Thermal and Calorimetric Evaluations of Polyacrylonitrile Containing Covalently-Bound Phosphonate Groups


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Unsaturated Acrylic Phosphonates
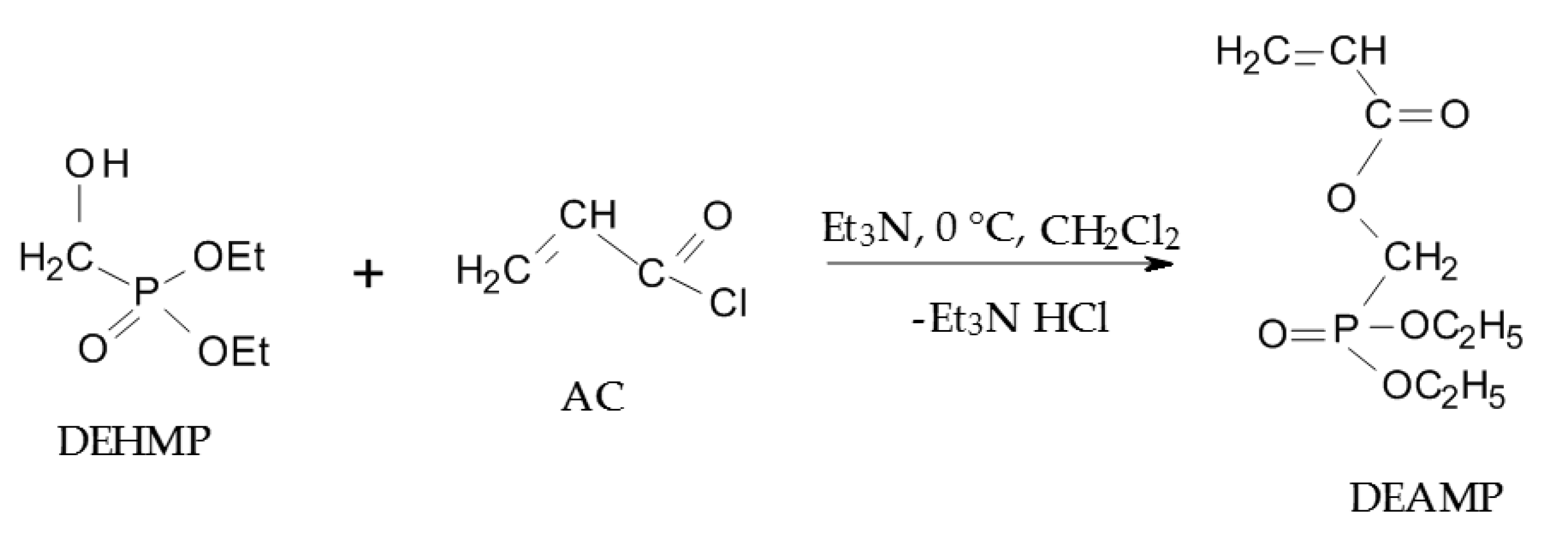
2.2.1. Synthesis of Diethyl(acryloyloxymethyl)phosphonate (DEAMP)
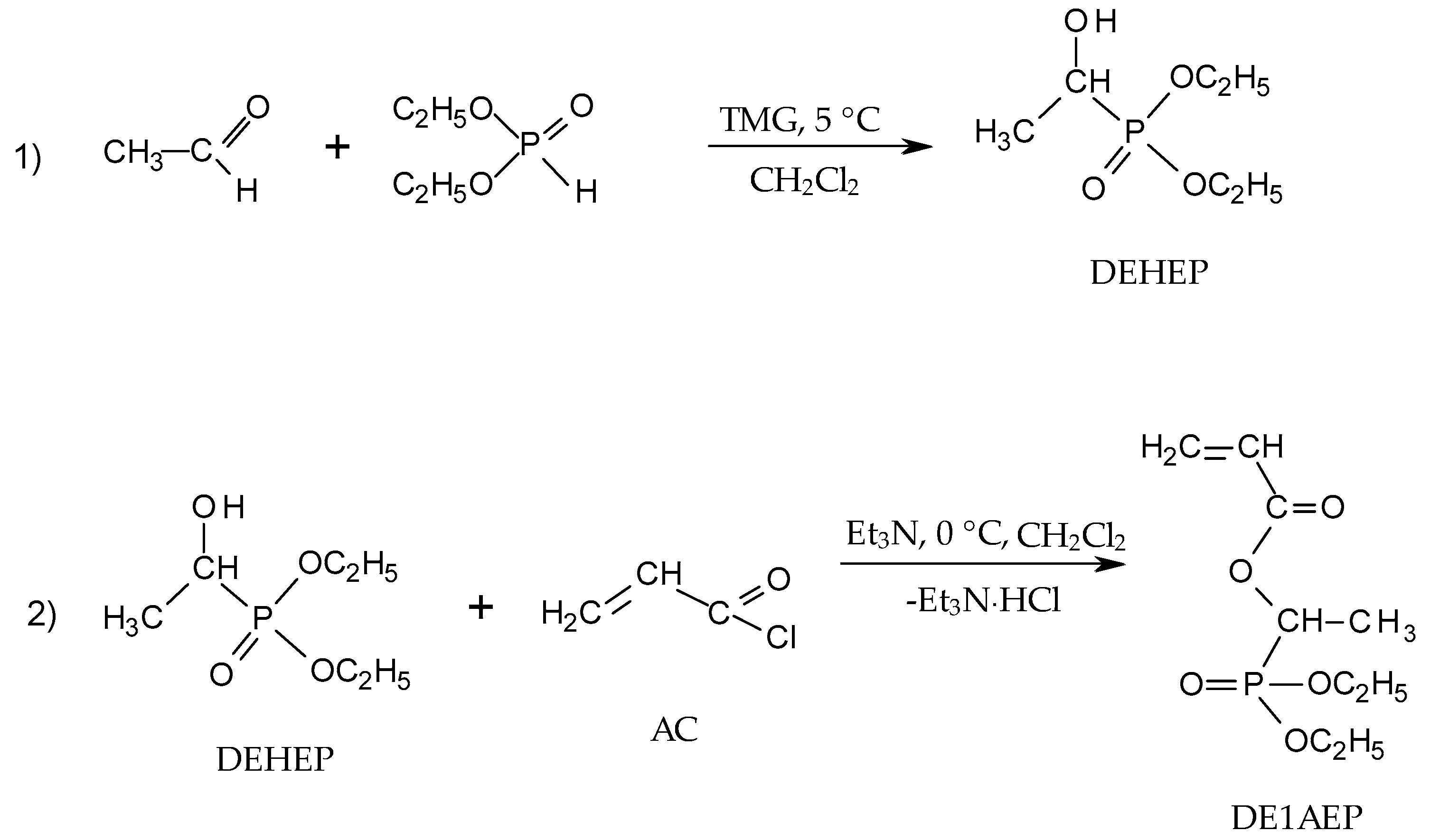
2.2.2. Synthesis of Diethyl(1-acryloyloxyethyl)phosphonate (DE1AEP)
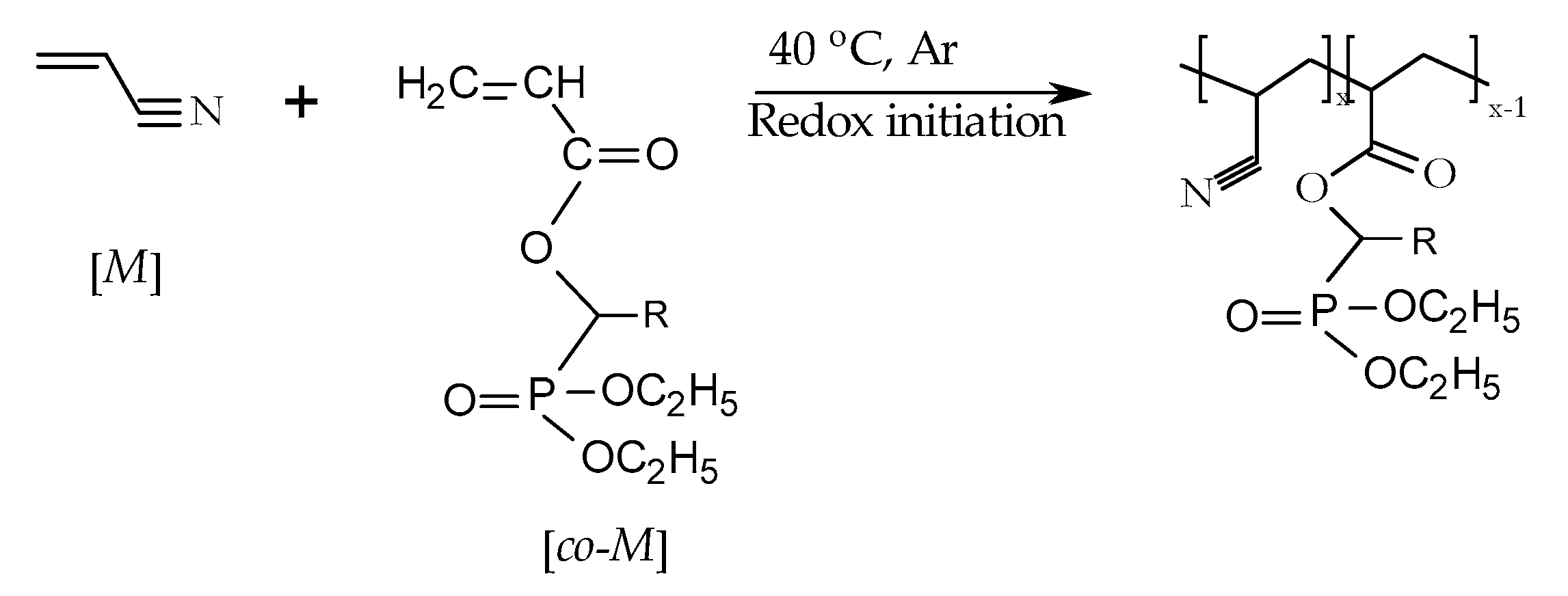
2.3. Chemical Modification of Polyacrylonitrile (PAN)
2.4. Characterisation Techniques
3. Results and Discussion
3.1. Synthetic Approaches
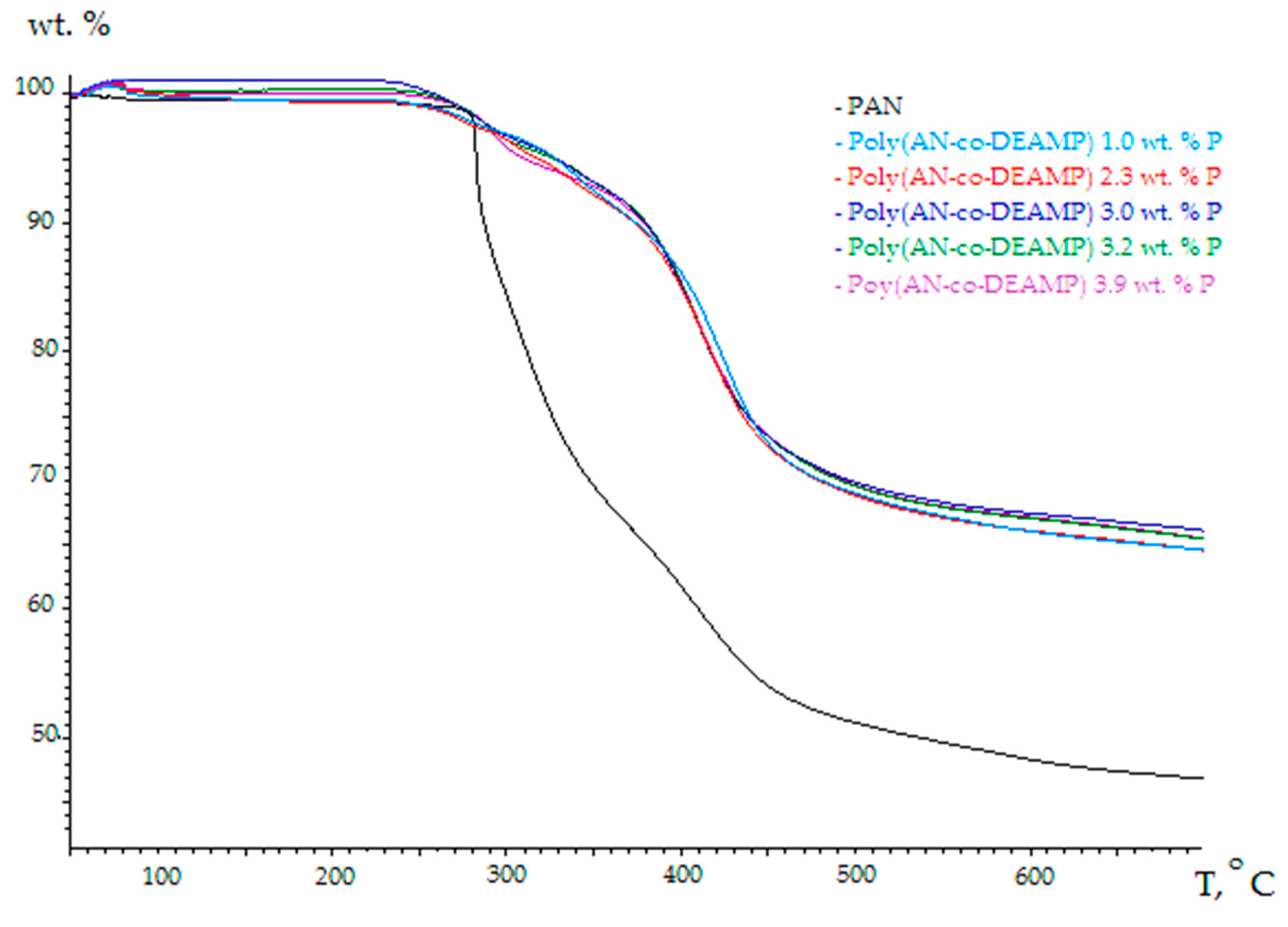
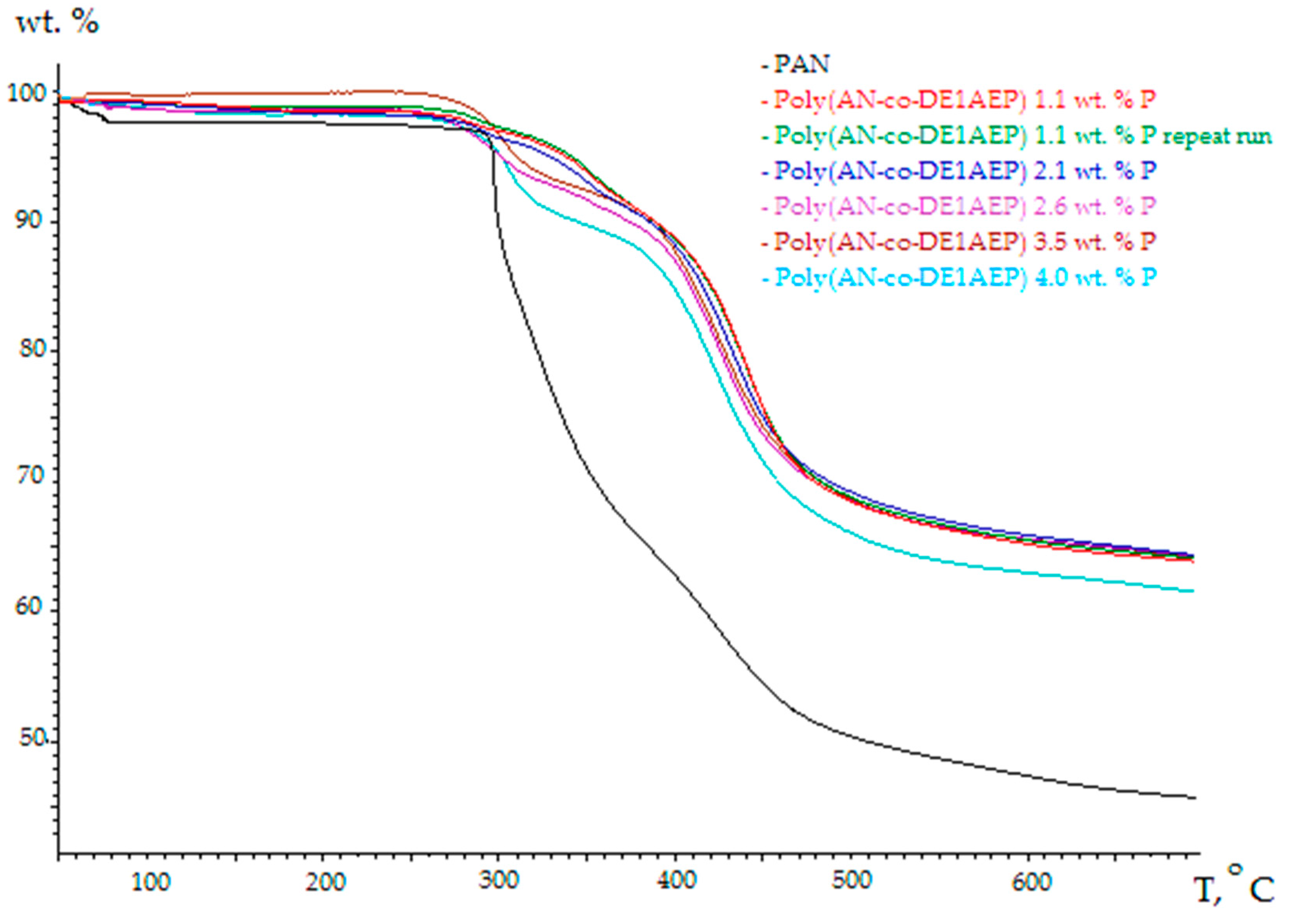
3.2. Thermo-Gravimetric Analyses (TGA)
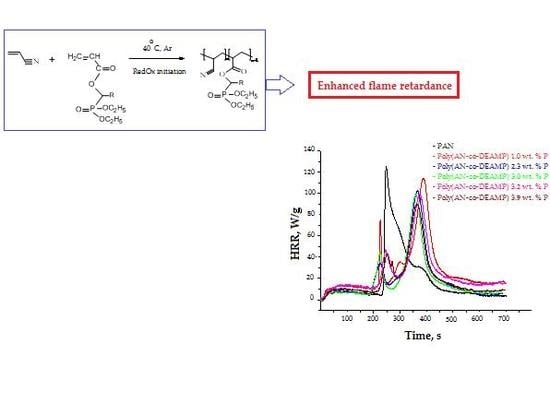
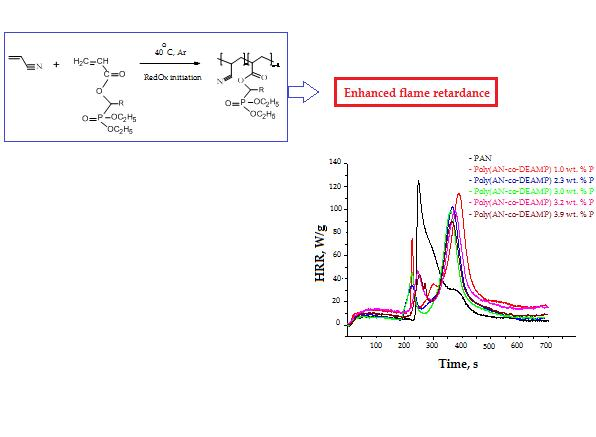
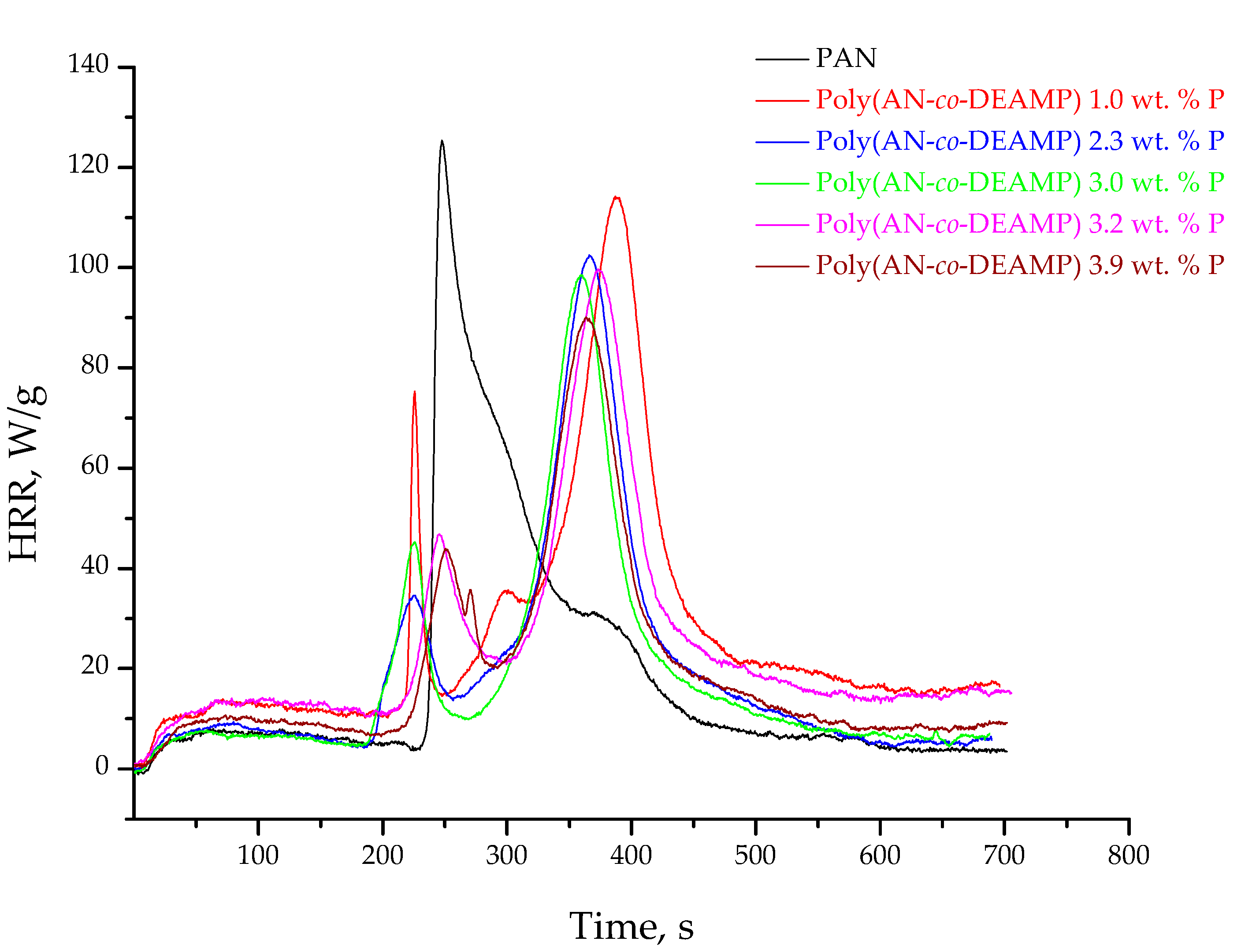
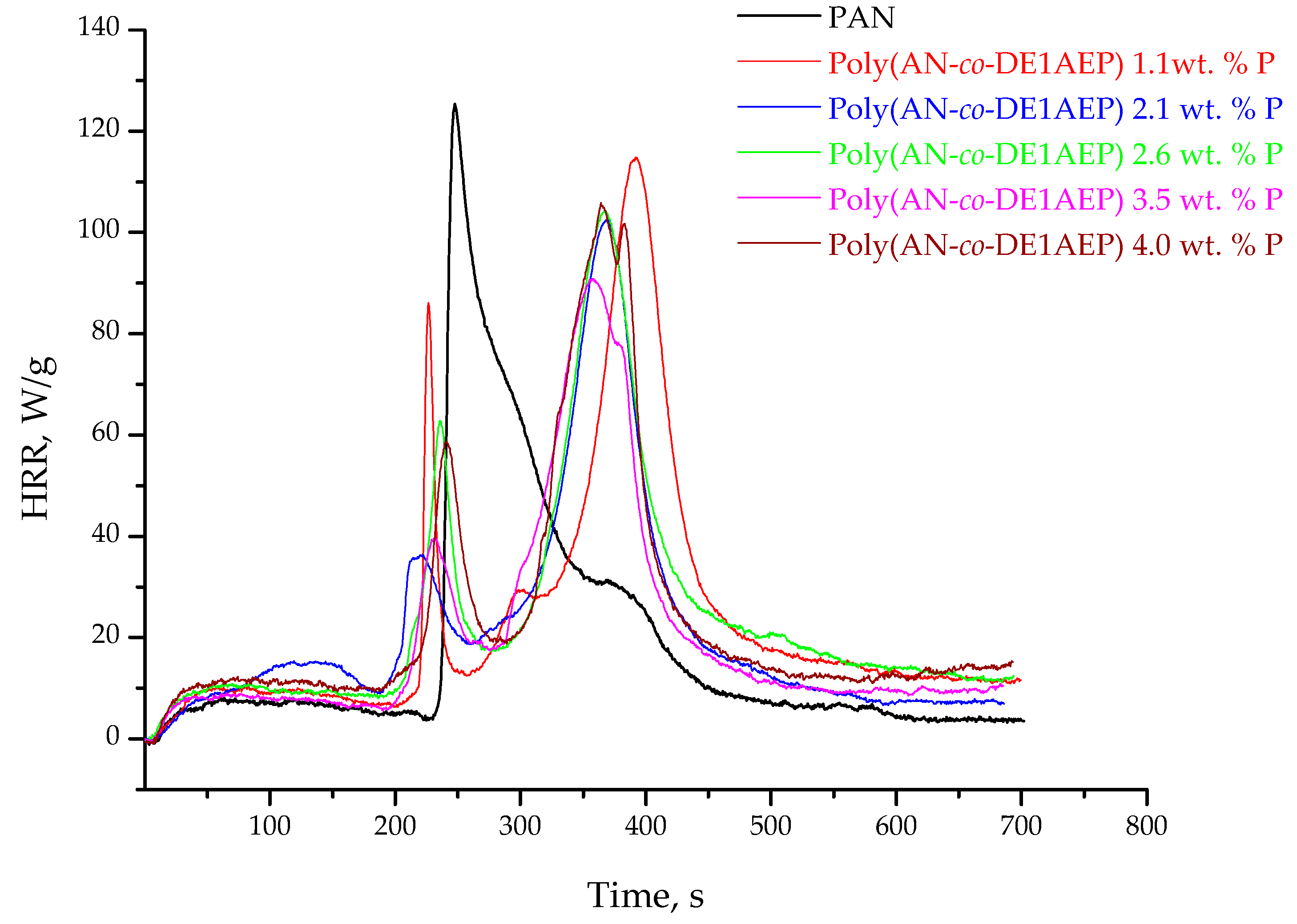
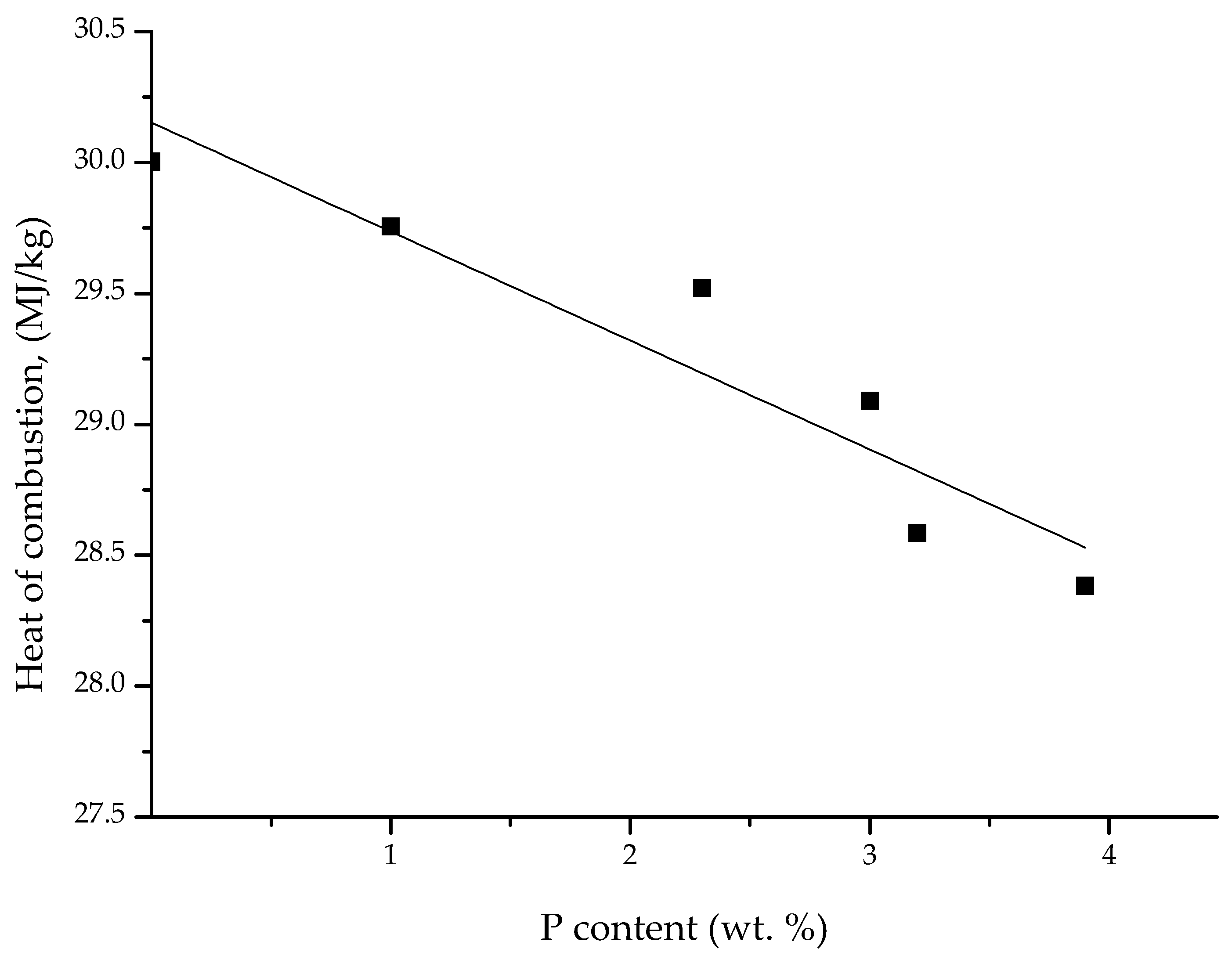
3.3. Pyrolysis Combustion Flow Calorimetric Evaluations
3.4. Mechanistic Aspects of Flame Retardance
