1. Introduction
Self-healing materials are under intense development and have been recently studied by many researchers as a new and improved route toward the safety, an increased lifetime, better energy efficiency, and improved environmental impact of manmade materials [
1,
2,
3]. The self-healing process can be efficiently utilized in a wide range of applications, such as biological muscles, tissue engineering, 3D/4D printing, and soft actuators [
4,
5,
6]. Several self-healing studies have been reported for polymer systems, such as the requirement of healing agents (including light or heat) [
7] and the use of reversible and irreversible covalent bonds, as well as dynamic noncovalent bonding interactions (such as hydrogen bonds, π-π stacking, ionomers, or metal-ion interactions) [
8,
9] as the healing motifs [
2]. The healing capability of a given material does not depend solely on the bonding characteristics of the molecular structures, but also on the chain dynamics, which govern properties, such as the mechanical or reaction kinetics [
8].
Although many self-healing strategies have been studied, certain problems remain regarding how to improve the strength and make long-lasting self-healing materials that can self-repair multiple times, without an additional external stimulus or an incidental performance [
7]. Metal–ligand complexes have the potential for dynamic healing design motifs, considering the flexibility of its wide-range of thermodynamic and kinetic parameters, leading to the possibility of developing highly tunable materials [
2]. In addition, metal–ligand interactions are less sensitive to moisture than hydrogen bonds, which should be beneficial for practical applications [
2]. Several self-healing gels and solid materials have been reported to use reversible metal−ligand interactions, for which the most commonly utilized ligand system involves multidentate nitrogen-based aromatic ligands, such as terpyridine [
9]. For example, Pierre et al. reported a bis-terpyridine cyclam (BTC) that can exhibit self-healing features with Ni
2+ ions forming Ni
2BTC metallo suprapolymer gels [
10]. Bode et al. studied self-healing polymer films through the crosslinking of terpyridine, containing polymethacrylate copolymers with different cadmium salts. The crosslinking with cadmium(II) halogenide (i.e., chloride, bromide, and iodide) presumably forms ionic clusters consisting of metal complexes. The presence of these clusters contributes to the healing of these networks [
11].
To prepare functionalized polymers, several methods have been established, such as the ionic and free radical polymerization of vinyl monomers, group transfer polymerization (GTP), and more recently, ring-opening metathesis polymerization (ROMP) [
12]. Among these methods, ROMP is an attractive method because it is robust, produces an absolutely linear material, and is amenable to the formation of various copolymers of a controlled architecture [
13,
14]. Polymers produced using ROMP can have a variety of chelating groups including terpyridine moieties, which can act as an anchor group in the binding of metal complexes with a polymer chain [
14]. A member of our group recently synthesized a terpyridine-based norbornene polymer using ROMP, and conducted visible-light induced photocatalytic oxidation of benzylamine into imine, to confirm its applicability as a regenerative and repairable photocatalyst. The metallo photo-active complex polymer, which can bind to a ruthenium(II) complex (Ru(bpy)
2(bpy–tpy)Cl
2) through metal ions, was applied to a reversible photo-catalyst [
13]. Other researchers have reported a chemodosimeter for chemical warfare agents (CWAs), using norbornene-derived terpyridine materials [
15]. Although terpyridine-based norbornene polymers have been reported in the application of photocatalysts [
13], chemo-sensors [
15], and anion exchange membranes [
16], they have yet to be reported in studies on self-healing.
Herein, we conducted experiments using transition metal ions and a terpyridine ligand complex gel to confirm not only its reversible properties but also its self-repair capabilities. More importantly, such gels can self-recover without the use of external stimuli or the delivery of healing agents. This approach demonstrates a promising route toward the design of strong, ideal, and self-healing materials that can repeatedly self-repair without degradation or the need for external stimuli.
