Thermal and Mechanical Characterization of EMA-TEGDMA Mixtures for Cosmetic Applications


 ,
,  , and
, and 
Abstract
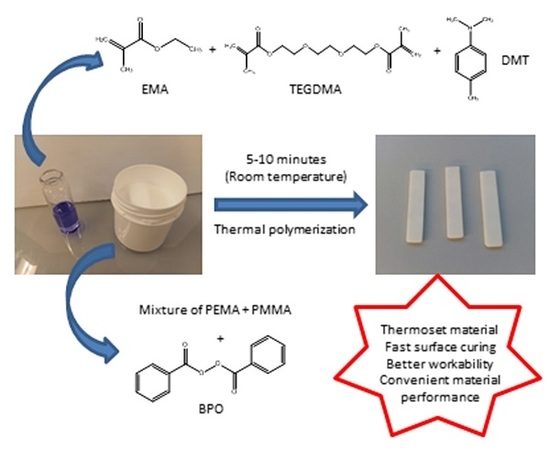
:
1. Introduction
2. Experimental Section
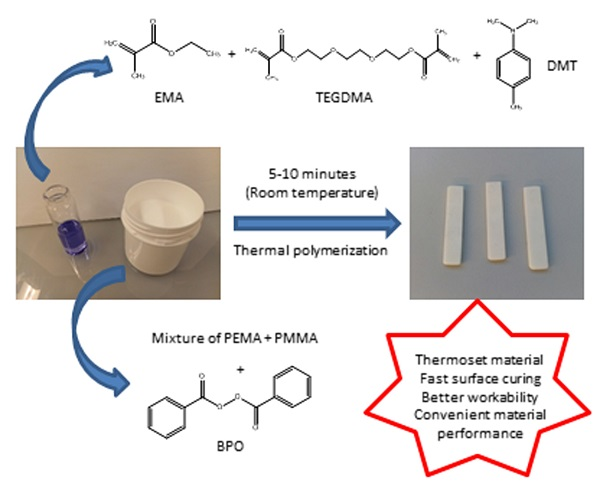
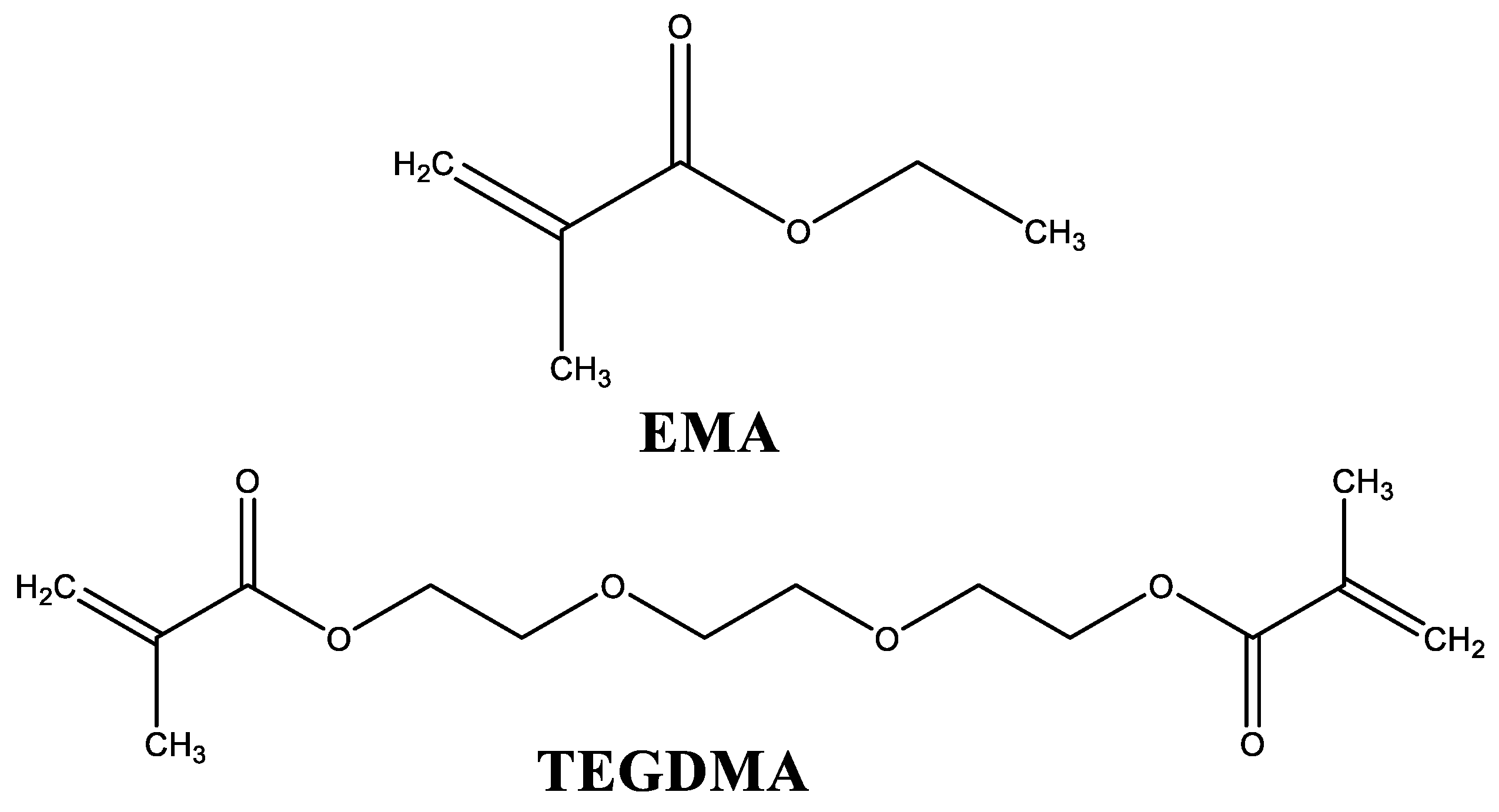
2.1. Materials
2.2. Experimental Procedures
2.2.1. Procedure for Checking the Curing Process for Liquid Formulations
- In a vial, about 200 mg of a liquid formulation + about 2 mg of BPO were weighted and quickly dissolved at room temperature;
- When the solution was complete, the curing process started, so this vial needed to be kept in an ice/salt bath at –15 °C in order to stop this polymerization process;
- While the DSC device was equilibrated at –50 °C, a sample of the solution was prepared in the crucible in order to introduce it quickly into the DSC oven;
- Finally, after sample introduction into the DSC oven, it was equilibrated at –50 °C a few minutes more and then a dynamic experiment was performed up to 200 °C, in order to check the curing process of the liquid mixture.
2.2.2. Procedure for Preparing Samples of Final Materials for Mechanical Testing
- In a vial, around 10 mL of each liquid formulation was dropped with around 6 g of POW;
- This mixture was stirred quickly at room temperature with a glass bar;
- When the solution achieved the dough state, the mixture was poured over the silicone mold in order to fill it completely;
- The mixture was kept at room temperature overnight in the silicone mold in order to complete the curing process of the materials.
