Crystal Structures of the 43 kDa ATPase Domain of Xanthomonas Oryzae pv. Oryzae Topoisomerase IV ParE Subunit and its Complex with Novobiocin



Abstract
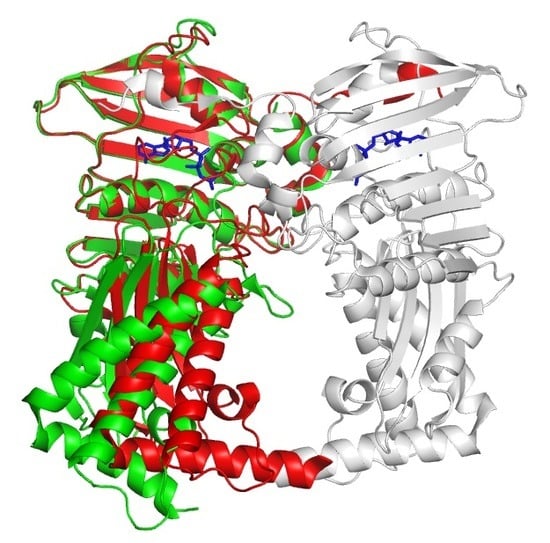
:
1. Introduction
2. Materials and Methods
2.1. Cloning and Protein Preparation
2.2. Crystallization, Data Collection, and Structure Determination
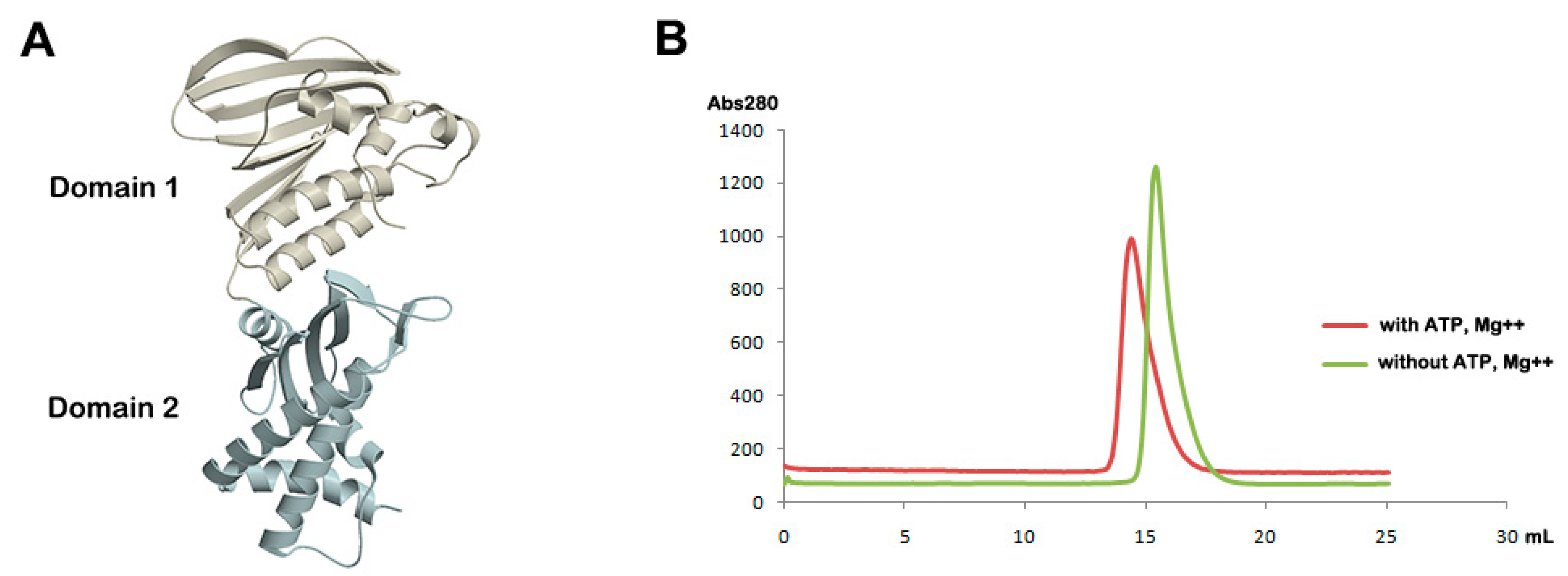
2.3. Analytical Gel Filtration
3. Results
3.1. Overall Structure of Apo Form
3.2. Structural Comparison of Xoo ParE and Other ParE Proteins
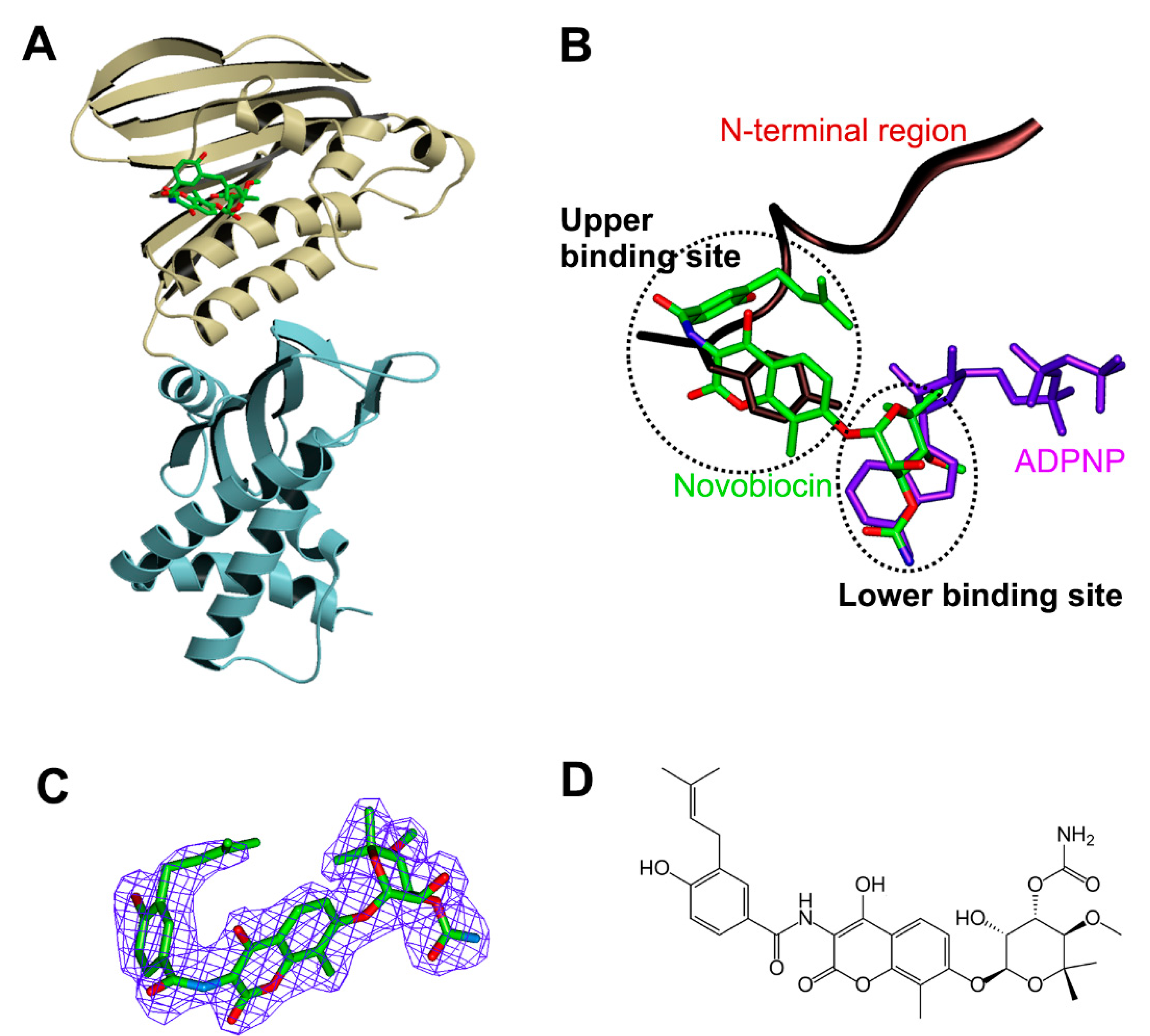
3.3. Structure of Xoo ParE in Complex with Novobiocin
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic DNA topoisomerases in transcription, replication and genome stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeffler, A.J.; Berger, J.M. DNA topoisomerases: Harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008, 41, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, K.; Fisher, L.M.; O’Dea, M.H.; Gellert, M. DNA gyrase action involves the introduction of transient double-strand breaks into DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Schoeffler, A.J.; Thomsen, N.D.; Berger, J.M. The structural basis of substrate specificity in DNA topoisomerase IV. J. Mol. Biol. 2005, 351, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Ann. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef]
- Crisona, N.J.; Strick, T.R.; Bensimon, D.; Croquette, V.; Cozzarelli, N.R. Preferential relaxation of positively supercoiled DNA by E. coli topoisomerase IV in single-molecule and ensemble measurements. Genes Dev. 2000, 14, 2881–2892. [Google Scholar] [CrossRef]
- Ullsperger, C.; Cozzarelli, N.R. Contrasting enzymatic activities of topoisomerase IV and DNA gyrase from Escherichia coli. J. Biol. Chem. 1996, 271, 31549–31555. [Google Scholar] [CrossRef]