3. Results and Discussion
To realize self-healing properties, a polymer should possess a reversible binding ability, such as an assembly. As terpyridine is known to be a metal-binding ligand enabling a reversible assembly [
11], we incorporated terpyridine groups into the polymer to investigate its self-healing behavior. To prepare the terpyridine-functionalized polymers, norbornene derivatives were chosen as monomers. Norbornene is widely utilized for functional group modifications, and can be easily polymerized through a ring-opening metathesis polymerization (ROMP), using Grubbs’ catalysts [
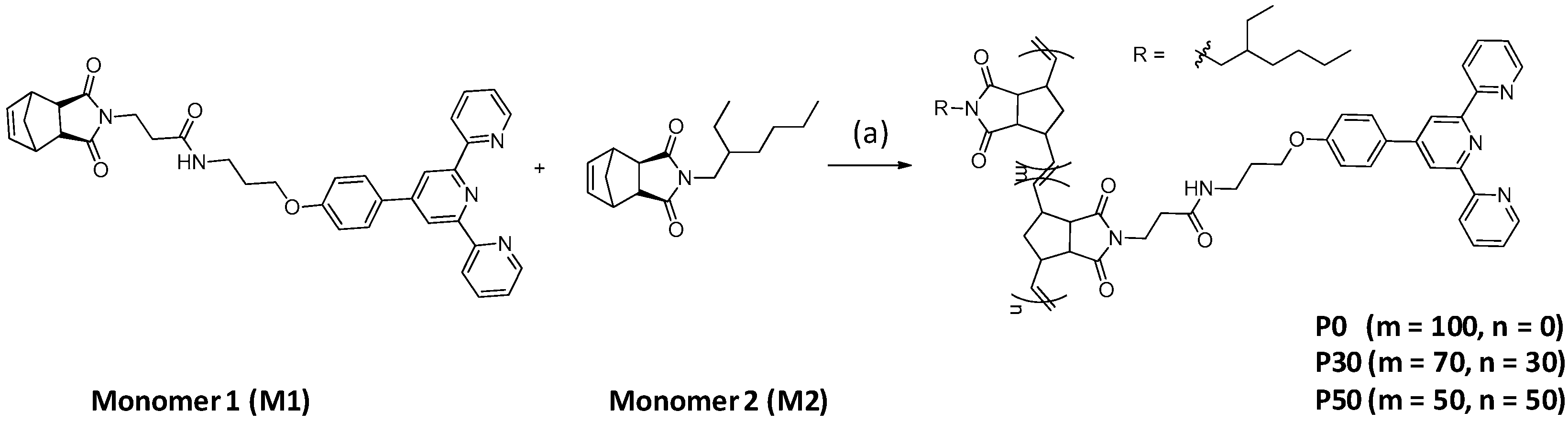
17]. In this study, we utilized Grubbs’ second-generation catalyst to produce a random linear copolymer. The synthetic approach for the norbornene-derived monomers is described in
Scheme 1. First, terpyridine- (M1), and ethylhexyl-functionalized norbornenes (M2) were synthesized, starting from
cis-5-norbornene-
exo-2,3-dicarboxylic anhydride, through a condensation reaction and an amide coupling reaction, as reported elsewhere [
13]. Subsequently, the linear random copolymers P0, P30, and P50 were synthesized with different ratios of M1, M2, using Grubbs’ second-generation catalyst, at room temperature, for 1 h, with ROMP (
Table 1). Grubbs’ second-generation catalyst had a sufficiently high activity in ROMP, while providing polymers with slightly larger PDIs than Grubbs’ third-generation catalyst, owing to the slower initiation step [
18]. As a result, the terpyridine-functionalized polymers of P30 and P50 were easily synthesized, using ROMP, with terpyridine contents of 30 and 50 mol%, respectively. For comparison, we prepared polynorbornene without a terpyridine moiety (P0), only from M2. Interestingly, it was found that the molecular weight of the polynorbornenes decreased as the mole ratio of terpyridine increased. We suspected that the active Ru catalyst might bind with the terpyridine moiety, during ROMP polymerization, becoming inactive. The higher terpyridine-containing polymer has a higher probability of a Ru complex with terpyridine. In the UV-vis spectrum of P30, a slight absorption was observed at 483 nm, which could be attributed to the singlet–singlet metal-to-ligand charge transfer (MLCT) band from the Ru-terpyridine complex (
Figure S1) [
19]. Nevertheless, terpyridine did not completely disturb the synthesis of polynorbornenes, when using ROMP with a Grubbs’ catalyst, despite the negative influence on the molecular weight.