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Joseph, P.; Tretsiakova-McNally, S. Mechanistic aspects of flame retardation by phosphorus-containing groups of some chain growth polymers. In Fire and Polymers VI: New Advances in Flame Retardant Chemistry and Science; Morgan, A.B., Wilkie, C.A., Nelson, G.L., Eds.; American Chemical Society: Washington, DC, USA, 2012; pp. 38–50. ISBN 978-0-8412-2780-4. [Google Scholar]
- Hall, M.E.; Horrocks, A.R.; Zhang, J. The flammability of polycrylonitirle and its copolymers. Polym. Degrad. Stab. 1994, 44, 379–386. [Google Scholar] [CrossRef]
- Joseph, P.; Ebdon, J.R. Flame-retardant polyester and polyamide textiles. In Polyesters and Polyamides; Deopura, B.L., Alagiruswamy, R., Joshi, M., Gupta, B., Eds.; Woodhead Publishing Limited: Cambridge, UK, 2008; pp. 306–324. ISBN 978-1-84569-298-8. [Google Scholar]
- Holder, K.M.; Smith, R.J.; Grunlan, J.C. A review of flame retardant nanocoatings prepared using layer-by-layer assembly of polyelectrolytes. J. Mater. Sci. 2017, 52, 12923–12959. [Google Scholar] [CrossRef]
