies of 1.0–1.7 eV, comparable with the free electron polaron and bound hole polaron energies. A recombination of free polarons with bound holes, however, can be clearly excluded as a source of visible luminescence, as the bound hole energy of 2.5 eV would leave only a luminescence energy between 0.4 and 1.8 eV. Even the largest calculated bandgap of 5.4 eV [
48] is not large enough to yield the peak energy of 2.6 eV.
As mentioned in
Section 2.2, a 2.5 eV absorption band (observed at 20 K under 532 nm picosecond pump pulse excitation) has been assigned to the bound hole polarons and vanishes under heating up to room temperature or under additional HeNe laser illumination [
8,
9]. An explanation for this disappearance may be an excitation of the bound hole polarons releasing them as free hole polarons from the defect trap.
The first measurements of this luminescence have been made by Koppitz et al. [
16] and Klose et al. [
15] and revealed a strong dependence on Mg doping, but also on stoichiometry and temperature. These measurements have been carried out for different stoichiometries and Mg dopings as well as in a temperature range from room temperature down to 77 K. The samples have been excited with X-rays [
16] or with a a Xenon lamp as well as with an excimer laser (249 nm, 20 ns) for lifetime measurements [
15], respectively.
The following behavior of the luminescence in Mg-doped CLN has been measured:
At 77 K a strong increase of the luminescence intensity as a function of the doping concentration has been found. The strongest increase by almost 2 orders of magnitude was between 2 and 4 mol % Mg, accompanied by a weaker increase between 4 and 9 mol % [
15].
Also measured at 77 K: the luminescence intensity of 6 mol % Mg:LNO for a range of different
ratios, showing an increase of 2–3 orders of magnitude between 42.5% (strong Li deficiency) and CLN stoichiometry [
15].
The luminescence is also strongly temperature-dependent [
16]. The ratio of luminescence intensities at 80 K and at 295 K reaches values of almost 200 for Mg:CLN with dopings of 4 mol % and higher. This means, at these dopings the luminescence increases by a factor of about 200 when cooling down to 77 K. At low dopings of 2 mol % and below, this ratio is quite lower. This low value has not been exactly displayed in the concerning diagram, but seems to be in the order of 10. The increase of this ratio between 2 and 5 mol % is clearly related to the doping threshold and the replacement of
anti-sites.
The temporal behavior of the luminescence showed also to be strongly influenced by temperature and doping. Using a pulsed excimer laser ( = 249 nm) with 20 ns pulses, the lifetimes of the luminescence were measured for different temperatures between 20 and 300 K and dopings up to 9 mol %:
Undoped CLN showed a constant lifetime of almost 10−7 s at all temperatures.
At the low temperatures the lifetimes grow with the Mg doping, reaching about 4 × 10−7 s at 20 K around the doping threshold.
From 20 to 200 K the luminescence lifetimes of the doped samples fall almost linearly, reaching the value of the undoped CLN at 200 K. Between 200 and 300 K all lifetimes are almost constant at the value of undoped CLN.
It has been assumed that below the doping threshold nonradiative decay channels quench the luminescence, while these channels are suppressed around the doping threshold [
15,
16,
17]. This behavior of the nonradiative decay channels has been attributed to variations of the electron-phonon interaction [
15,
16,
17], which appears to be probable as Xin et al. found a moderate reduction of the electron-phonon interaction by 50% around the doping threshold [
42]. As nonradiative decay in a solid is related with the emission of phonons, this reduction will probably affect the luminescence quenching as well. However, a reduction by 50% is not sufficient at all to explain the huge variations of the luminescence intensity by about two orders of magnitude. On the other hand, the doping and stoichiometry dependencies indicate that the luminescence will be suppressed through the presence of
anti-site defects. In comparison to the moderate variations of the electron-phonon coupling, the complete removal of the
anti-sites is obviously the ultimate cause of the strong luminescence enhancement. This indicates that these intrinsic defects are very important for the non-radiative decay channels. Hence, reducing the defect concentration by doping around the threshold increases the luminescence and its lifetime, while an increase of the lithium deficiency reduces the luminescence intensity again by restoring the
anti-sites [
15].
Note that there is still the strong temperature dependence of the luminescence above the doping threshold, indicating the existence of different luminescence-quenching channels being independent from anti-sites. If such channels are responsible for the luminescence quenching at higher temperatures, they are obviously very sensitive to temperature.
The effect of defect concentration on luminescence has also been reported for the luminescence from dopants as Cr
3+ and Er
3+ [
85,
86]. Although these cases deal not with polaronic luminescence itself we will discuss them in the next paragraphs because they provide some deeper insights into the luminescence quenching mechanism.
As the luminescence spectra from dopant ions originate from their radiative transitions, this radiation is obviously excited by the transition of electrons from the conduction band (or from neighbored polarons) over unoccupied states of the dopant ion to the valence band—via at least one radiative transition. In principle these radiative transitions can be quenched by the same mechanisms that also quench radiative electron and hole polaron recombinations—via concurring nonradiative channels returning excited charge carriers back to the valence band, bypassing all radiative transitions. The first example are the strong variations of the Cr
3+ luminescence in Cr-doped LNO under varying stoichiometry, as there is a strong luminescence quenching in non-stoichiometric LNO depending on the number of defects [
85]. A similar example is the enhancement of Er
3+ luminescence in Er-doped CLN at higher Mg dopings, as reported by Tang et al. [
86]. Absorption measurements showed the presence of bipolarons in these materials and a reduction of this presence through Mg doping. As the measurements showed no detectable sign of single bound polarons, the authors proposed this concurring decay to be mediated by bipolaron formation and subsequent recombination with hole polarons [
86], as depicted in
Figure 1. This decay channel depends immediately on the presence of
anti-sites that can form bipolarons. It is conceivable that not the whole bipolaron has to be recombined with two hole polarons at once, but only one of the two constituting electron polarons with one hole polaron. This would leave behind the other electron polaron as a bound polaron, where a new free electron polaron can aggregate. At any case, this nonradiative decay would require the presence of
anti-sites as hosts. As these anti-sites are surrounded by lithium vacancies, it is possible that the nonradiative recombination takes place between bipolarons and hole polarons that have been trapped as bound hole polarons in the neighborhood of these lithium vacancies. For the visible polaronic luminescence, this quenching mechanism may explain well the luminescence quenching in LNO samples with a higher amount of
, as in CLN samples with low Mg doping. When at low or zero dopings very much anti-sites are present, the nonradiative recombination might also take place via single bound polarons instead of bipolarons; the important point is again the presence of
anti-sites. Furthermore, it is also possible that the niobium interstitials
show physically the same behavior, thus contributing comparably to the nonradiative recombination. The temperature-dependent luminescence quenching occurring in highly doped Mg:CLN at room temperature, however, cannot explained in that way due to the absence of
anti-sites. As already stated above, the reason for that quenching must be found in different concurring nonradiative recombination processes, maybe mediated by Mg-containing defect complexes.
The temperature dependence of luminescence in higher doped CLN has also parallels in the luminescence from dopant ions. For Tb
3+ luminescence in 2.8 mol % Tb:CLN, temperature-dependent quenching was observed that sets in above 150 K. Its temperature dependence shows an Arrhenius-like behavior with an activation energy of 0.22 eV [
87]. This value lies within the range of the polaron hopping energy, thus indicating an involvement of polaron hopping processes in the luminescence quenching. Note that a 2.8 mol % Tb
3+ doping may approach the doping threshold for Tb
3+, leaving only few
anti-sites in the material. In that case we would have an analogy to the temperature-dependent luminescence quenching in Mg:CLN with doping concentrations around the threshold. On the other hand, the temperature-dependent behavior of Cr
3+ luminescence in Cr-doped SLN showed to be determined by the electron-phonon coupling. In a range from 77 to 300 K the temperature dependence of the Cr
3+ luminescence agreed well with calculations based on a quantitative model of electron-phonon interaction [
88], thus leaving no space for any indications for an involvement of polaron hopping. Probably the nonradiative decay channel exists in the Cr
3+ ions themselves and is directly related with the emission of phonons without any energy transfer to polarons.
These examples demonstrate a variety of possible luminescence quenching mechanisms in (doped) LNO. As we will discuss in the next section, the temperature dependence of the visible polaronic luminescence in highly Mg-doped CLN shows also an Arrhenius-like behavior with activation energies in the range of the polaron hopping energy, thus being comparable to the case of Tb3+ luminescence.