- Dekker, N.H.; Rybenkov, V.V.; Duguet, M.; Crisona, N.J.; Cozzarelli, N.R.; Bensimon, D.; Croquette, V. The mechanism of type IA topoisomerases. Proc. Natl. Acad. Sci. USA 2002, 99, 12126–12131. [Google Scholar] [CrossRef] [Green Version]
- Roca, J.; Berger, J.M.; Harrison, S.C.; Wang, J.C. DNA transport by a type II topoisomerase: Direct evidence for a two-gate mechanism. Proc. Natl. Acad. Sci. USA 1996, 93, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Roca, J. The path of the DNA along the dimer interface of topoisomerase II. J. Biol. Chem. 2004, 279, 25783–25788. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agent. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide, and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Sohi, M.K.; Veselkov, D.A.; Pan, X.S.; Sawhney, R.; Thompson, A.W.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009, 16, 667–669. [Google Scholar] [CrossRef]
- Ali, J.A.; Jackson, A.P.; Howells, A.J.; Maxwell, A. The 43-kilodalton N-terminal fragment of the DNA gyrase B protein hydrolyses ATP and binds coumarin drugs. Biochemistry 1993, 32, 2717–2724. [Google Scholar] [CrossRef]
- Bellon, S.; Parsons, J.D.; Wei, Y.; Hayakawa, K.; Swenson, L.L.; Charifson, P.S.; Lippke, J.A.; Aldape, R.; Gross, C.H. Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): A single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase. Antimicrob. Agents Chemother. 2004, 48, 1856–1864. [Google Scholar] [CrossRef]
- Lu, J.; Patel, S.; Sharma, N.; Soisson, S.M.; Kishii, R.; Takei, M.; Fukuda, Y.; Lumb, K.J.; Singh, S.B. Structures of Kibdelomycin Bound to Staphylococcus Aureus Gyrb and Pare Showed a Novel U-Shaped Binding Mode. ACS Chem. Biol. 2014, 9, 2023–2031. [Google Scholar] [CrossRef]