2.3. Measurement Techniques
3. Results and Discussion
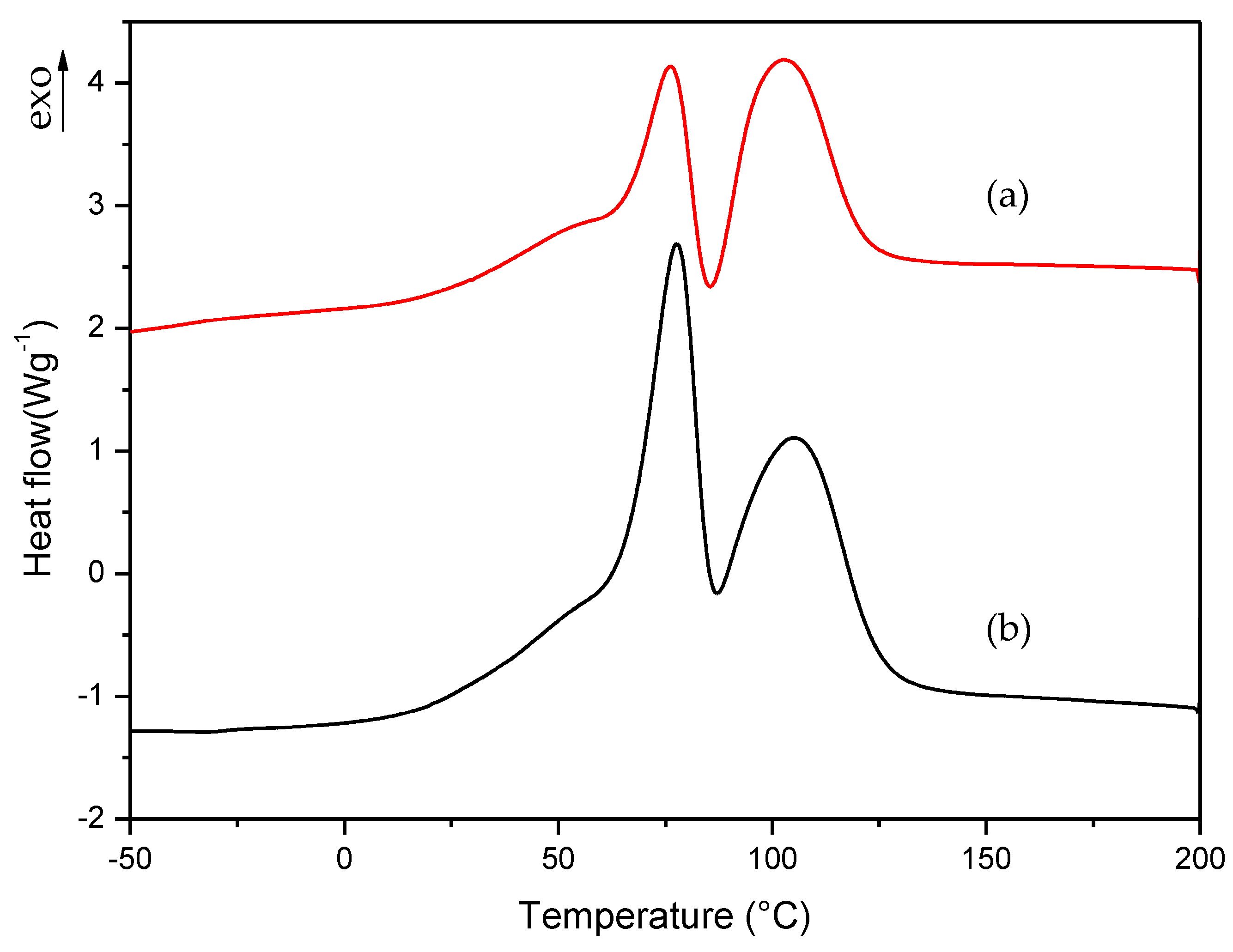
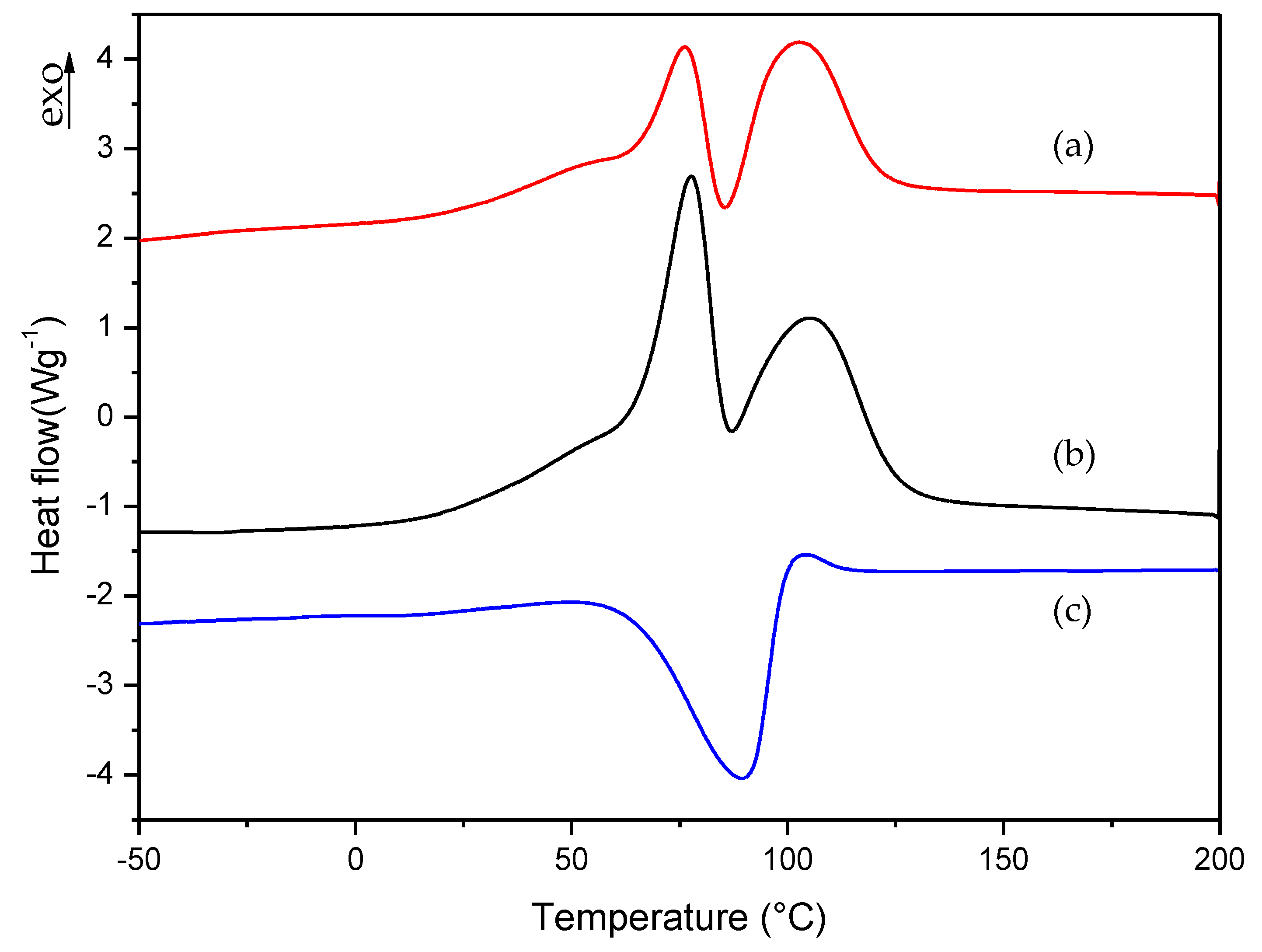