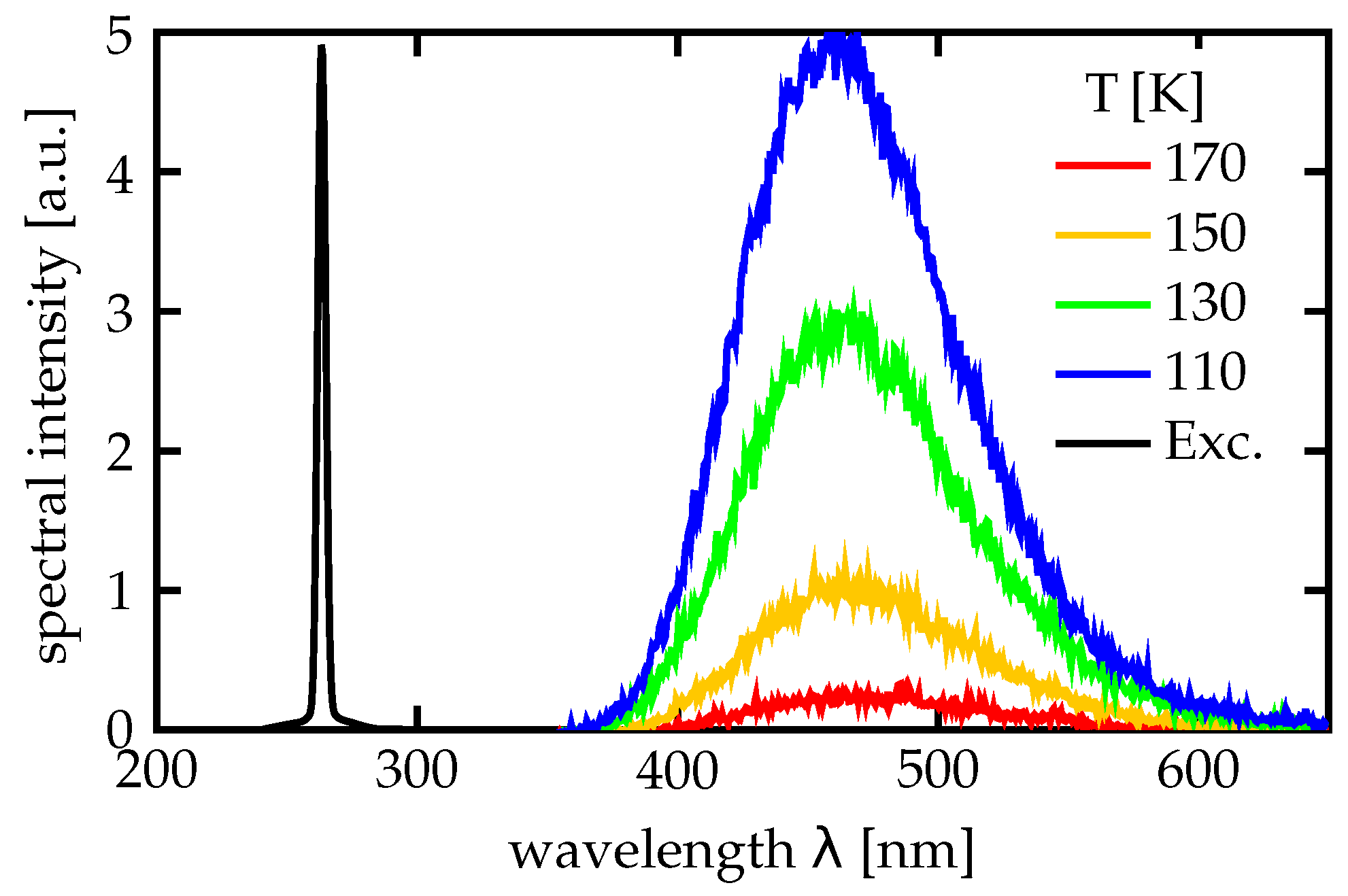
Our first investigations on this visible photoluminescence [
17,
18] mainly dealt with the investigation of its domain contrast to be discussed in the next part. We detected the luminescence spectrum with a maximum around 2.6 eV and a width of almost 0.35 eV upon a three-photon excitation. For that we focused a near-infrared femtosecond laser beam from a Ti:Sa laser (100 fs at 790 nm and with a repetition rate of 75 MHz) through a high N.A. immersion objective onto 0.5 to 1 mm thick z-cut samples. To avoid the pyrolysis of the immersion oil, the focus was kept 10 µm inside the sample bulk. To overcome the bandgap, a three-photon process is necessary for this excitation wavelength, as has also been indicated by power-dependent measurements of the luminescence intensity [
17]. The luminescent light was collected by the same objective and detected by a spectrometer after being coupled out by a beamsplitter and after blocking of the 790 nm stray light. We also applied a spectral intensity calibration for both the spectrometer as for the detection path. However, the excitation wavelengths of our pulsed lasers (and their higher harmonics) were too far off the effective wavelengths for the NIR luminescence excitation. Therefore our investigations have been concentrated on the visible luminescence band.
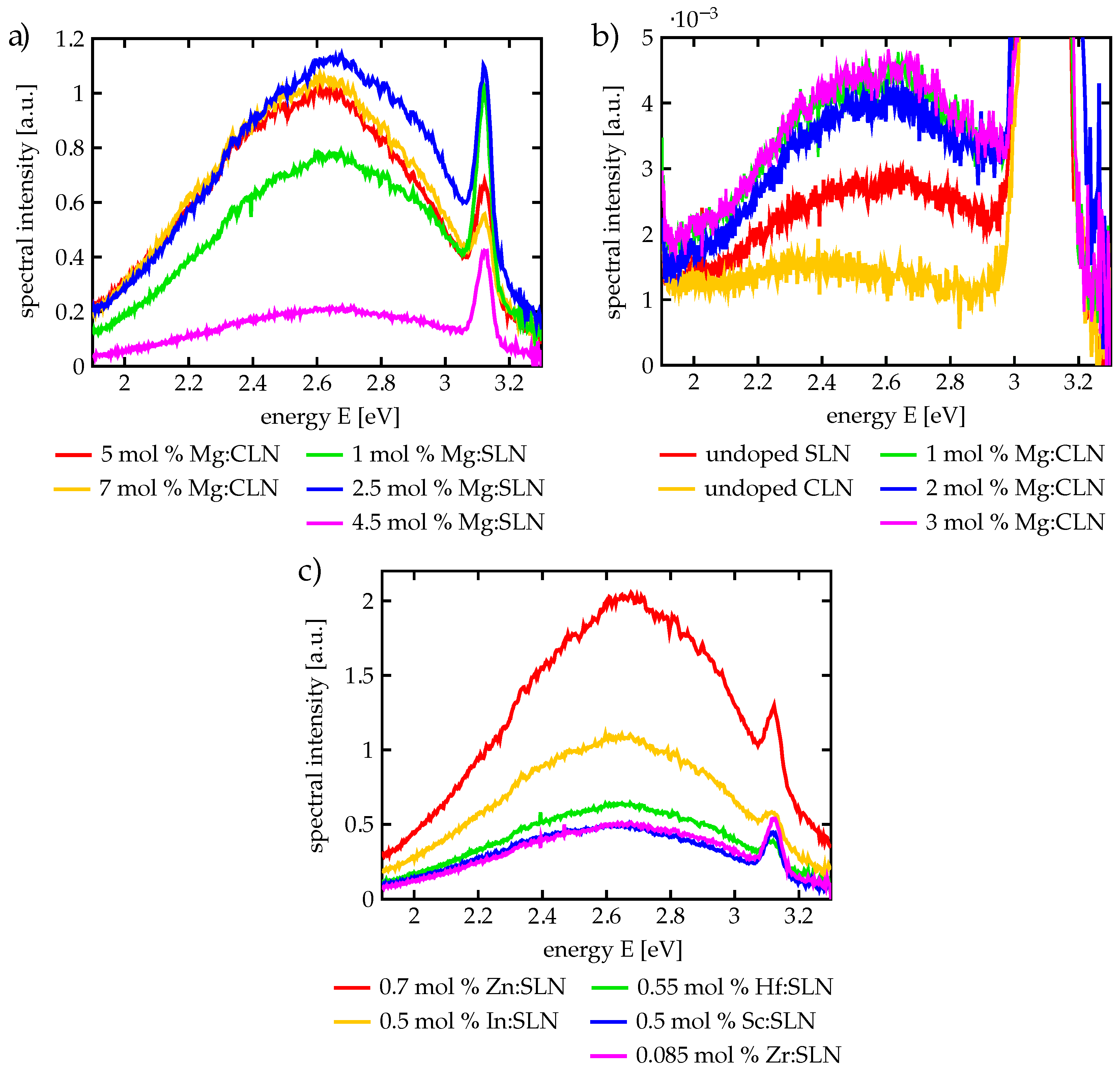
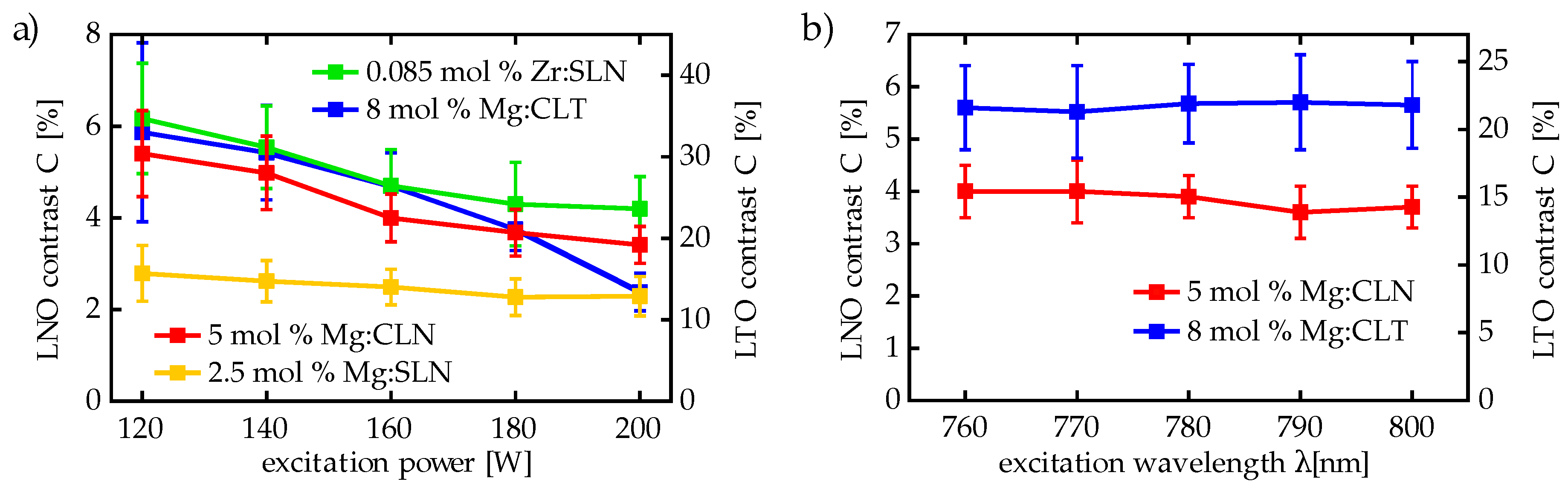
Our luminescence measurements have confirmed the strong dependence on Mg doping, especially on the 5 mol % doping threshold: For CLN samples of 5 and of 7 mol % Mg doping, respectively, high luminescence intensities have been observed, while for 3 mol % and below the luminescence intensity was lower by at least two orders of magnitude, as shown in
Figure 4.
For this review we have carried out additional measurements of the luminescence spectrum of stoichiometric lithium niobate (SLN) samples to prove the importance of the doping threshold for different dopings and stoichiometries, too. For this purpose SLN samples have been prepared in the Wigner Research Center for Physics in Budapest, where SLN has already been prepared for investigations on the bandgap [
43]. The SLN samples prepared for our luminescence investigations had been doped with different Mg concentrations as well as with other optical damage resistant dopants like Zn, In, Sc, Hf, and Zr. The dopant concentrations were chosen to be always above the threshold, which depends on the growth process and the valence state of the dopant [
89,
90]. Our aim was to take a further look on the luminescence quenching not only by a different stoichiometry but especially by different dopants than Mg. Note that due to the still non-perfect stoichiometry of usual SLN there is still a finite doping threshold of about 1 mol % for Mg doping [
36], while improved approaches to perfect stoichiometry yielded a further reduction of the Mg doping threshold to as low as about 0.2 mol % Mg [
37].
Figure 4a shows the luminescence spectra of Mg:CLN and Mg:SLN. Both 5 and 7 mol % Mg:CLN and 1, 2.5 and 4.5 mol % Mg:SLN show a luminescence intensity in the same order of magnitude. The decrease in luminescence at 4.5 mol % doping in Mg:SLN may be a consequence of an increasing number of defects that far above the doping threshold. Moreover, all samples show a strong second-harmonic peak around 3.1 eV.
Figure 4b shows the weak three-photon luminescence from undoped CLN and SLN, as well as from CLN doped below the threshold. The luminescence is at least two orders of magnitude smaller than for threshold and super-threshold doping, thus confirming the findings presented above. As the SLN crystals still show a certain Li deficiency, they contain enough luminescence-suppressing
defects to keep the luminescence on the same low level as for the weakly doped Mg:CLN samples. In accordance with our expectations, the luminescence signal from undoped CLN is lowest as these crystals contain the highest concentrations of