- Carosio, F.; Alongi, J. Influence of layer by layer coatings containing octapropylammonium polyhedral oligomeric silsesquioxane and ammonium polyphosphate on the thermal stability and flammability of acrylic fabrics. J. Anal. Appl. Pyrolysis 2016, 119, 114–123. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Reactive modifications of some chain- and step-growth polymers with phosphoruscontaining compounds: Effects on flame retardance—A review. Polym. Advan. Technol. 2011, 22, 395–406. [Google Scholar] [CrossRef]
- Wyman, P.; Crook, V.; Ebdon, J.; Hunt, B.; Joseph, P. Flame-retarding effects of dialkyl-p-vinylbenzyl phosphonates in copolymers with acrylonitrile. Polym. Int. 2006, 55, 764–771. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Combustion behaviours of chemically modified polyacrylonitrile polymers containing phosphorylamino groups. Polym. Degrad. Stab. 2012, 97, 2531–2535. [Google Scholar] [CrossRef]
- Tretsiakova-McNally, S.; Joseph, P. Pyrolysis Combustion Flow Calorimetry studies on some reactively modified polymers. Polymers 2015, 7, 453–467. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Chemical modification of polyacrylonitrile (PAN) with phosphate groups: Effects on flame retardance. Polym. Mater. Sci. Eng. 2012, 106, 22–23. [Google Scholar]