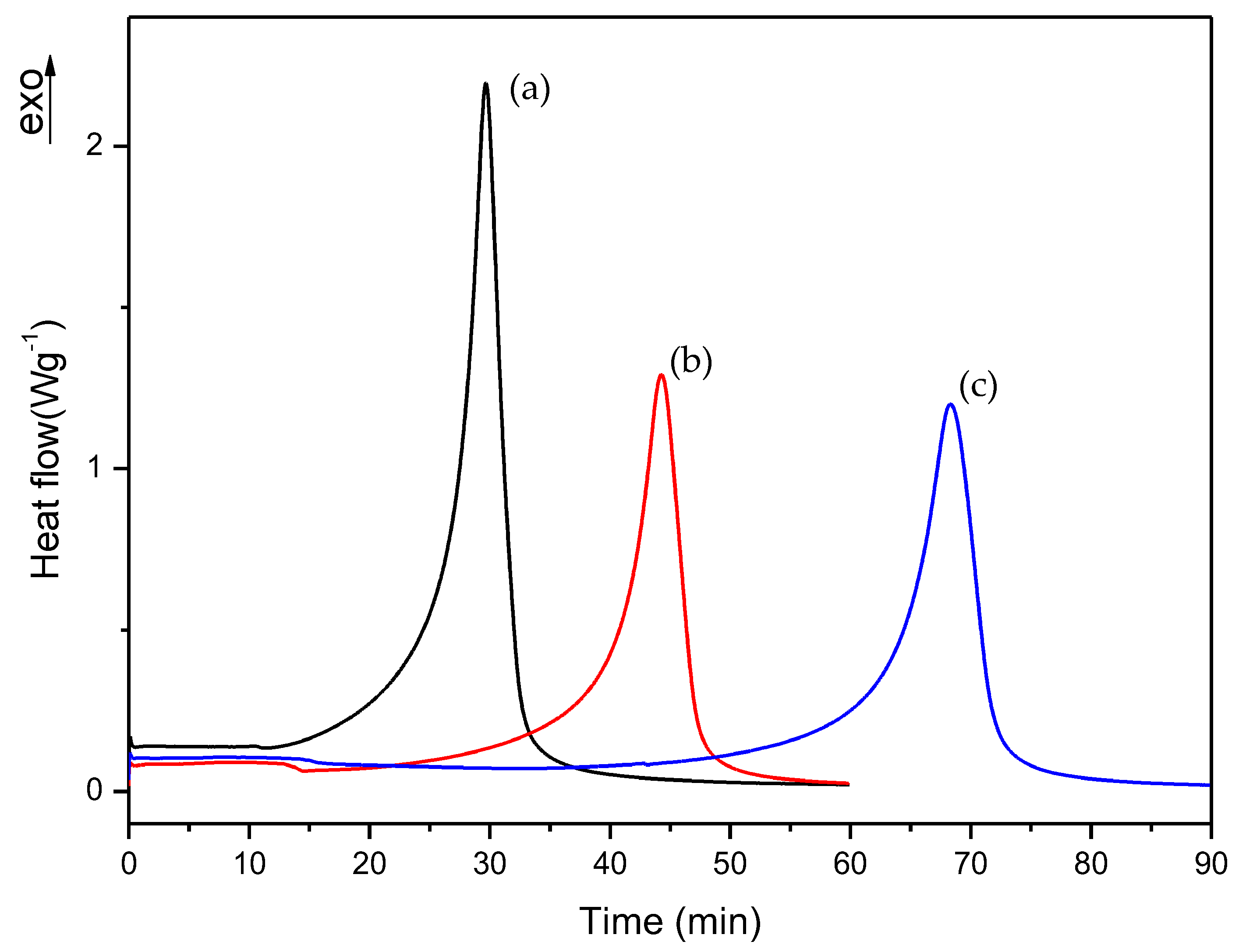
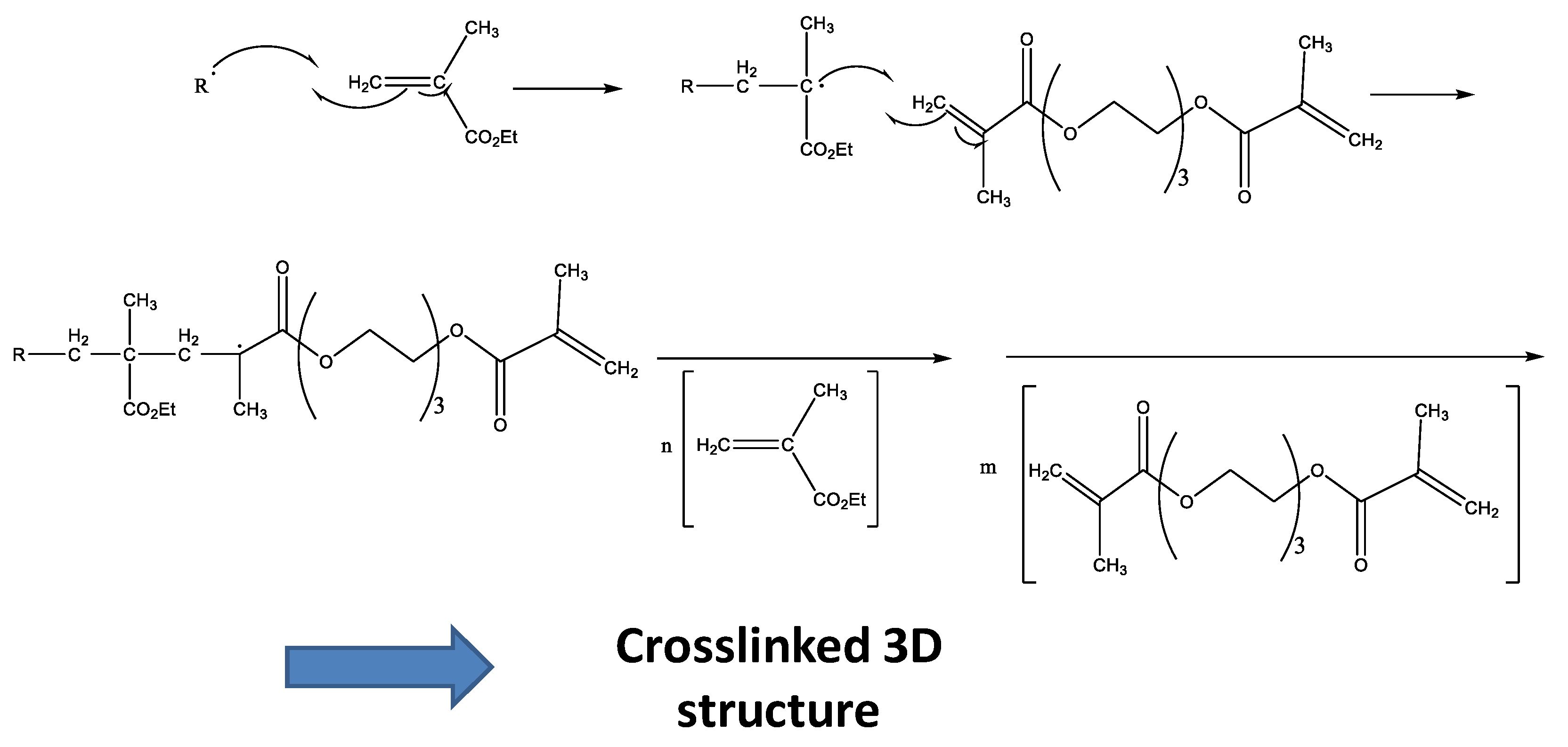
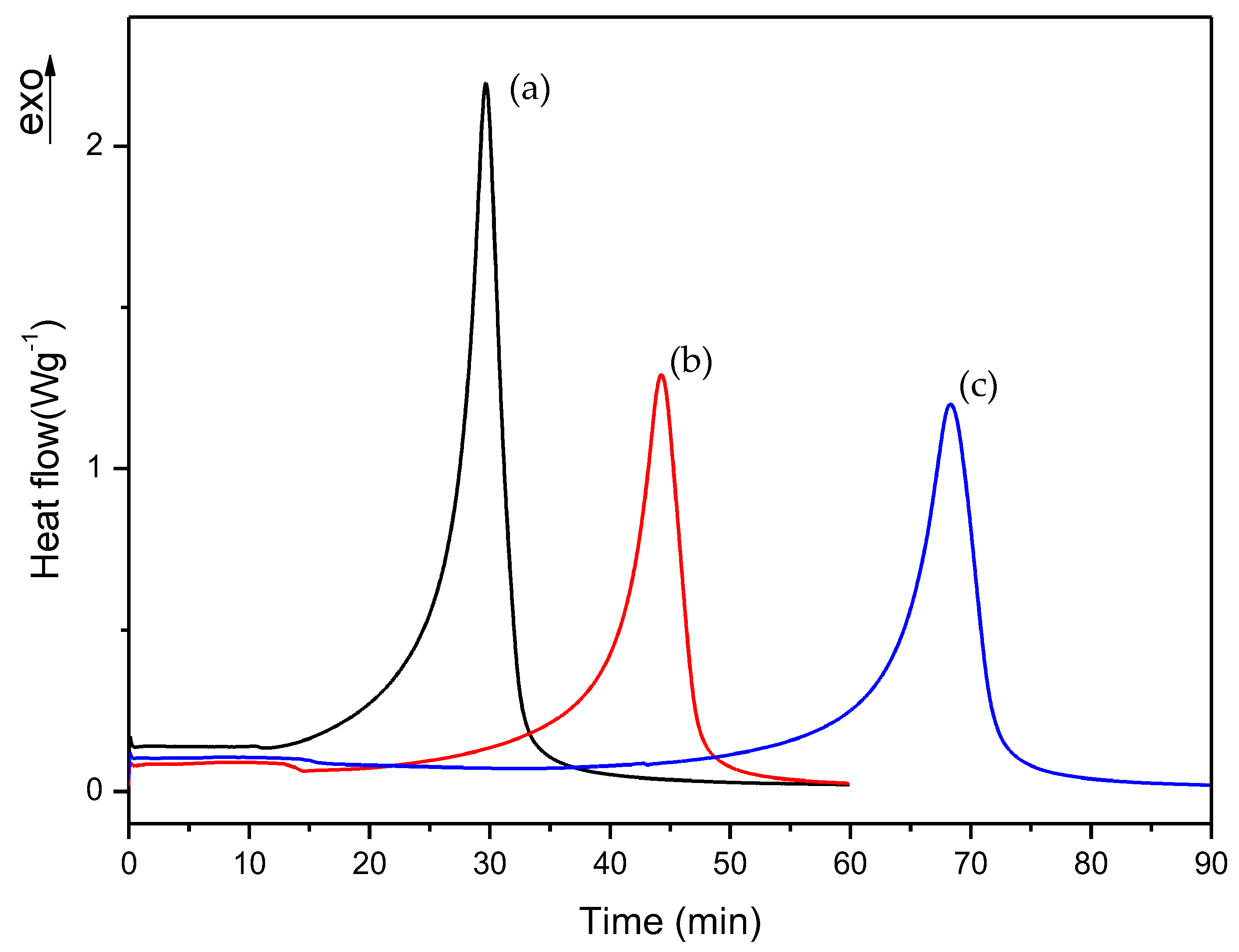
3.1. Dynamic DSC Study of the Thermal Curing Process