.
Figure 4c shows photoluminescence spectra of SLN with different damage-resistant dopants: Hf, Zr, Zn, Sc and In, that all leave the SLN transparent. The luminescence strength is indeed of the same order of magnitude as for the 1 mol % Mg-doping around the threshold. The considerable luminescence at dopings of around 0.5 mol % for Sc, In and Hf, as well as the stronger luminescence for 0.7 mol % Zn, compared to the weak luminescence of undoped SLN as shown in
Figure 4b, confirms the influence of the low threshold of SLN on the luminescence quenching. An outstanding example, however, is the low Zr doping being accompanied with a luminescence intensity as high as for the 0.5 mol % doping of other dopants. Doping SLN with Zr, however, showed indeed a comparably low doping threshold of only 0.085 mol % [
91] instead of 0.2–1 mol % as in the case of Mg doping [
36,
37]. Moreover, even for congruent lithium niobate a rather low doping threshold of only 2 mol % has been found for Zr doping [
92]. Note that the dopings of Zn [
93] as well as for Hf and Zr have been directly measured, while all other concentrations have been determined from the melt. Deviations between the doping concentrations in the melt and in the crystal as well as slightly different doping thresholds may explain e.g., the larger luminescence intensity from 0.7 mol % Zn compared to 1 mol % Mg or of 0.5 mol % In compared to 0.5 mol % Sc or 0.55 mol % Hf.
Thus, we have confirmed that also the doping-induced luminescence enhancement is really independent from the specific properties of Mg defect ions and works in the same way with different optical damage resistant dopants. Note that all these luminescence spectra show the same shape with the maximum around 2.6 eV and a width of almost 0.35 eV, independent from the specific kind of dopant. This underlines again the polaronic origin of that luminescence instead of being caused by any specific dopant defect centers (like the Er
3+ luminescence in Er:LNO [
94,
95]), and it confirms the identical mechanism of luminescence enhancements by doping with these dopants which leads to the replacement of
anti-sites.
In addition to these damage-resistant dopings that leave the samples transparent, we have also made measurements with SLN and CLN samples doped with Fe, Mn or Er. These impurities—that change the absorption in the visible range drastically—do not yield any sign of an enhanced 2.6 eV photoluminescence. This is probably due to their strong absorption or by providing new strong decay channels. Only the Er dopants show the known Er luminescence [
86,
94,
95]. Especially Fe does not increase the visible luminescence peak at all; even a sample with all Fe ions being brought into the Fe
3+ state (changing the sample color from dark red to pale yellow) did not show any luminescence peak around 2.6 eV.
2.5. Time Dependent Measurements of the Visible Luminescence Band
To obtain a deeper insight into the nature of the visible photoluminescence we carried out time-dependent measurements that have been published in a separate paper [
19]. In these measurements we used a pulsed UV laser light source at 266 nm with 4 ns pulses up to 5 mJ. The setup allowed to measure luminescence lifetimes up to the range of ms. Strongly enhanced luminescence signals were obtained by cooling the sample down to a temperature range of 100 to 200 K. The sample surfaces were illuminated under 45° and the luminescence was detected under 90° to the exciting beam. Time-averaged luminescence spectra measured from 7 mol % Mg:CLN for different temperatures are shown in
Figure 5, showing a strong decrease of the luminescence with temperature. The strong temperature dependence of the luminescence has thus been confirmed for dopings around the threshold, indicating the existence of further luminescence quenching mechanisms not being mediated by
anti-sites.