- Armarego, W.L.F.; Chai, C.L. Purification of Laboratory Chemicals, 5th ed.; Elsevier Science: Cornwall, UK, 2003. [Google Scholar]
- Liepins, R.; Surles, J.R.; Morosoff, N.; Stannett, V.; Duffy, J.J.; Day, F.H. Localized radiation grafting of flame retardants to polyethylene terephthalate. II. Vinyl phosphonates. J. Appl. Polym. Sci. 1978, 22, 2403–2414. [Google Scholar] [CrossRef]
- Lyon, R.E.; Walters, R.N. Pyrolysis combustion flow calorimetry. J. Anal. Appl. Pyrolysis 2004, 71, 27–46. [Google Scholar] [CrossRef]
- Ebdon, J.R.; Hunt, B.J.; Joseph, P.; Wilkie, T.K. Flame retardance of polyacrylonitriles covalently modified with phosphorus- and nitrogen containing groups. In Fire Retardancy of Polymers: New Strategies and Mechanisms; Hull, T.R., Kandola, B.K., Eds.; Royal Society of Chemistry: Cambridge, UK, 2009; pp. 331–364. [Google Scholar]
- Ebdon, J.R.; Hunt, B.J.; Joseph, P.; Konkel, C.S.; Price, D.; Pyrah, K.; Hull, T.R.; Milnes, G.J.; Hill, S.B.; Lindsay, C.I.; et al. Thermal degradation and flame retardance in copolymers of methyl methacrylate with diethyl(methacryloyloxymethyl)phosphonate. Polym. Degrad. Stab. 2000, 70, 425–436. [Google Scholar] [CrossRef]