All synthesized terpyridine-functionalized polynorbornenes were confirmed using
1H-NMR and FT-IR spectroscopic methods. The
1H-NMR analysis was carried out by calculating the ratio of terpyridine peak, corresponding to the newly generated olefin in
1H-NMR (
Figure S2). Based on the results obtained from the
1H-NMR analysis, no significant differences were shown between the feed ratios of terpyridine M1 and ethylhexyl M2 and the analytical composition in the synthesized polynorbornenes, which meant that the two monomers showed a similar activity toward the Ru catalyst, when applying ROMP (
Table 1).
To confirm that terpyridine had been indeed functionalized well in P30 and P50, we implemented an FT-IR analysis, for a comparison with P0. All polymers were purified using re-precipitation, several times, to remove any unreacted monomers. In the FT-IR spectra of the synthesized polymers, intensities of 2900 cm
−1 (C-H stretch in aromatics), 1470 cm
−1 (C-H stretch in aromatic rings), and 1126 cm
−1 (C-N stretch in amine), and overtone peaks, which correspond to the signature peaks of the terpyridine moiety, increased with respect to the 1700 cm
−1 C=O peak owing to the increasing proportion of terpyridine moiety, in the polymers, from 0.55 (P30) to 4.01 (P50) (
Figure S3). According to the above results, it could be confirmed that the terpyridine moiety was successfully incorporated into the polynorbornenes, when using ROMP.
Interestingly, we found that the content of terpyridine in the polynorbornenes affected their glass transition temperatures (
Tg).
Tg was measured with heating and cooling rate of 10 K/min and was determined as the midpoint of the transition on the second cooling cycle. As summarized in
Table 1, the measured
Tg values of P30 and P50 (86 °C and 103 °C, respectively) were higher than those of P0 (68 °C), and the more terpyridine that was incorporated, the higher the
Tg that was observed (
Figure S4). We suspected that the presence of the terpyridine moiety increased the
Tg of the polymers because it caused intermolecular associations (e.g., a π-π interaction), thereby, restricting the torsional mobility of the polymer chains. Enke et al. also reported increased
Tg values of polymers with a terpyridine ligand [
20]. To verify the possible existence of the π-π interaction, we investigated the
1H-NMR spectra of terpyridine-incorporated polynorbornenes (P30 and P50) and terpyridine monomer (M1). At different concentrations of P30 and P50, we were not able to find the peak-shifting of the terpyridine moiety. However, we found that terpyridine protons in M1 appeared to shift upfield, when incorporated in polynorbornenes (