However, at these deep temperatures a layer of microscopic ice crystals was formed by sublimated water, which affects the reflection properties of the sample surfaces. As this effect increased with lower temperatures the measured intensities could not be used for an exact quantitatively analysis but only for a simple demonstration of the increase of luminescence quenching with temperature. For more precise measurements another setup would be necessary keeping the sample in vacuum or within a completely dry nitrogen atmosphere. Additionally, like with our thee-photon femtosecond setup no near-infrared luminescence has been detected with the UV laser pulse setup.
Consequently, the time-dependent measurements of luminescence have been carried out at 150 K, using the advantage of reduced luminescence suppression and making also measurements at sub-threshold dopings feasible. Like in the case of the non-radiative decay of bound polarons into the Fe defect centers, a stretched-exponential decay of the luminescence can be found. This confirms the polaron recombination as the luminescence source, as the recombination of spatially randomly distributed polarons yields a superposition of different exponential decays [
15,
19]. The intensity of the luminescence hence has to obey the equation
corresponding to the time-derivative of the stretched-exponential decay of the number of excited charge carriers:
which is typical for transitions that form an ensemble of emitters with distributed decay rates [
19,
71,
78]. The closer
to 1, the nearer the decay comes to a single exponential. A small
, however, indicates a broader distribution of decay times. After passing of the exciting pulse the relaxation of excited free charge carriers into polarons takes place within picoseconds [
50,
51,
52], so that at later times only recombining electron and hole polarons are present and their dynamics can be described just by Equation (
2).
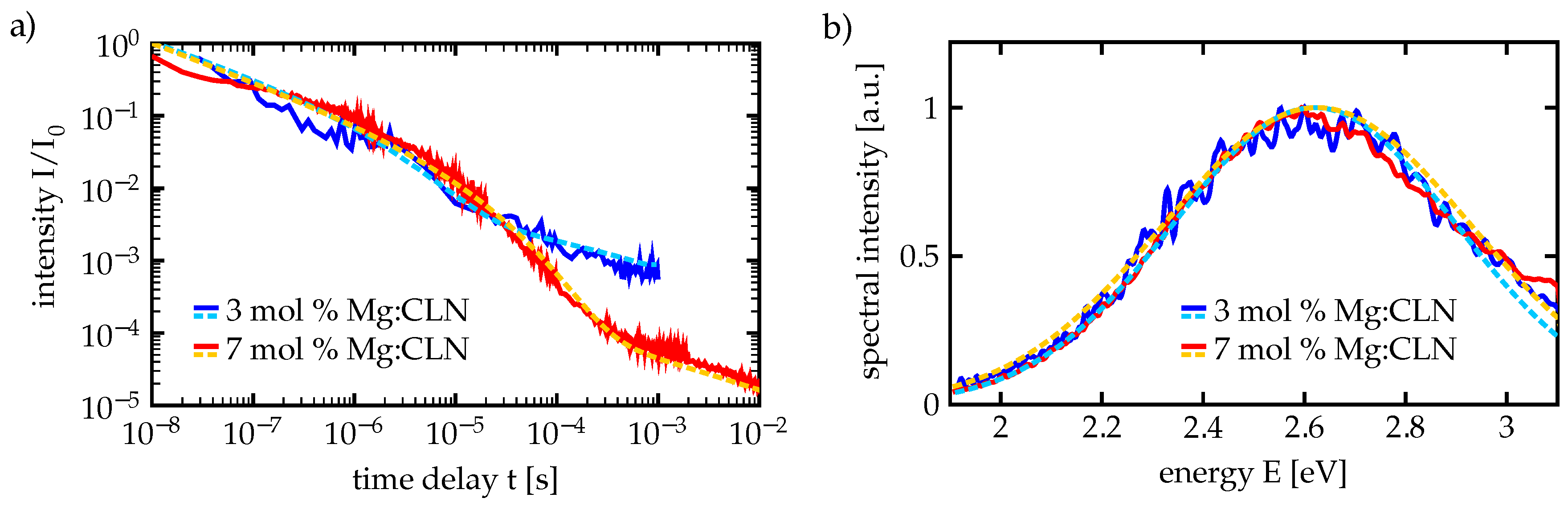
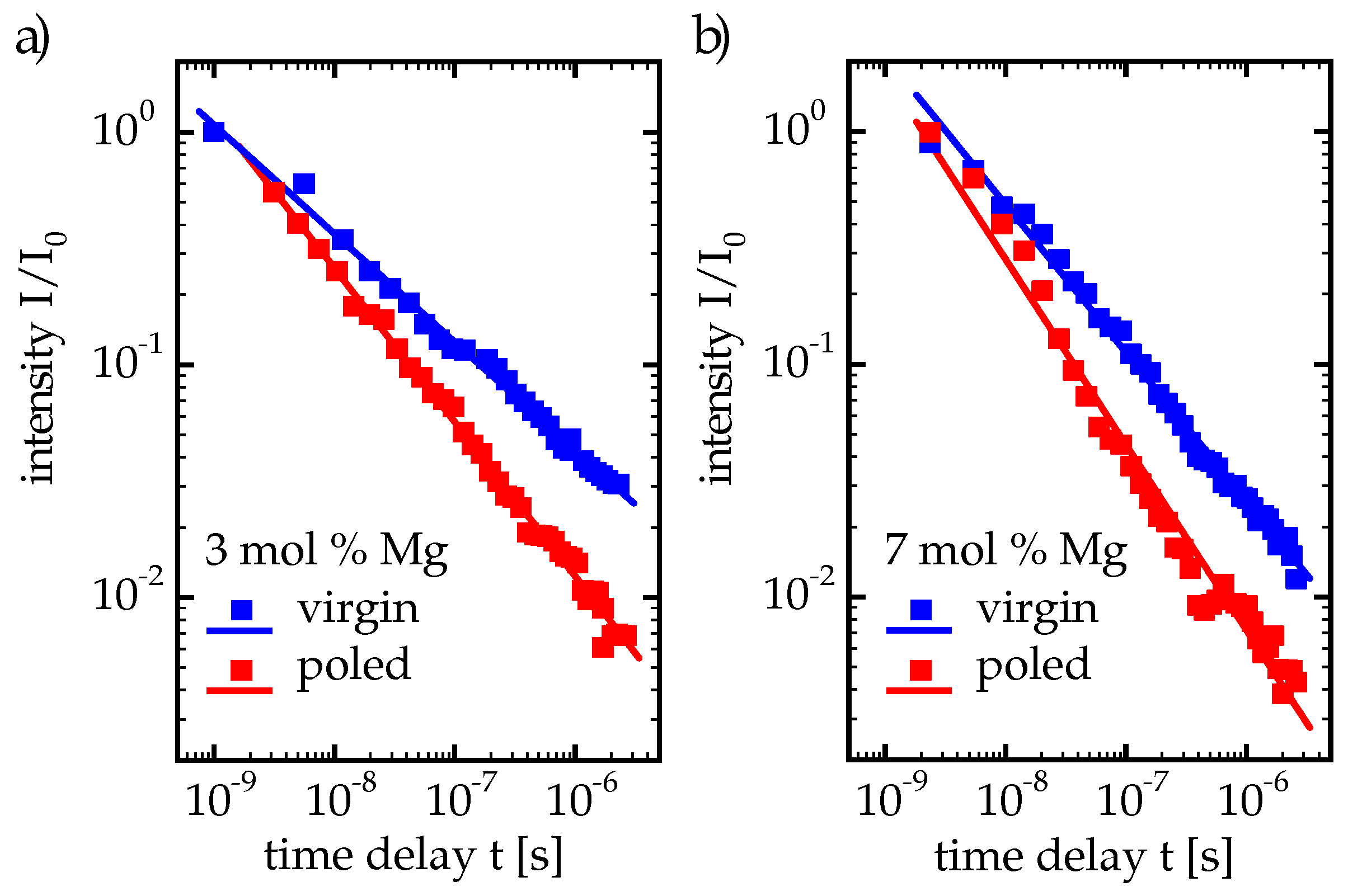
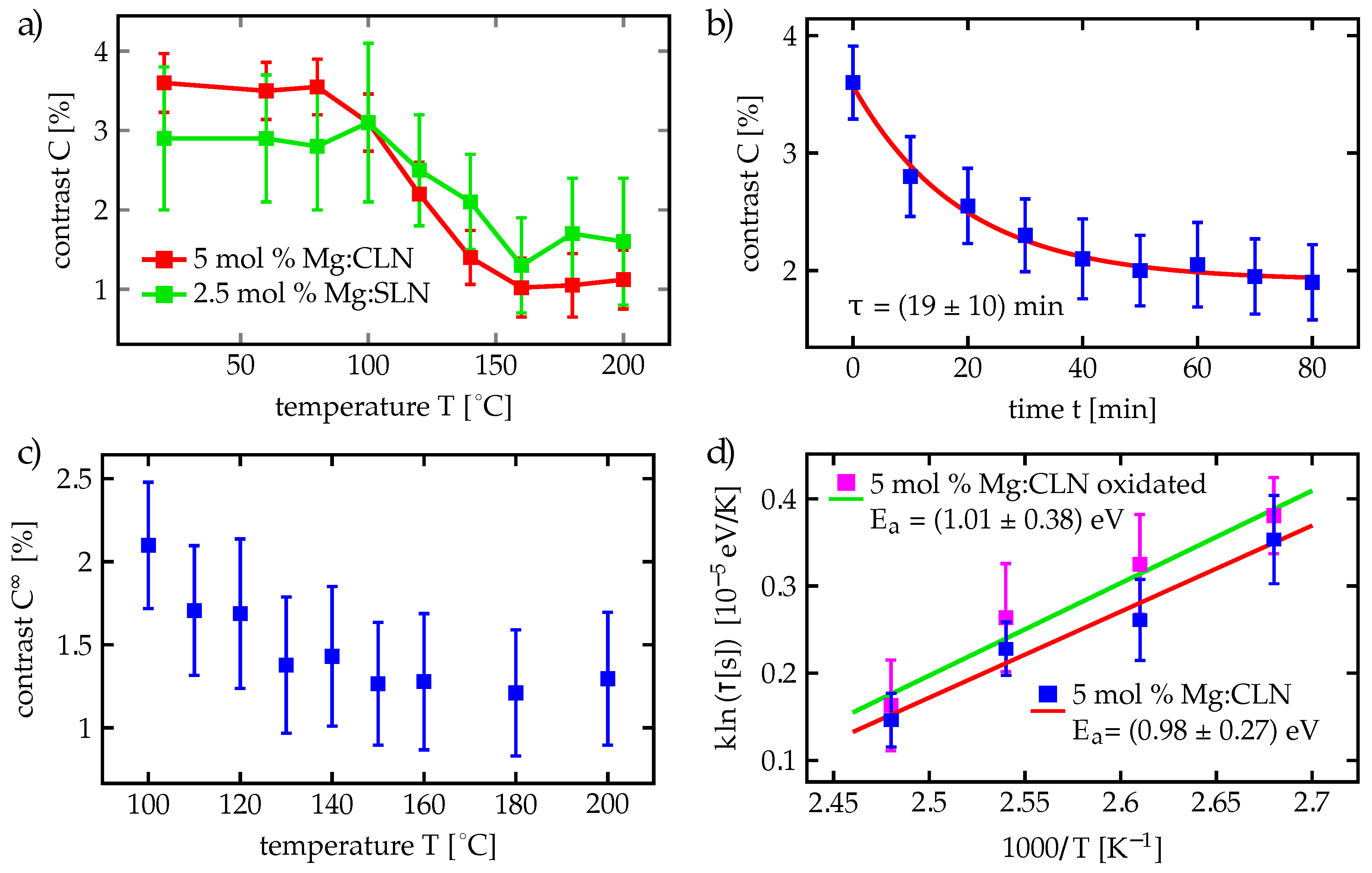
We carried out our time-dependent measurements at CLN samples with dopings above and below the doping threshold, namely at 3 mol % and 7 mol % Mg content, to investigate the behavior in these two regimes. While the visible luminescence of 3 mol % Mg:CLN is very weak at room temperature, the cooled nanosecond excitation setup provides the possibility to observe the increased luminescence intensities at low temperatures. This follows from the measurements in Ref. [
16], showing a strong temperature depence of luminescence already at 3 mol % Mg doping, which allows good luminescence measurements for both dopings at low temperatures.
In both cases of 3 and 7 mol % Mg doping we found that the temporal decay has to be fitted with a superposition of two stretched exponentials, resulting in each two values
,
(with
in the order of µs and
in the order of 10 ms to 1 s) and
,
(between 0.5 and 0.7), as shown in