- Ebdon, J.R.; Price, D.; Hunt, B.J.; Joseph, P.; Gao, F.; Milnes, G.J.; Cunliffe, L.K. Flame retardance in some polystyrenes and poly(methyl methacrylate)s with covalently bound phosphorus-containing groups: Initial screening experiments and some laser pyrolysis mechanistic studies. Polym. Degrad. Stab. 2000, 69, 267–277. [Google Scholar] [CrossRef]
- Ebdon, J.R.; Huckerby, T.N.; Hunter, T.C. Free-radical aqueous slurry polymerizations of acrylonitrile: 1. End-groups and other minor structures in polyacrylonitriles initiated by ammonium persulfate/sodium metabisulfite. Polymer 1994, 35, 250–256. [Google Scholar] [CrossRef]
- Crook, V.; Ebdon, J.; Hunt, B.; Joseph, P.; Wyman, P. The influence of comonomers on the degradation and flammability of polyacrylonitrile: Design input for a new generation of flame retardants. Polym. Degrad. Stab. 2010, 95, 2260–2268. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comonomer | [M] 1 (g) | [co-M] 2 (g) | Ratio [M]/[co-M] in the Feed (Mole Fraction) | Na2S2O5/(NH4)2S2O8 | Water (cm3) | Yield (wt %) |