- Tari, L.W.; Trzoss, M.; Bensen, D.C.; Li, X.; Chen, Z.; Lam, T.; Zhang, J.; Creighton, C.J.; Cunningham, M.L.; Kwan, B.; et al. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE). Part I: Structure guided discovery and optimization of dual targeting agents with potent, broad-spectrum enzymatic activity. Bioorg. Med. Chem. Lett. 2013, 23, 1529–1536. [Google Scholar] [CrossRef]
- Laponogov, I.; Pan, X.S.; Veselkov, D.A.; Skamrova, G.B.; Umrekar, T.R.; Fisher, L.M.; Sanderson, M.R. Trapping of the transport-segment DNA by the ATPase domains of a type II topoisomerase. Nat. Commun. 2018, 9, 2579. [Google Scholar] [CrossRef] [PubMed]
- Verdier, V.; Vera Cruz, C.; Leach, J.E. Controlling rice bacterial blight in Africa: Needs and prospects. J. Biotechnol. 2012, 159, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Yun, M.; Song, J.Y.; Kim, H.J.; Heo, Y.S. Crystallization and X-ray diffraction data collection of topoisomerase IV ParE subunit from Xanthomonas oryzae pv. oryzae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Mani, N.; Gross, C.H.; Parsons, J.D.; Hanzelka, B.; Muh, U.; Mullin, S.; Liao, Y.; Grillot, A.L.; Stamos, D.; Charifson, P.S.; et al. In vitro characterization of the antibacterial spectrum of novel bacterial type II topoisomerase inhibitors of the aminobenzimidazole class. Antimicrob. Agents Chemother. 2006, 50, 1228–1237. [Google Scholar] [CrossRef]
- Charifson, P.S.; Grillot, A.L.; Grossman, T.H.; Parsons, J.D.; Badia, M.; Bellon, S.; Deininger, D.D.; Drumm, J.E.; Gross, C.H.; LeTiran, A.; et al. Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: Intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J. Med. Chem. 2008, 51, 5243–5263. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apo | Novobiocin Complex | |
|---|---|---|
| Data Collection | ||
| X-ray source | PLS 5C | PLS 5C |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Space group | P42212 | P42212 |