Table 1. The decay curves are displayed double-logarithmically in
Figure 6a, corresponding spectra are shown in
Figure 6b, the latter confirming the peak position at 2.6 eV and the width around 0.3 eV. For the time-dependent measurements the luminescence spectrum has been integrated over a small range around the spectral maximum. Especially the luminescence decay curve from the 7 mol % Mg sample shows a distinct separation into two regimes below
s and around
s that have indeed to be fitted with two stretched exponentials.
are the prefactors of the stretched exponentials and give the contributions of these exponentials to the intensity
at
0. The subscript “max” denotes that the measured time-dependent intensity is obtained by integration around the spectral maximum, to which the stretched exponentials are fitted. In
Table 1,
and
are normalized for each sample to the respective larger value. For 7 mol % Mg they have a small ratio of
of about 3.5
, i.e., the intensity from the first stretched exponential is considerably higher than from the second one. For the 3 mol % Mg:CLN, however, the opposite is the case: the inverse ratio
is very small at about 2.3
and the first exponential shows a rather weak contribution. As a consequence, the kink between the two regimes is much less pronounced.
The separation of the time development into two regimes for 7 mol % Mg:CLN may be explained by the time development of hole polaron and electron polaron density. Within some hundred fs after each exciting pulse the excited charge carriers relax into polaron states [
47,
50,
51], and free electron and hole polarons occur in such a density that each charge carrier finds a partner for recombination in its nearest neighborhood. Thus, many recombinations involve none or only few electron/hole polaron hoppings to neighbored sites because adjacent electron and hole polarons can recombine directly. This yields the first stretched exponential as a superposition of shorter lifetimes from direct recombinations about small distances and only few hoppings. These conditions change when these charge carriers—the majority of excited charge carriers—have recombined and the remaining charge carriers have to move over longer distances by hopping. This results in a different superposition of decay times where the contributions from the recombinations are negligible compared to the longer hopping times [
19]. These different contributions of direct recombinations and hopping times may explain the existence of two regimes that make two stretched-exponential fits sensible.
For the 3 mol % Mg:LNO, however, the relative intensity of the first exponential is much weaker than of the second. This behavior is probably caused by the stronger nonradiative decay below the doping threshold: Most of the electron and hole polarons created through the exciting pulse recombine nonradiatively, which obviously causes a very strong drain of the first regime.