|---|---|---|---|---|---|---|
| - | 10.32 | - | 1.00/0.00 | 2.86 | 290 | 88 |
| DEAMP | 10.24 | 0.90 | 0.98/0.02 | 2.86 | 290 | 86 |
| DEAMP | 10.00 | 1.78 | 0.96/0.04 | 2.86 | 290 | 86 |
| DEAMP | 9.84 | 2.68 | 0.94/0.06 | 2.86 | 290 | 89 |
| DEAMP | 9.68 | 3.58 | 0.92/0.08 | 2.86 | 290 | 80 |
| DEAMP | 9.37 | 4.48 | 0.90/0.10 | 2.86 | 290 | 85 |
| DE1AEP | 10.24 | 0.96 | 0.98/0.02 | 2.86 | 290 | 84 |
| DE1AEP | 10.00 | 1.88 | 0.96/0.04 | 2.86 | 290 | 85 |
| DE1AEP | 9.84 | 2.88 | 0.94/0.06 | 2.86 | 290 | 83 |
| DE1AEP | 9.68 | 3.78 | 0.92/0.08 | 2.86 | 290 | 81 |
| DE1AEP | 9.37 | 4.76 | 0.90/0.10 | 2.86 | 290 | 80 |
| Polymer System | [co-M], Mole Fraction | P, wt % | TG Residue at 700 °C in Nitrogen, wt % | TG Residue at 700 °C in Air, wt % | TG Residue at 700 °C in Oxygen, wt % |
|---|---|---|---|---|---|
| PAN | 0 | 0 | 46.8 | 43.5 | 1.1 |
| Poly(AN-co-DEAMP) | 0.018 | 1.0 | 64.5 | 46.1 | 4.2 |
| Poly(AN-co-DEAMP) | 0.044 | 2.3 | 64.5 | 48.0 | 10.0 |
| Poly(AN-co-DEAMP) | 0.060 | 3.0 | 66.1 | 49.0 | 16.9 |
| Poly(AN-co-DEAMP) | 0.066 | 3.2 | 65.4 | 51.0 | 15.4 |