Figure S5). This upfield shifting seems to accord with previous reports on the π-π interaction of terpyridine moieties [
21], which may support our hypothesis for the increase of
Tg, with increasing amount of terpyridines.
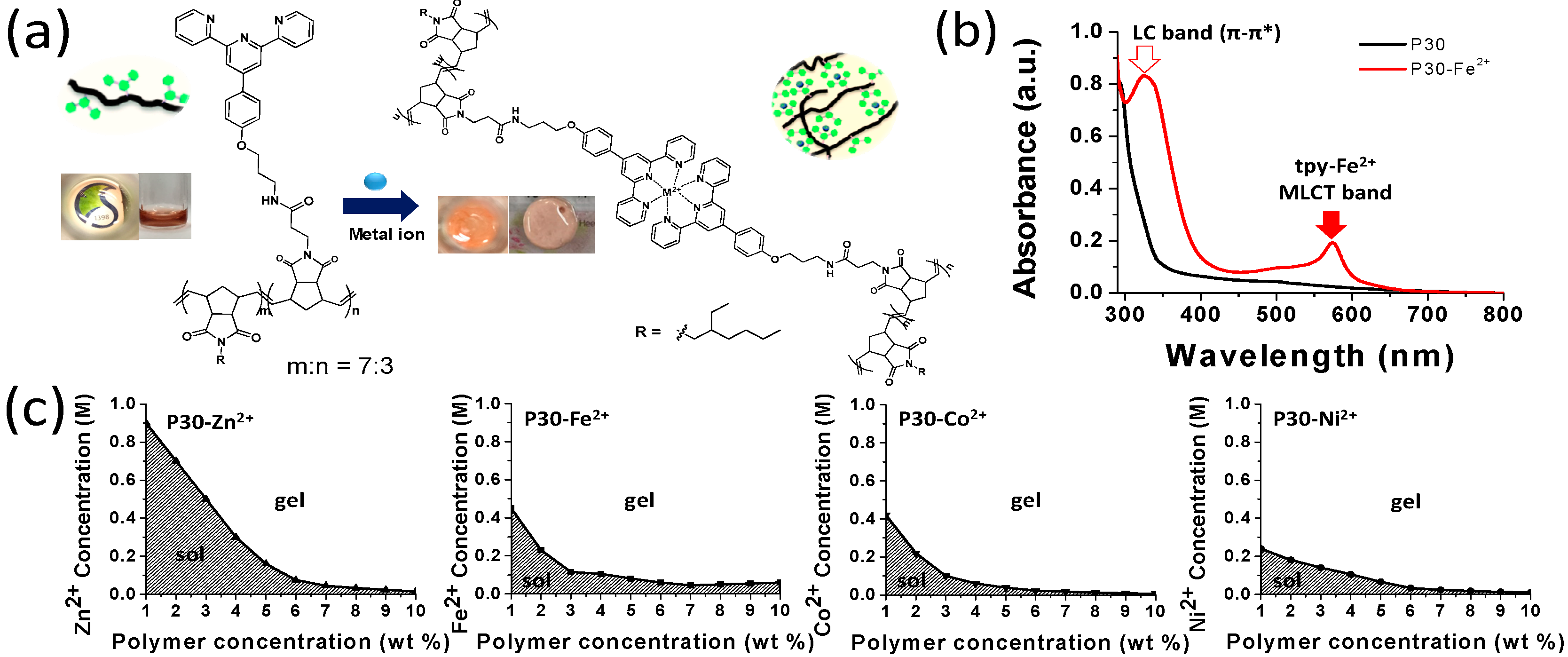
We investigated the sol-gel transition and self-healing properties using terpyridine-functionalized polynorbornenes. When a selected transition metal perchlorate (Fe(ClO
4)
2, Co(ClO
4)
2, Ni(ClO
4)
2, or Zn(ClO
4)
2) was added to each polymer solution of P30, gelation occurred because of the terpyridine moiety in the polymer combined with the metal ions (
Figure 1a). The UV-vis spectroscopic measurements confirmed that a metal–ligand coordination was indeed formed; ligand planarization and metal-to-ligand charge-transfer (MLCT) bands were observed (
Figure 1b). The MLCT bands of the transition metal complexes resulted from the shift in charge density from a molecular orbital with a metal-like characteristic to one with a ligand-like characteristic. In fact, in most cases, a metal-ion induced self-assembly was readily identified, through a visual inspection, owing to the strong coloration of the solution upon metal-ion coordination. For instance, the Fe(II)-terpyridine complex showed a new absorption, at approximately 580 nm, which can be attributed to the MLCT band. As shown in
Figure 1b, upon the addition of Fe
2+, the MLCT band (~580 nm) was clearly observed. In addition, the ligand-centered (LC) band was shifted from ~280 to ~340 nm, owing to the planarization of the terpyridine ligand, upon the binding of Fe
2+ ions.
To determine the gelation conditions depending on the polymer concentration and type of metal ions, we prepared metal complex gels with varying concentrations of polynorbornene P30 and metal perchlorates (Fe
2+, Co
2+, Ni
2+, and Zn
2+) (
Figure 1c). Each metal ion was continuously added to a given concentration of P30 (e.g., 1 wt % in DMF), up to the point at which the sol began to be converted into a gel. Other polymer solutions of different concentrations (2 to 10 wt %) were subjected to the same procedure described above. As shown in
Figure 1c, each gelation point, at which the polymer sol sate was converted into a gel without a flow, was connected, and the shaded areas under the line indicate the sol state of the polymer solution, whereas the area above the line represents the gel state.