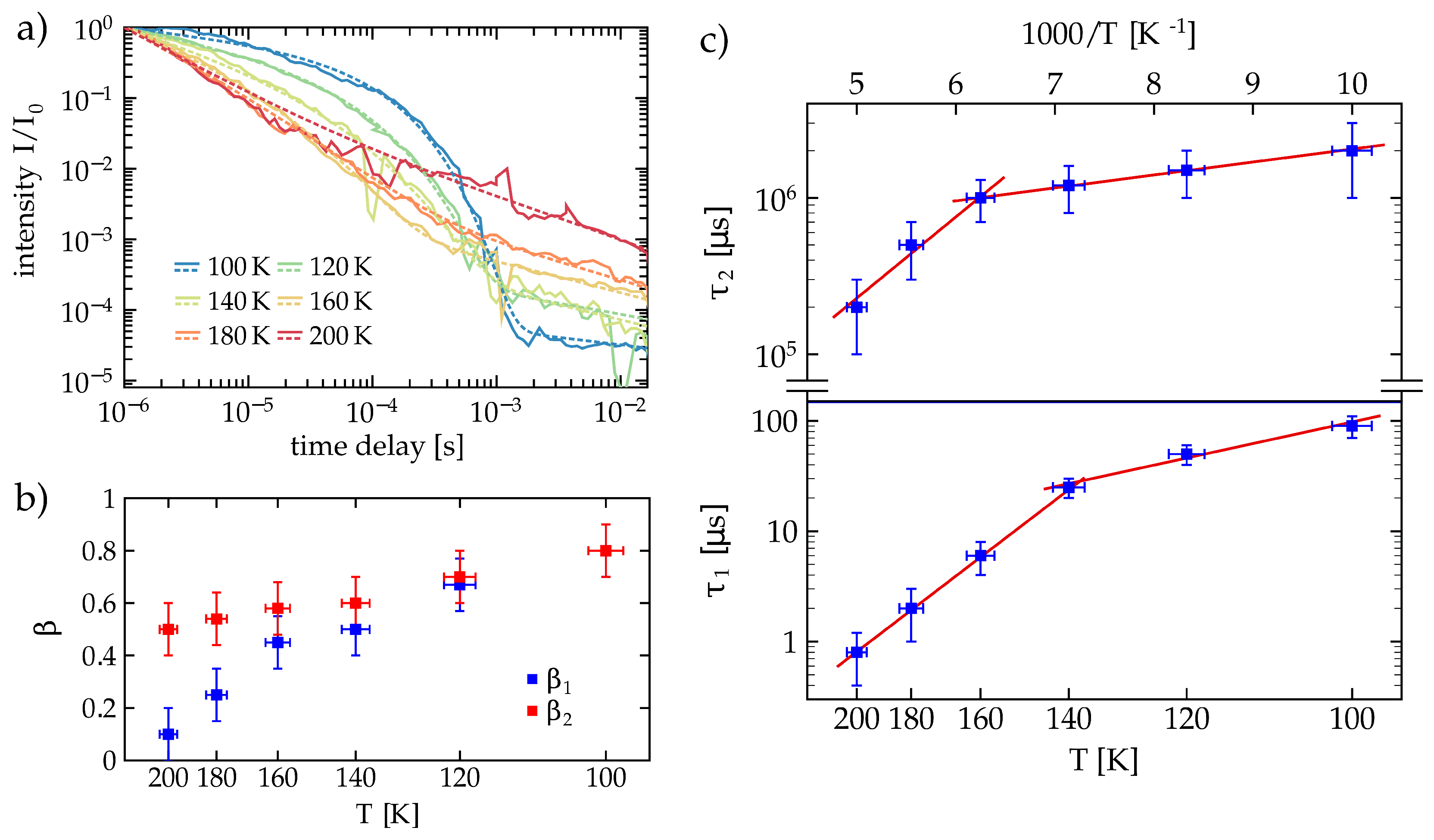
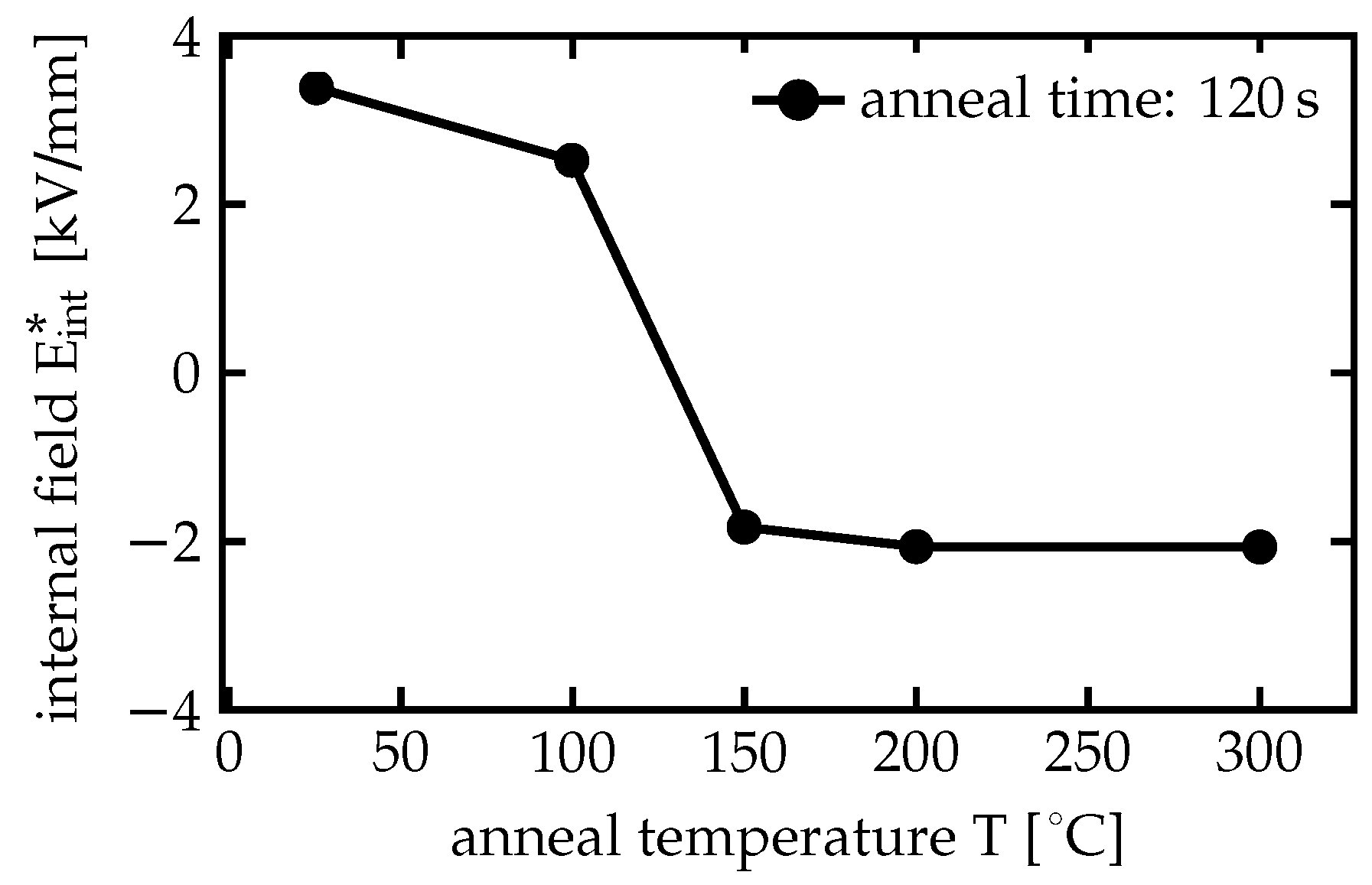
After these first time-dependent measurements at the 3 mol % and the 7 mol % Mg:CLN samples, the measurements on the 7 mol % Mg:CLN sample—the one with the larger luminescence—have been extended to different temperatures between 100 and 200 K. These measurements are shown in
Figure 7a, revealing a strong temperature dependence of the decay curves and their parameters.
Figure 7b shows the dependence of
and
on the temperature,
Figure 7c the temperature dependence of the lifetimes
and
. Although higher temperatures yield a faster recombination and lower decay times, the higher energy of the crystal and its constituents appear to yield a broader distribution of the decay times resulting in stretched exponentials with lower
values. Note that the curves from the lower temperatures show the kink which distinctly separates the two regimes with different
’s and
’s, while at 200 K this kink has disappeared. The higher
values at the lower temperatures render the curves more exponential-like, yielding a stronger bending in the double-logarithmic plot that makes the kink between the two regimes more pronounced. This is related with a narrower distribution of decay times in the two regimes, indicated by the
parameters closer to 1. The vanishing of the kink at 200 K, however, coincides with smaller
and may indicate that the broadening of these decay time distributions yields an overlap of these distributions that dissolves the clear separation of the two regimes.
Moreover, the ratio
of the second exponential intensity to the first showed a strong temperature dependence: at 100 K
is about
while it rises to the factor 10 at 200 K [
19]. Like in the case of 3 mol % Mg:CLN this behavior probably stems from the luminescence quenching that increases with temperature and drains the first regime, thus lowering
in relation to
. Note that due to the sublimation of water on the cooled sample surface a sensible quantitative evaluation of the absolute intensities
and
was not possible, while the division of
by
at each temperature cancels this unwanted effect.
The luminescence lifetimes
and
, shown in
Figure 7c, drop by 1–2 orders of magnitude from 100 to 200 K. They show an Arrhenius-like behavior with two distinct regimes below and above 150 K. Using the Arrhenius-like law
the activation energies
can be obtained through linear fits by plotting the logarithm of
over
(Arrhenius plot). In that kind of plot two regimes with constant slopes appear. All resulting activation energies are shown in
Table 2. Fitting the lifetimes
in the higher temperature range between 150 K to 200 K yields activation energies of (139 ± 22) meV for
and (103 ± 48) for
. For temperatures below 150 K, however, the fits yield much lower activation energies of
and
.