for Liquid Formulations:
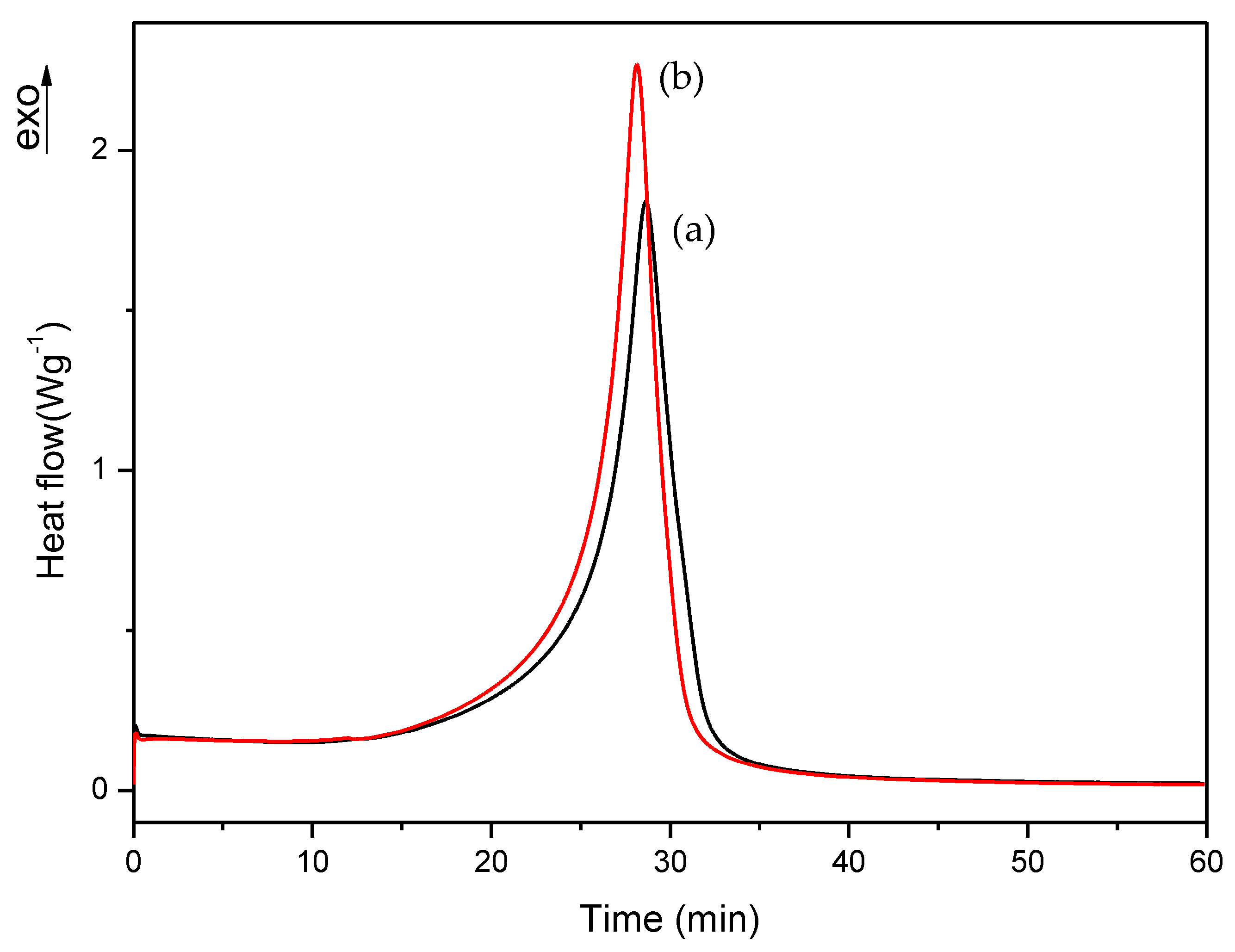
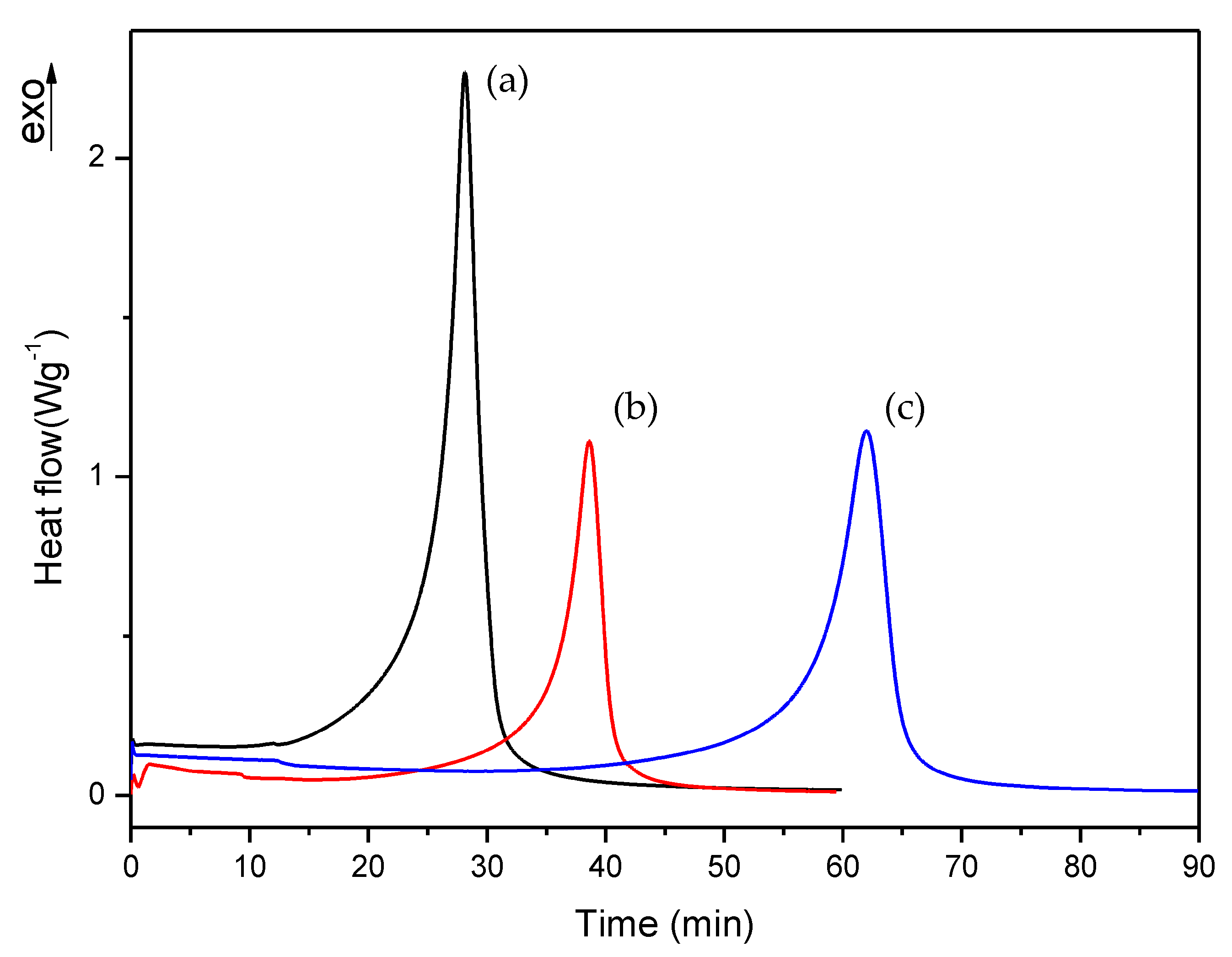
3.2. Isothermal DSC Study of the Thermal Curing