When the metal ions Fe
2+, Ni
2+, and Co
2+ were added to a polymer solution, the sol state was rapidly converted into a gel through metal–ligand interactions. In contrast, in the case of the Zn
2+ ions, it took a relatively long time to obtain a Zn-complex gel, and required a larger amount of zinc ions for the given polymer concentration. We suspect that these trends are related to the metal binding affinity. The generally known binding affinity of metal ions to terpyridine occurs in the order of Fe
2+ > Ni
2+ > Co
2+ > Zn
2+ [
22]. In this study, the Zn
2+ ions appeared to have the weakest affinity, among the metal-bis-terpyridine complex. We confirmed that the larger the binding constant between terpyridine and the metal ion, the smaller the concentration of the metal ions necessary for the sol-gel transition. In addition, a faster sol-gel transition was observed for the metal ions with a higher binding constant. These observations showed that a sufficient amount of ligands and metal ions are required to form a gel, and the different metal–ligand affinity directly affects the metallogel generation. When we measured the storage moduli (G’) using a rheometer (
Figure S6), the G’ of the Fe-complex gel appeared to be higher than that of the Zn-complex gel because of the stronger binding affinity of Fe
2+ to terpyridine, than to Zn
2+. However, the stronger binding strength might have reduced the rapid and reversible exchange of metal–ligand interactions, which would have hindered the fast self-healing property. Therefore, we carried out investigations on self-healing by Zn
2+, since zinc bis-terpyridine complexes exist in dynamic equilibrium with mono-complexes and, thus, exchange rapidly [
2,
23].
We investigated the self-healing properties of the metallogels and found that the Zn-complex gel of P30 showed a good self-healing behavior through metal–ligand interactions. The higher content of terpyridine in polynorbornene (P50) rendered the metallogels rather compact and less flexible, which was consistent with the higher
Tg of P50 (
Table 1).
The stronger metal–ligand interaction (metallogels with Fe
2+, Ni
2+, or Co
2+) also results in a decrease in the flexibility of the polymer chain and the self-exchange rate of the metal–ligand complex [
20]. These results might indicate that, for self-healing, it is very important to ensure the molecular flexibility of polynorbornenes, which could be controlled based on the ratio of terpyridine and the choice of metal ions.