These results raise the question about the origin of this Arrhenius-like behavior with these activation energies. For a possible answer we will discuss now a hypothetical picture. Note that this picture is based on the assumption that the bandgap energy is around 4 eV, yielding a rather low free hole polaron energy around 0.3 eV. This low energy is important for the following discussion. A higher bandgap and a higher free hole polaron energy, however, would result a completely different picture. Despite this ambiguity we present the following considerations as an example in what manner these recombination processes could be modeled.
If only a radiative recombination would occur, the radiative lifetime
would be determined by the mobilities of free electron and hole polarons. Assuming the considerably lower free polaron energy of 0.3 eV would also yield a stronger delocalization, resulting into a lower activation energy and a higher mobility. Due to their higher mobility the recombination dynamics is dominated by the dynamics of the free hole polarons, because the hole polarons move faster and do not have to wait for the slower free electron polarons. The temperature dependence of the radiative recombination lifetime
should then be determined by the low hole polaron activation energy. With real electron polaron hopping energies in a range of 0.1–0.24 eV [
58,
64,
65] associated to an electron polaron energy of 1 eV, the hopping energies of hole polarons (with 0.3 eV polaron enery) can accordingly be estimated to be in a range of 0.03–0.08 eV. The activation energy of
for
T< 140 K—almost 40 meV—lies at the lower end of that range. This is consistent with the assumption that the radiative recombination is dominant in the range between 100 and 150 K.
Around 150 K, however, another regime with an activation energy of 139 meV appears. As demonstrated by
Figure 5, the luminescence intensity drops also strongly with the temperature, indicating a concurring nonradiative recombination. As the luminescence intensity has dropped by more than two orders of magnitude at 200 K, this process can be considered as dominant for
150 K and the higher activation energy can be assigned to this nonradiative recombination. This activation energy fits well with the measured activation energies obtained from electrical DC conductivity [
4,
58,
64,
65], indicating that the nonradiative recombination process is mediated by free polaron hopping. The activation energies of
are smaller than for
, but
behaves still quite similar to
.
As there are no anti-sites in 7 mol % Mg:CLN, the nonradiative recombination cannot be mediated by bipolarons (or bound polarons). As there are still lithium vacancies in the substrate, especially as parts of the + defect clusters, the recombination of free polarons with bound hole polarons may be a promising candidate for a nonradiative recombination channel. As the bound hole polarons are trapped at the lithium vacancies, the recombination dynamics would be governed by the hopping of the free electron polarons that have to meet these bound holes, thus resulting an activation energy according to the electron polaron hopping energy. Thus, the + defect clusters would, in a certain sense, take over the role of the anti-sites in undoped or weakly doped LNO. However, in Mg-doped LNO also interstitials that occupy regularly empty octahedra, including associated lithium vacancies, would probably contribute to luminescence quenching as well by providing similar nonradiative recombination channels. In this hypothetical model, the mechanisms of hole polarons by lithium vacancies would be the same.
A similar behavior of the temperature-dependent quenching of Tb
3+ luminescence has already been discussed in the last section [
87]. The activation energy of 0.22 eV matching to polaron hopping has been found at temperatures above 150 K, at lower temperatures the luminescence was almost constant. This indicates that the Tb
3+ luminescence is quenched by the same nonradiative recombination channel. Unfortunately we were not able to round the picture off by an exact evaluation of the luminescence intensities over different temperatures.
We note again that this speculative model requires a low free hole polaron energy and high mobility, based on a bandgap of 4 eV. As the question of the exact bandgap value being relevant for the photoluminescence is not yet finally resolved, proper modeling of the recombination physics requires a reliable knowledge of the energy and other properties of free hole polarons. These data should be obtained by independent measurements.
A comparison with the lifetime measurements made by Klose et al. [
15] reveals some further unclear points. Note that the presented lifetime measurements are made with similar nanosecond-pulsed laser light (20 ns pulses at 249 nm in the measurements of Klose et al. and 4 ns at 266 nm in our measurements). Klose et al. did not differentiate between
and
; as they measured lifetimes in the range below 10
−6 s it is sensible to compare their measurements with our
values. When we can thus compare the lifetime measurements by Klose et al. with our
values, the discussion of the results shows that the following points deserve a deeper investigation:
Doping CLN with Mg reduces the luminescence quenching induced by
and increases the lifetimes [
15]. This indicates that the radiative recombination and the nonradiative recombination via
live indeed from the same reservoir of (freely moving) polarons, because the presence of the additional nonradiative recombination reduces the total lifetime of
and therefore also the luminescence lifetime. While the replacement of
by Mg doping increases the lifetime, however, the temperature-dependent quenching at the higher dopings brings the lifetime back just to the value at zero doping. Is that accordance only accidentally or based on deeper reasons? The answer to this question requires a deeper understanding of the radiative and nonradiative recombination processes.
At zero Mg doping the lifetime measured by Klose et al. remained constant between 100 and 300 K. The strong luminescence suppression without doping underlines the dominance of the nonradiative recombination mediated by the anti-sites. Obviously the dynamics of this recombination is rather insensitive to temperature.
However, it seems to be difficult to find a consistent explanation for that. When the nonradiative recombination process probably lives on the same polaron reservoir like the radiative recombination, it is difficult to understand why not at least the hole polaron activation energy should occur in the luminescence lifetime, yielding at least a temperature sensitivity as in 7 mol % Mg:CLN below 150 K.
Although the nonlinear recombination via bipolarons or bound polarons at sites looks very probable, a clear model of this recombination, that can easily explain the lifetime behavior, is still missing.
In these lifetime measurements the luminescence lifetimes decrease roughly linearly in the temperature range up to 200 K. Plotting
from
Figure 7c not logarithmically, but in a linear scale over
T, it also decreases roughly linearly up to 160 K and nestles to the
T axis at higher temperatures. However, this nestling occurs already at 160, and not at 200 K. Moreover,
covers a range from almost 100 to about 1 µs, being reduced to about 1/100 of the value at
T = 100 K. The lifetimes from Klose et al., on the other hand, are in the order of not more than 5 µs, and around 200 K they stay constant at still 1/5 of the maximum at 80 K.
We could not explain these differences as in both measurement series Mg-doped CLN and similar kinds of laser light have been used. Future investigations should therefore also take a look if and how further parameters take stronger influence on the luminescence dynamics. Especially the influence of excitation intensity and pulse energy should be checked.
At all, the lifetime measurements of and should be expanded to measurement series from undoped CLN to 7 mol % Mg:CLN in steps of 1 mol % Mg doping. Furthermore, not only different dopings but also stoichiometries are probably interesting for investigations. Especially the behavior of the activation energies and the temperature of transition between the different regimes (with different activation energies) would be of interest.
Moreover, clearer measurements of the temperature dependence of the luminescence intensities and are necessary for a sensible modeling of the recombination processes. These measurements should, of course, cover the same range of Mg dopings and stoichiometries.
The discussed model includes the influence of the temperature-dependent polaron hopping. However, nonradiative transition processes in solids require the emission of phonons (or, in general, similar neutral quasiparticles as e.g. magnons). Thus a thorough understanding of the temperature dependence of the radiative and nonradiative recombination rates for different dopings comprises necessarily the knowledge of the temperature and doping dependence of the electron-phonon coupling as well. An interesting question may be if the electron-phonon coupling contributes significantly to the strong increase of the temperature dependence of luminescence around the doping threshold. On the one hand, the
doping dependence of this interaction, as given by Xin et al. [
42], is too moderate to explain the large doping dependence of the luminescence. On the other hand, the influence of electron-phonon coupling on the
temperature dependence of luminescence is unclear. The discussed Arrhenius plots of the lifetimes did not allow to separate the influence of electron-phonon coupling from the influence of polaron hopping. To separate the influence of the electron-phonon coupling onto the luminescence from other processes like polaron hopping, not only further lifetime measurement series but also additional investigations with different methods are necessary.
The electron-phonon coupling is possibly not the only further influence that has to be taken into consideration. As shortly mentioned above, the polaron model breaks down at sufficient deep temperatures due to a transition to an itinerant polaron band [
66,
67,
68,
69]. However, we have not found preciser information on this temperature range for LNO. In the relevant temperature range, this transition would probably have a great impact on the luminescence and its dynamics.
To conclude this discussion, the nonradiative recombination via bipolarons/bound electron polarons at and via bound hole polarons at + defect clusters may provide good models for the luminescence quenching through low doping or at higher temperatures. To really understand the radiative and nonradiative recombination dynamics much more and deeper investigations are necessary. These investigations will allow to prove the proposed theories; if they are successful, the models could be developed towards an accurate agreement with the measured data.