| Poly(AN-co-DEAMP) | 0.085 | 3.9 | 65.4 | 50.8 | 31.3 |
| Poly(AN-co-DE1AEP) | 0.020 | 1.1 | 65.3 | 50.4 | 2.6 |
| Poly(AN-co-DE1AEP) | 0.042 | 2.1 | 65.5 | 55.2 | 10.3 |
| Poly(AN-co-DE1AEP) | 0.052 | 2.6 | 65.5 | 49.7 | 15.1 |
| Poly(AN-co-DE1AEP) | 0.075 | 3.5 | 65.3 | 46.9 | 20.2 |
| Poly(AN-co-DE1AEP) | 0.089 | 4.0 | 62.6 | 45.5 | 31.0 |
| Polymer System | [co-M], Mole Fraction | P, wt % | TPHRR 1, °C | PHRR, W/g | THR, kJ/g | HRC, kJ/g·K | PCFC Char Yield, wt % |
|---|---|---|---|---|---|---|---|
| PAN | 0 | 0 | 289 | 182.7 | 14.9 | 200.7 | 45.6 |
| Poly(AN-co-DEAMP) | 0.018 | 1.0 | 429 | 112.2 | 17.5 | 121.9 | 58.4 |
| Poly(AN-co-DEAMP) | 0.044 | 2.3 | 417 | 102.3 | 13.6 | 110.9 | 57.1 |
| Poly(AN-co-DEAMP) | 0.060 | 3.0 | 412 | 99.1 | 14.8 | 107.2 | 58.7 |
| Poly(AN-co-DEAMP) | 0.066 | 3.2 | 409 | 99.0 | 16.1 | 107.5 | 57.1 |
| Poly(AN-co-DEAMP) | 0.085 | 3.9 | 406 | 93.5 | 14.4 | 101.5 | 61.2 |
| Poly(AN-co-DE1AEP) | 0.020 | 1.1 | 432 | 121.8 | 16.8 | 131.7 | 55.8 |
| Poly(AN-co-DE1AEP) | 0.042 | 2.1 | 418 | 103.3 | 15.0 | 112.0 | 53.8 |