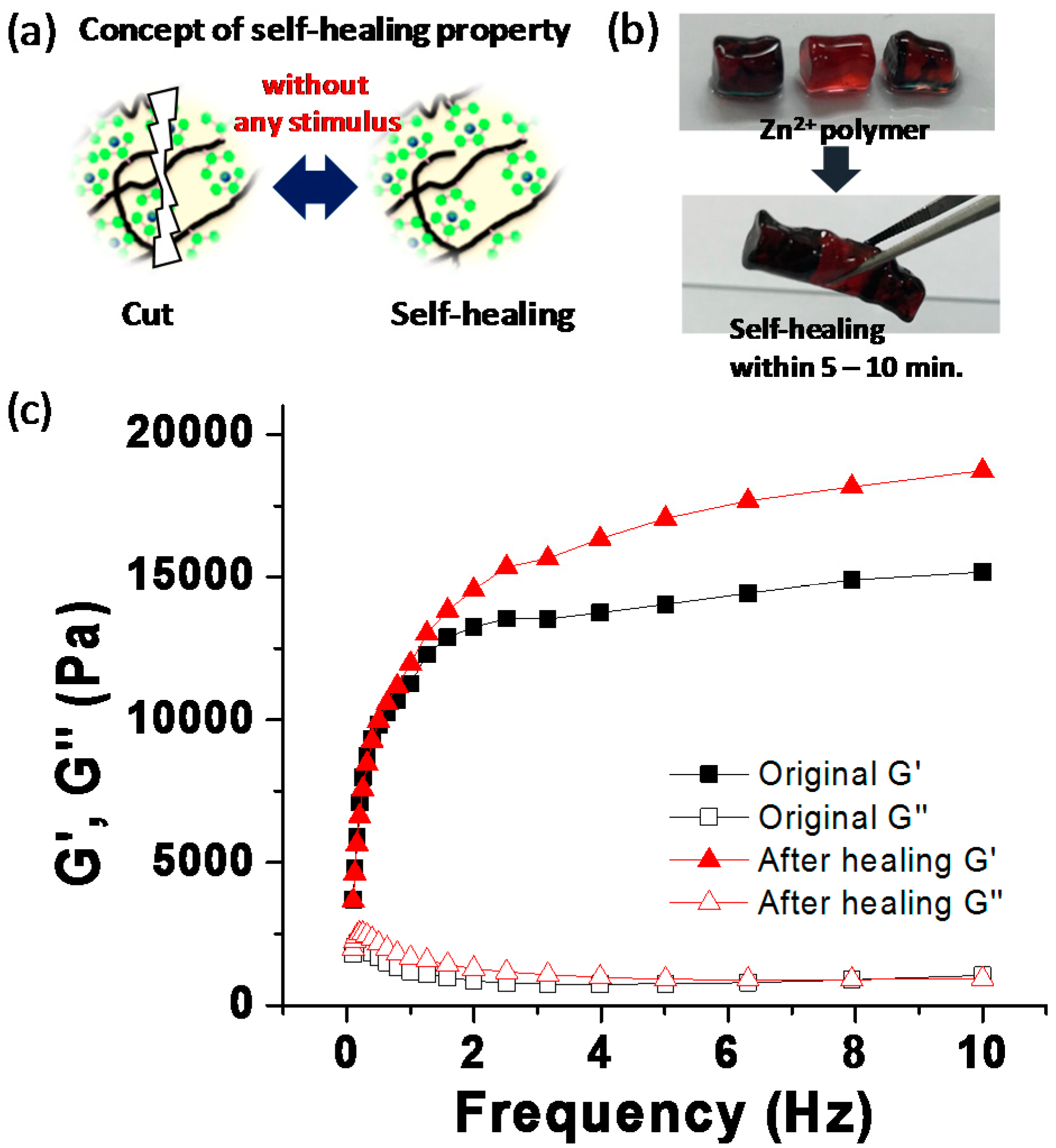
The self-healing performance was tested using a Zn-complex gel, which was cut into three pieces. The middle one was stained with allura red AC, and the other pieces were dyed with disperse blue 3 for easy visualization. When putting the pieces together, the gel was healed completely into a single piece, after 5–10 min at room temperature, without any external stimulus (
Figure 2b). To demonstrate the self-healing behaviors of the Zn-complex gel, we conducted rheological tests. The storage (G’) and loss (G’’) moduli of the original Zn-complex gel and the gel, after experiencing the cutting–healing process were measured under a fixed strain of 20%, within a frequency range of 0.1 to 10 Hz. As shown in
Figure 2c, the healed gel showed nearly the same values of G’ and G’’ as the original Zn-complex gel, which indicated a good self-healing capacity. These results suggested that the Zn-complex gel from P30 exhibited a rapid recovery in the presence of a dynamic metal–ligand interaction.
In order to compare the effect of counter-ions (anions), we investigated the gelation and self-healing properties from P30 and various Zn salts, with several counter-ions (ClO
4−, CN
−, Cl
−, SO
42−, Br
−). It should be noticed that all other conditions were exactly same, except for the type of the counter-ion. As shown in
Figure S7, the gelation time seemed to be greatly affected by the type of counter-ions, and accordingly, the time needed for self-healing. Although the trend was not completely accordance with the reported binding affinity of anions to Zn
2+ [
24], we confirmed that, in general, strong binding anions retarded the gel formation and self-healing, while weak or no binding species resulted in faster responses. For example, the gelation was instant and the self-healing was the fastest with ClO
4−, the anion of the weakest binding affinity. On the other hand, the stronger binding SO
42− showed rather slow gelation and self-healing. Interestingly, we found that the gelation with bromide was not effective and resulted in a very sticky, gum-like solid. We found indeed that the type of anions (counter-ions) significantly affected the gelation and the self-healing property, presumably due to the differences in binding strength to Zn
2+, size, and polarizability of the anions, although the exact mechanism needs further investigation.






{kind=link}
{kind=link}
{kind=link}
{kind=link}