Process for Liquid Formulations
3.3. Thermal and Mechanical Characterization of Materials Prepared with Each Liquid Formulation
3.3.1. Thermal Stability Studies of the Cured Materials by TGA
3.3.2. Thermo-Mechanical Characterization of the Cured Materials by DMTA
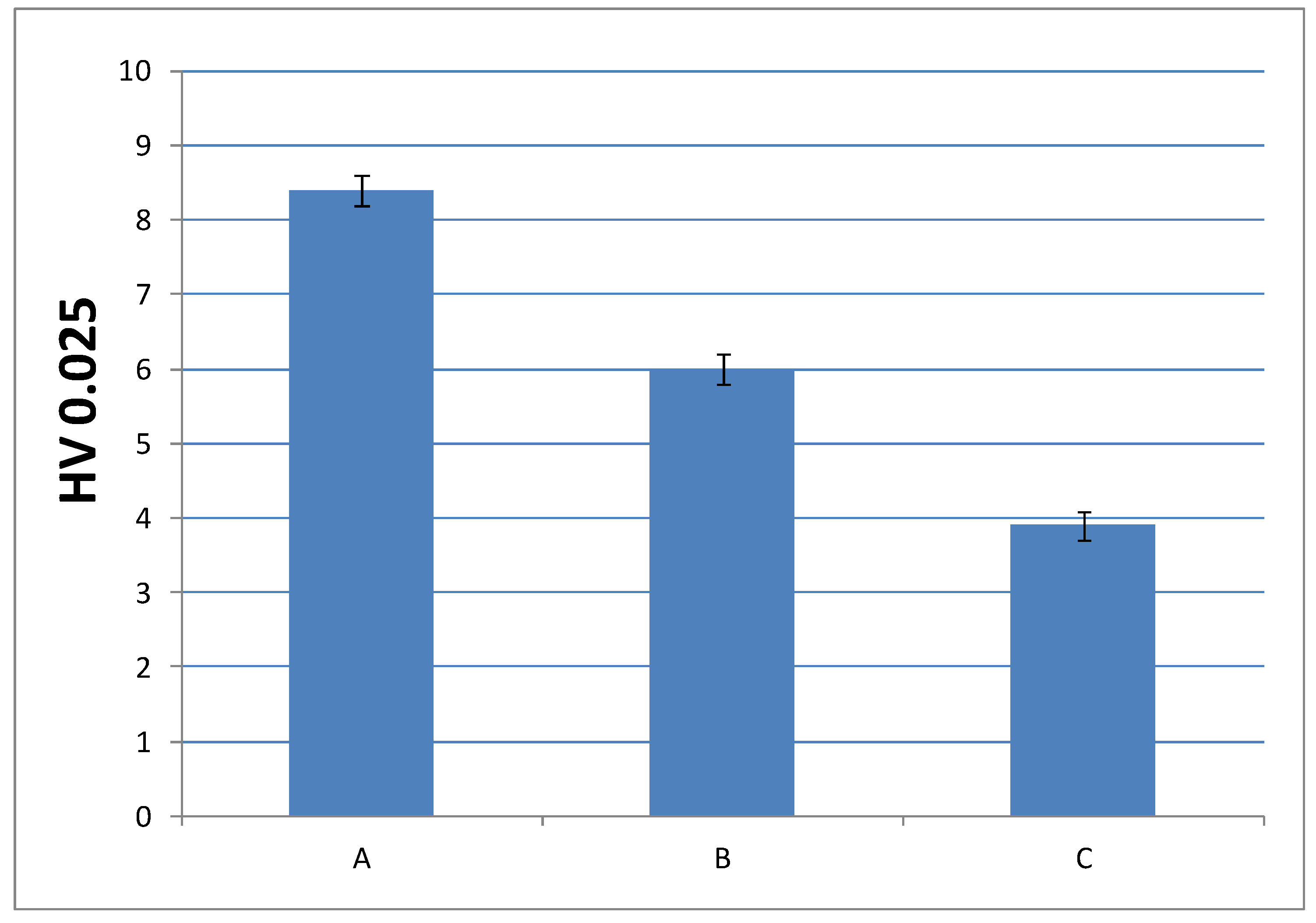