| Poly(AN-co-DE1AEP) | 0.052 | 2.6 | 412 | 96.7 | 15.9 | 105.5 | 60.1 |
| Poly(AN-co-DE1AEP) | 0.075 | 3.5 | 409 | 96.9 | 14.7 | 105.3 | 56.2 |
| Poly(AN-co-DE1AEP) | 0.089 | 4.0 | 411 | 93.4 | 15.5 | 102.1 | 56.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tretsiakova-McNally, S.; Joseph, P. Thermal and Calorimetric Evaluations of Polyacrylonitrile Containing Covalently-Bound Phosphonate Groups. Polymers 2018, 10, 131. https://doi.org/10.3390/polym10020131
Tretsiakova-McNally S, Joseph P. Thermal and Calorimetric Evaluations of Polyacrylonitrile Containing Covalently-Bound Phosphonate Groups. Polymers. 2018; 10(2):131. https://doi.org/10.3390/polym10020131
Chicago/Turabian StyleTretsiakova-McNally, Svetlana, and Paul Joseph. 2018. "Thermal and Calorimetric Evaluations of Polyacrylonitrile Containing Covalently-Bound Phosphonate Groups" Polymers 10, no. 2: 131. https://doi.org/10.3390/polym10020131
APA StyleTretsiakova-McNally, S., & Joseph, P. (2018). Thermal and Calorimetric Evaluations of Polyacrylonitrile Containing Covalently-Bound Phosphonate Groups. Polymers, 10(2), 131. https://doi.org/10.3390/polym10020131