| Cell dimensions | ||
| a,b,c (Å) | 105.30, 105.30, 133.76 | 105.12, 105.12, 135.97 |
| α, β, γ (˚) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 2.20 (2.25–2.20) * | 2.29 (2.34–2.30) |
| Rsym (%) | 7.8 (42.7) | 8.1 (48.6) |
| I/σI | 58.1 (2.3) | 55.1 (2.3) |
| Completeness (%) | 99.9 (99.0) | 97.2 (94.5) |
| Redundancy | 5.9 (2.5) | 5.8 (2.6) |
| Refinement | ||
| Resolution (Å) | 2.20 | 2.30 |
| No. reflections | 38,409 | 34,790 |
| Rwork/Rfree (%) | 23.6/25.7 | 22.8/25.3 |
| No. atoms | ||
| Protein | 2717 | 2717 |
| Water | 194 | 187 |
| Heterogen | 0 | 44 |
| R.m.s. deviation | ||
| Bond lengths (Å) | 0.005 | 0.007 |
| Bond angles (˚) | 1.31 | 1.23 |
| Ramachandran | ||
| Favored (%) | 96.76 | 96.76 |
| Allowed (%) | 2.95 | 2.95 |
| Outlier (%) | 0.29 | 0.29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.Y.; Heo, Y.-S. Crystal Structures of the 43 kDa ATPase Domain of Xanthomonas Oryzae pv. Oryzae Topoisomerase IV ParE Subunit and its Complex with Novobiocin. Crystals 2019, 9, 577. https://doi.org/10.3390/cryst9110577
Jung HY, Heo Y-S. Crystal Structures of the 43 kDa ATPase Domain of Xanthomonas Oryzae pv. Oryzae Topoisomerase IV ParE Subunit and its Complex with Novobiocin. Crystals. 2019; 9(11):577. https://doi.org/10.3390/cryst9110577
Chicago/Turabian StyleJung, Ha Yun, and Yong-Seok Heo. 2019. "Crystal Structures of the 43 kDa ATPase Domain of Xanthomonas Oryzae pv. Oryzae Topoisomerase IV ParE Subunit and its Complex with Novobiocin" Crystals 9, no. 11: 577. https://doi.org/10.3390/cryst9110577
APA StyleJung, H. Y., & Heo, Y. -S. (2019). Crystal Structures of the 43 kDa ATPase Domain of Xanthomonas Oryzae pv. Oryzae Topoisomerase IV ParE Subunit and its Complex with Novobiocin. Crystals, 9(11), 577. https://doi.org/10.3390/cryst9110577