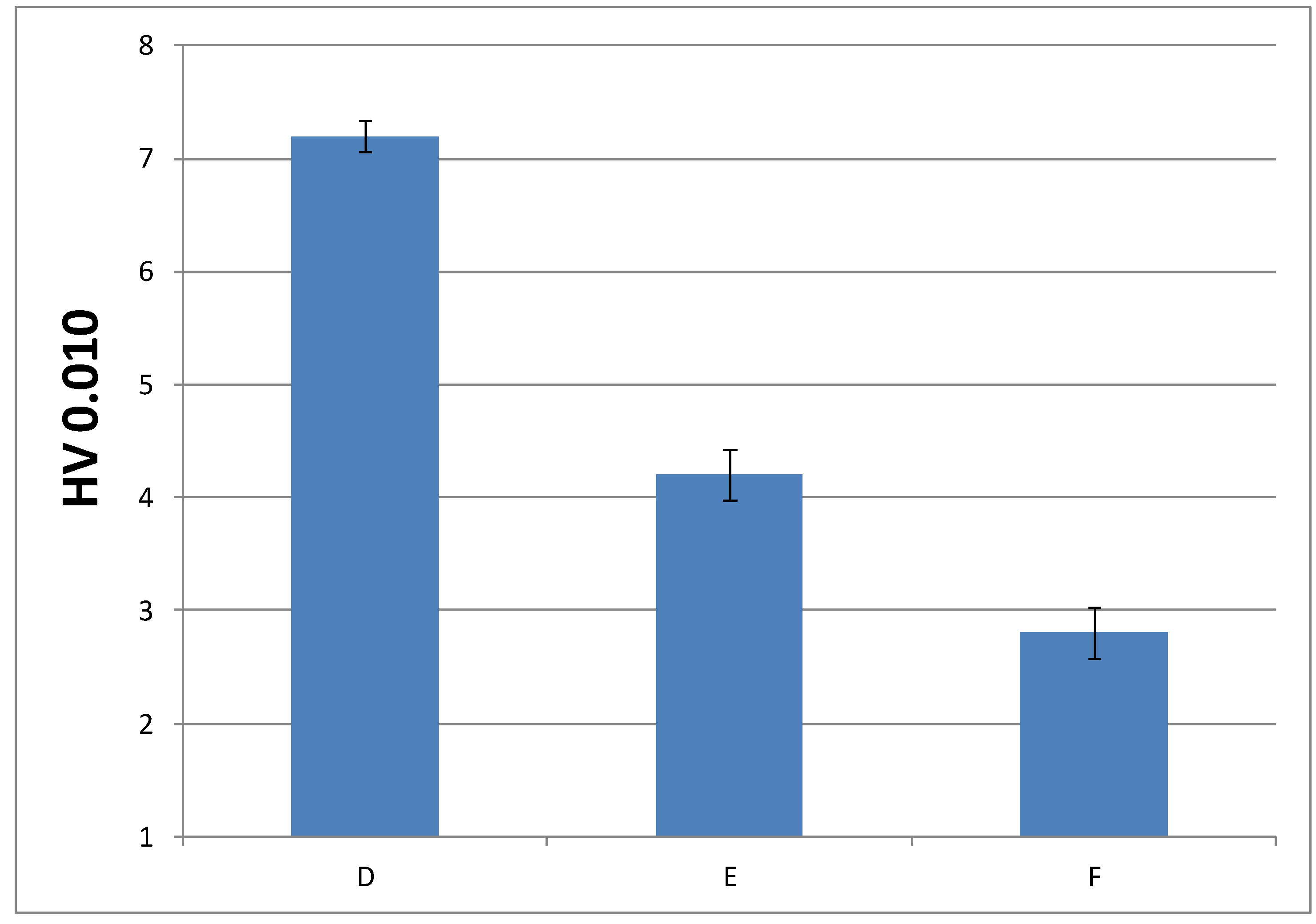
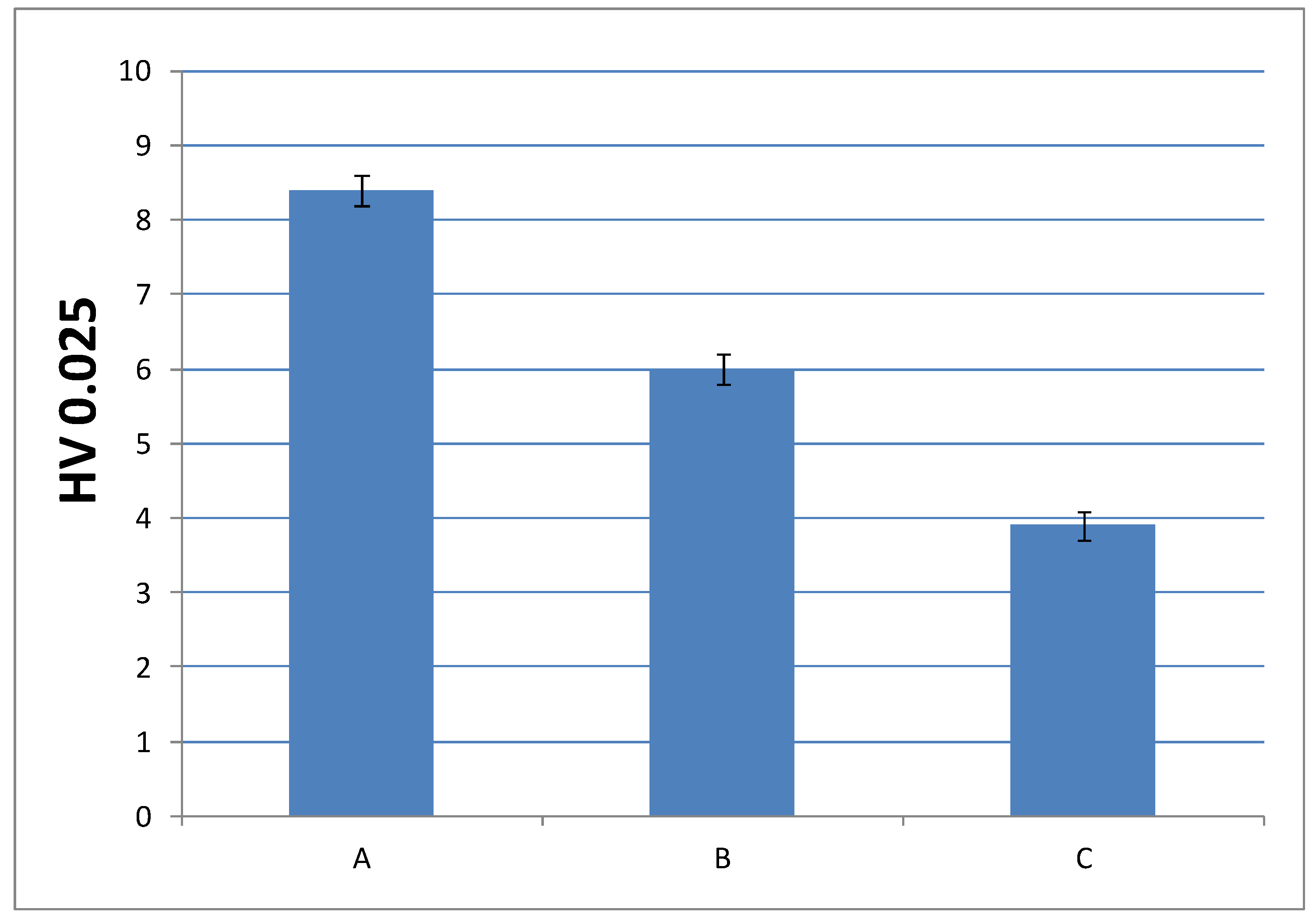
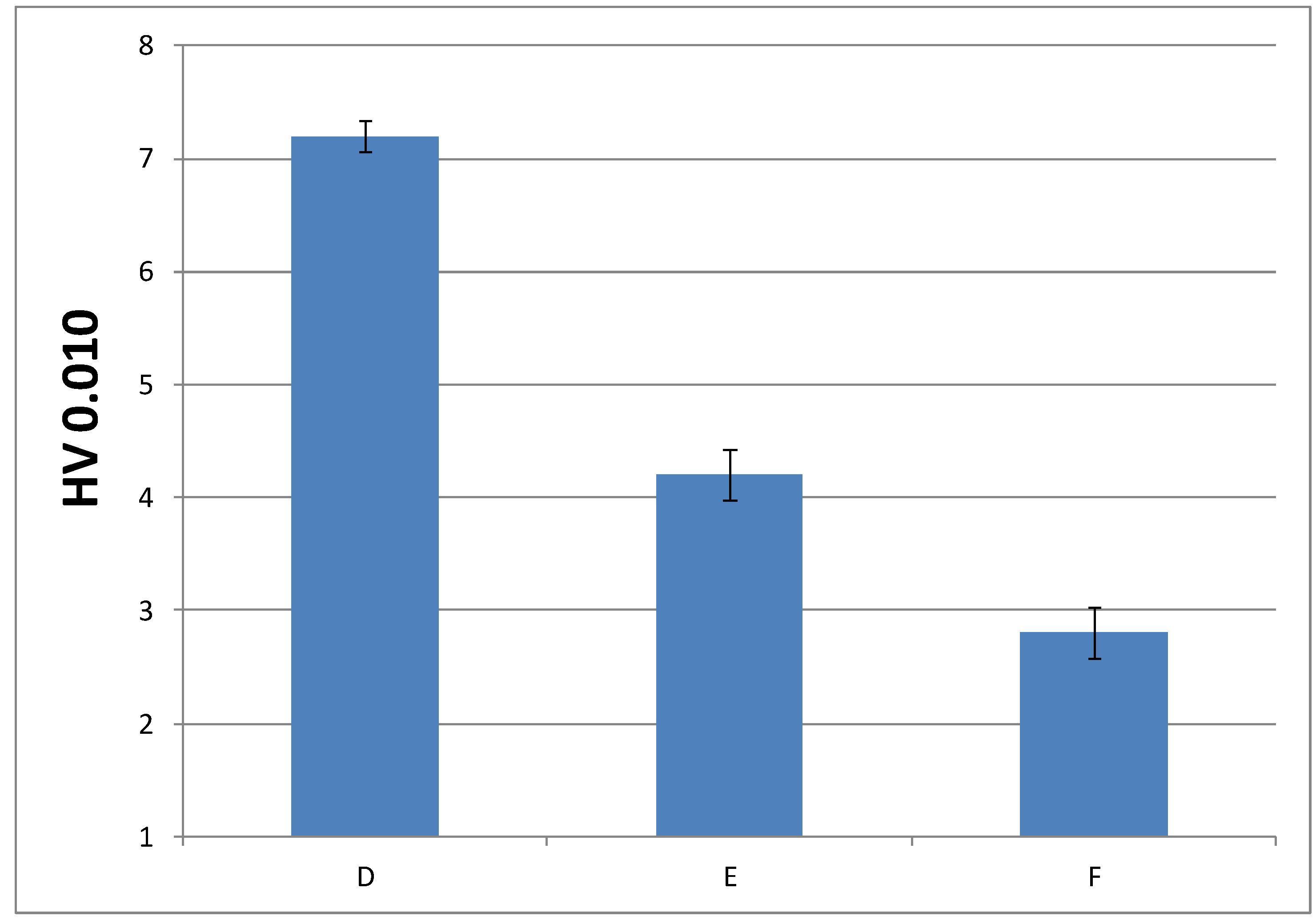
3.3.3. Microindentation Hardness