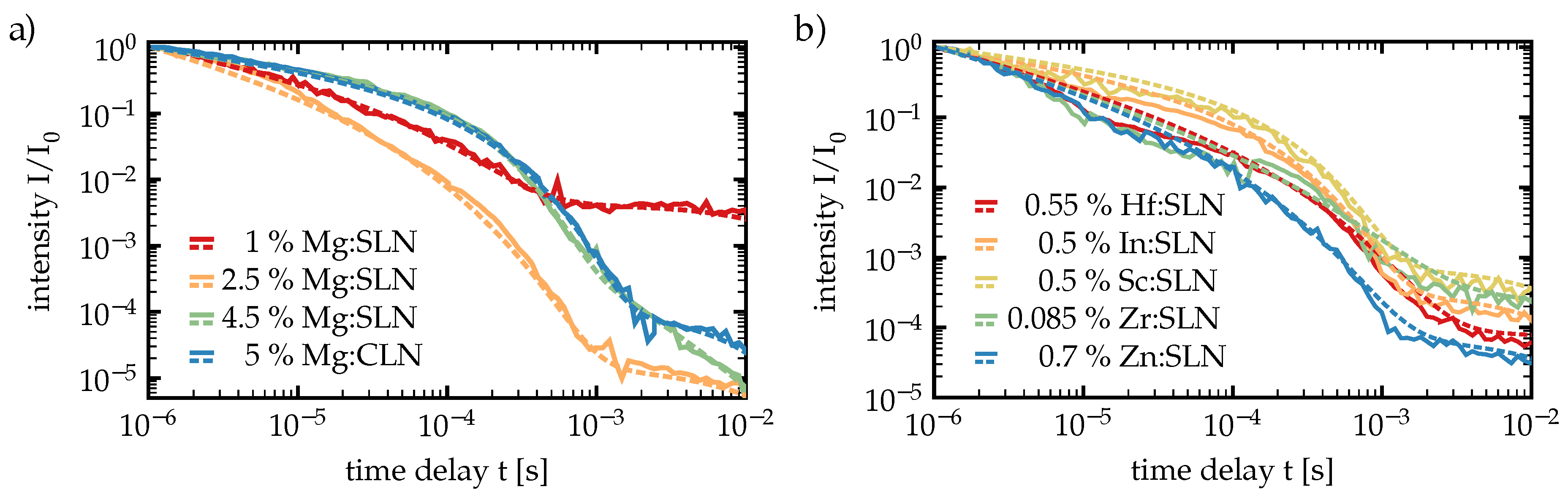
The time-and temperature dependent luminescence measurements on 3 and 7 mol % Mg:CLN, that have been thoroughly discussed in this section, have already been published in Ref. [
19]. For this review, further time-dependent measurements have been carried out with the same set of SLN samples that we already used to extend our results on luminescence spectra, together with 5 mol % Mg:CLN for comparison. We recorded the time-dependent luminescence now at 100 K, the results are shown in
Figure 8, the
values and lifetimes from these measurements are presented in
Table 3.
The lifetimes were mostly in the order of 10−5 – 10−6 s for and 0.01 s for , while most values lie between 0.4 and 0.8 and most in the range of 1. However, fitting of the second (long-time) stretched exponential turned out to be difficult in many of these cases, hence the corresponding and values suffer from larger uncertainties. The differences between these values for different dopings indicate a significant influence of the dopant on the luminescence decay. Most outstanding is the behavior of the 4.5 mol % Mg:SLN sample with strongly reduced and . This corresponds to the behavior of the curve for times > 10−3 s showing a much less distinct kink and a much stronger negative slope compared to the other curves, resulting in an extraordinary small . It seems that doping far above the SLN doping threshold may cause certain degrees disorder in the crystal structure with significant impacts on the temporal behavior of luminescence.
2.6. Summary of Part 2
In LNO the existence of electron and hole polarons has been proven. Excited electrons can occur
in free polaron states on sites with an absorption maximum at 1 eV,
in bound polaron states on anti-sites with an absorption maximum at 1.6 eV,
or in bound pairs of bound and free electron polarons, called bipolarons, with an absorption maximum at 2.5 eV.
To obtain the absorption spectra of these electron polarons, electrons have been brought into these polaron states by reduction of the crystal or by optical excitation of -doped samples. In this way the bound polaron (1.6 eV) and bipolaron (2.5 eV) have been found as well as the dissociation of bipolarons with 0.37 eV dissociation energy and the free electron polarons (around 1 eV) in samples with strongly reduced numbers of by Mg doping.
The hole polarons, however, have been found to occur at oxygen sites. In contrast to free hole polarons the oxygen sites of bound hole polarons adjoin immediately to defects, probably lithium vacancies [
2,
9,
11]. The absorption maximum of bound holes has been determined to 2.5 eV [
8,
9]. There is no similar data available on free holes. Due to the uncertainty of the LNO bandgap, the estimation of their energy based on the luminescence peak energy extends still over a larger range from 0.3 to 1.7 eV. The exact energy value, however, is important for the properties of hole polarons, as a small energy would be related with low activation energies, strong delocalization and high mobility. For a deeper understanding of the recombination processes, the energy level and other properties of the hole polarons should be determined precisely by independent experiments.
Two important photoluminescence bands have been revealed that are mediated by these polaron states: one in the near-infrared between 1 and 2 eV and another in the visible range around 2.6 eV. In the near-infrared actually two peaks have been observed: one at 1.5 eV and one at 1.3 eV. On the latter peak unfortunately no deeper physical investigations have been conducted so far. The 1.5 eV luminescence peak, however, shows a mono-exponential relaxation, indicating its origin from a single center without transitions between different, spatially randomly distributed defect states. This luminescence has been assigned to the radiative relaxation of an excited state of the bound polaron. The following properties support that interpretation: The intensity depends on the stoichiometry of the sample that determines the number of anti-sites and also on the number of ions from which electrons can be excited into a bound polaron state. It further strongly depends on temperature, likely due to a non-radiative decay channel that involves thermally induced free electron polaron hopping on neighboring sites.
In contrast, the visible luminescence at 2.6 eV originates from the recombination of free polarons and free hole polarons. This can be concluded from the typical recombination lifetimes in the µs range and from the stretched-exponential luminescence decay being characteristic for the recombination of spatially randomly separated charge carriers. The luminescence and their lifetimes show a strong dependence on temperature, and the luminescence is considerably increased at low temperatures below 200 K. The luminescence increases also strongly with Mg doping concentration up to the doping threshold. The likewise reduction of luminescence by reducing the Li content indicates the obvious importance of the number of
for creating luminescence-quenching non-radiative decay channels. The measurements using SLN doped with various optical damage resistant ions confirm the role of the doping threshold, as can be seen from the small luminescence intensities from undoped SLN and weakly doped CLN, while dopings around the low SLN threshold yield as large luminescence intensities as for the much higher doped 5 mol % Mg:CLN. The occurrence of this enhancement in SLN for different dopants verifies that the mechanism of luminescence enhancement does not depend on the specific properties of Mg, although luminescence intensities, lifetimes, and doping thresholds vary for the differently doped SLN samples. A promising model for the luminescence quenching at low dopings is the mediation of the non-radiative decay by the formation of bipolarons (as proposed for Er
3+-doped CLN [
86]) or bound polarons. This would explain the requirement of
anti-sites for the nonradiative recombination.
Furthermore, especially at higher Mg dopings above the threshold, a temperature-dependent luminescence quenching appears. By this quenching the luminescence intensity at room temperature is by about two orders lower than at 100 K and below. As almost all anti-sites are replaced around the doping threshold, while + defect clusters appear, obviously another concurring nonradiative recombination process occurs. A probable hypothesis is a nonradiative recombination with bound hole polarons being trapped at the lithium vacancies. However, a full and thorough understanding and modeling of all recombination processes is still missing—especially concerning the temperature dependence of intensities and lifetimes, and the influences from polaron hopping, electron-phonon coupling or further processes. More clarity is also needed about the contributions to luminescence quenching from Mg or Nb insterstitials not occupying Li sites but regularly empty oxygen octahedra. We have already suggested that they play probably a similar role like their pendants on Li sites, however, this has still to be confirmed. To solve all these questions, many further investigations would have to be carried out.

 ,
, 




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}