Characterization of the Cured Materials
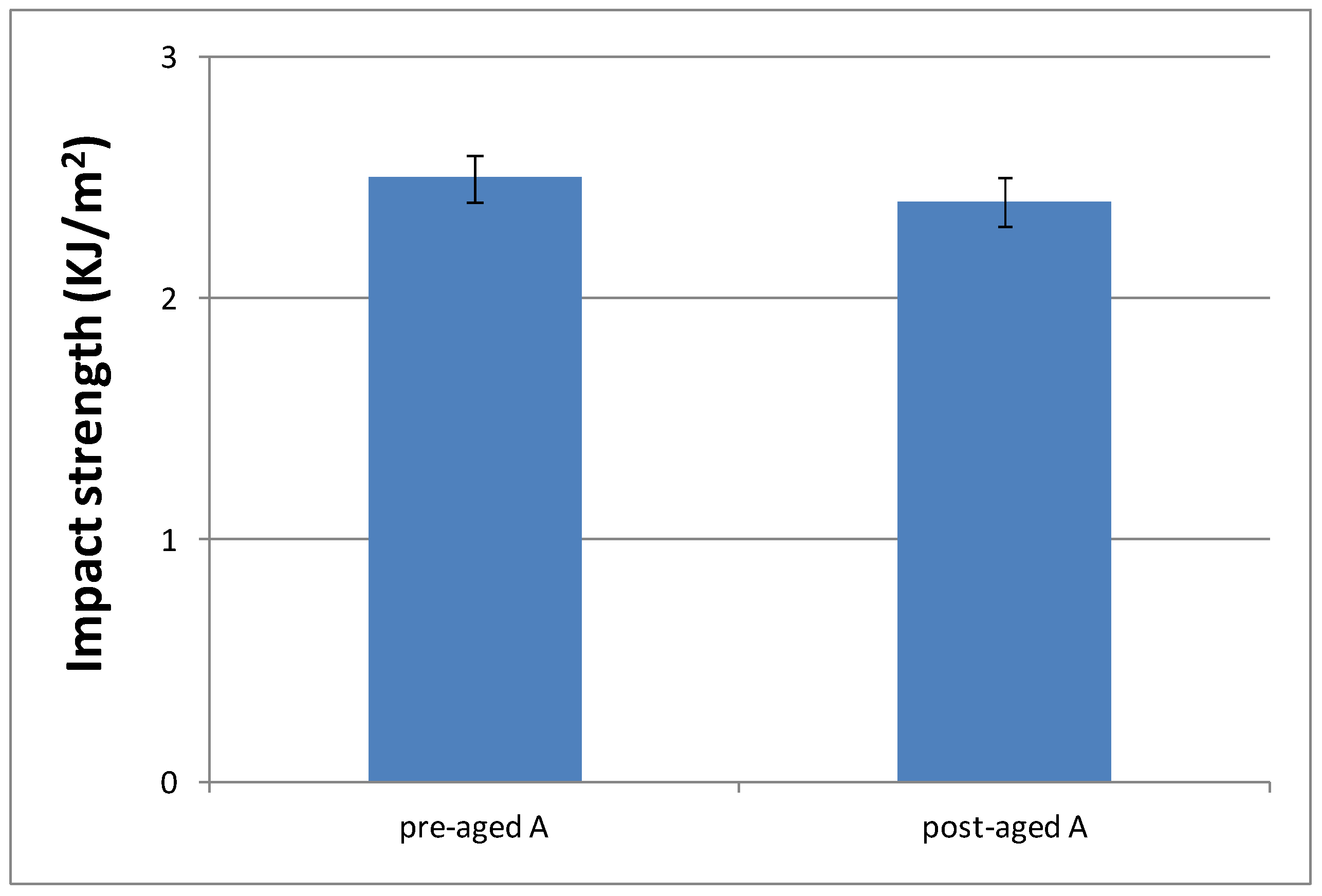
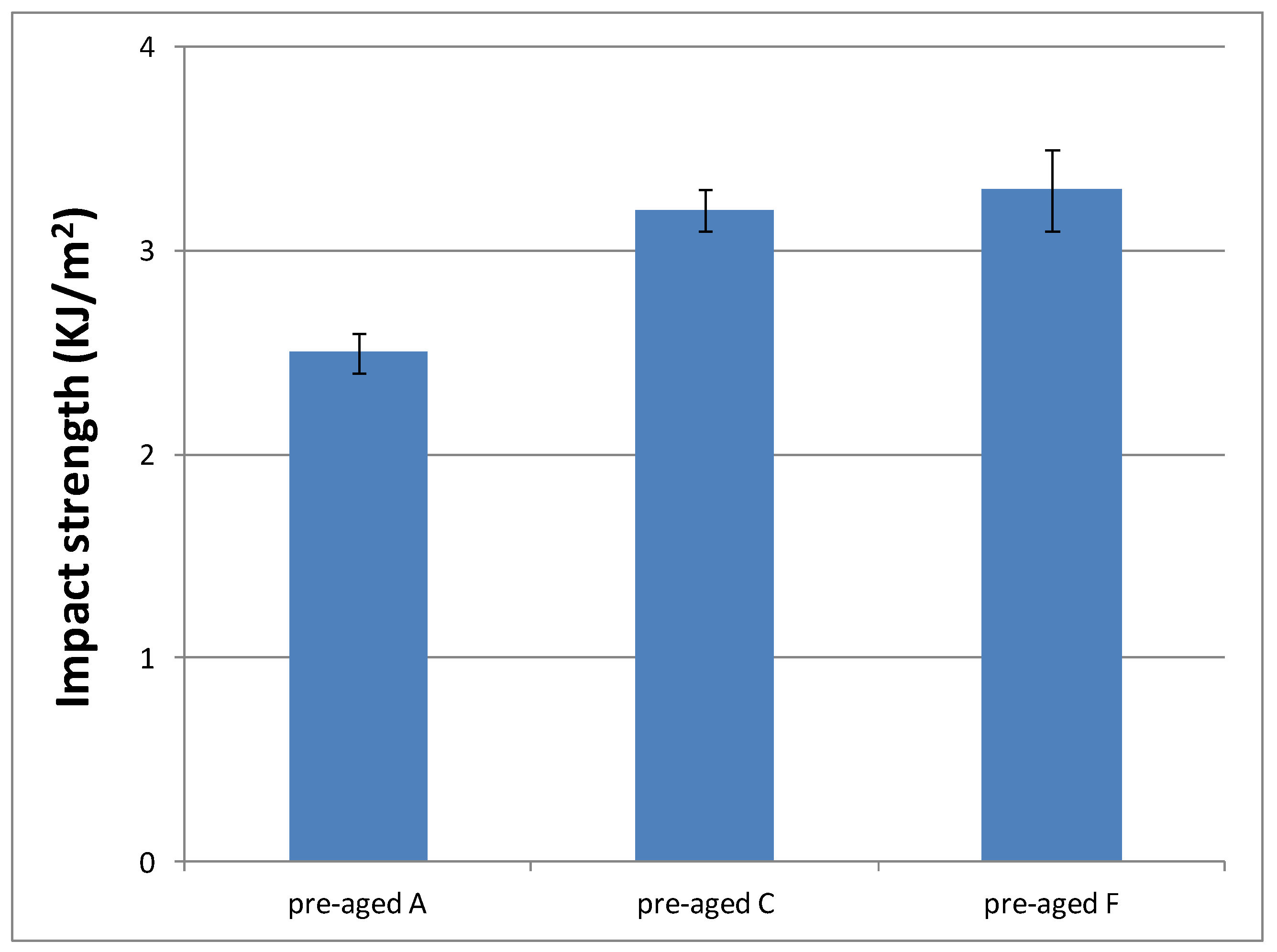
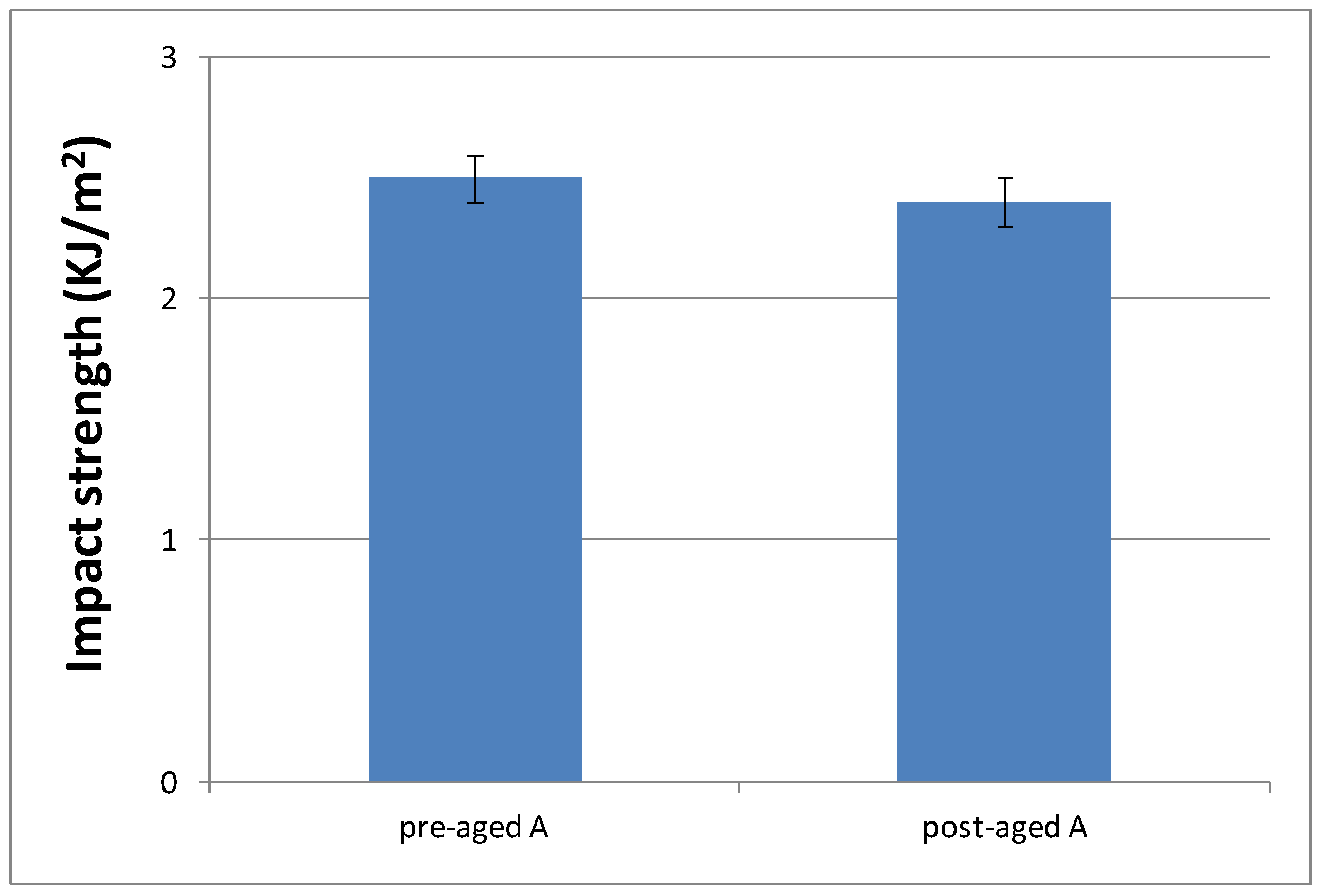
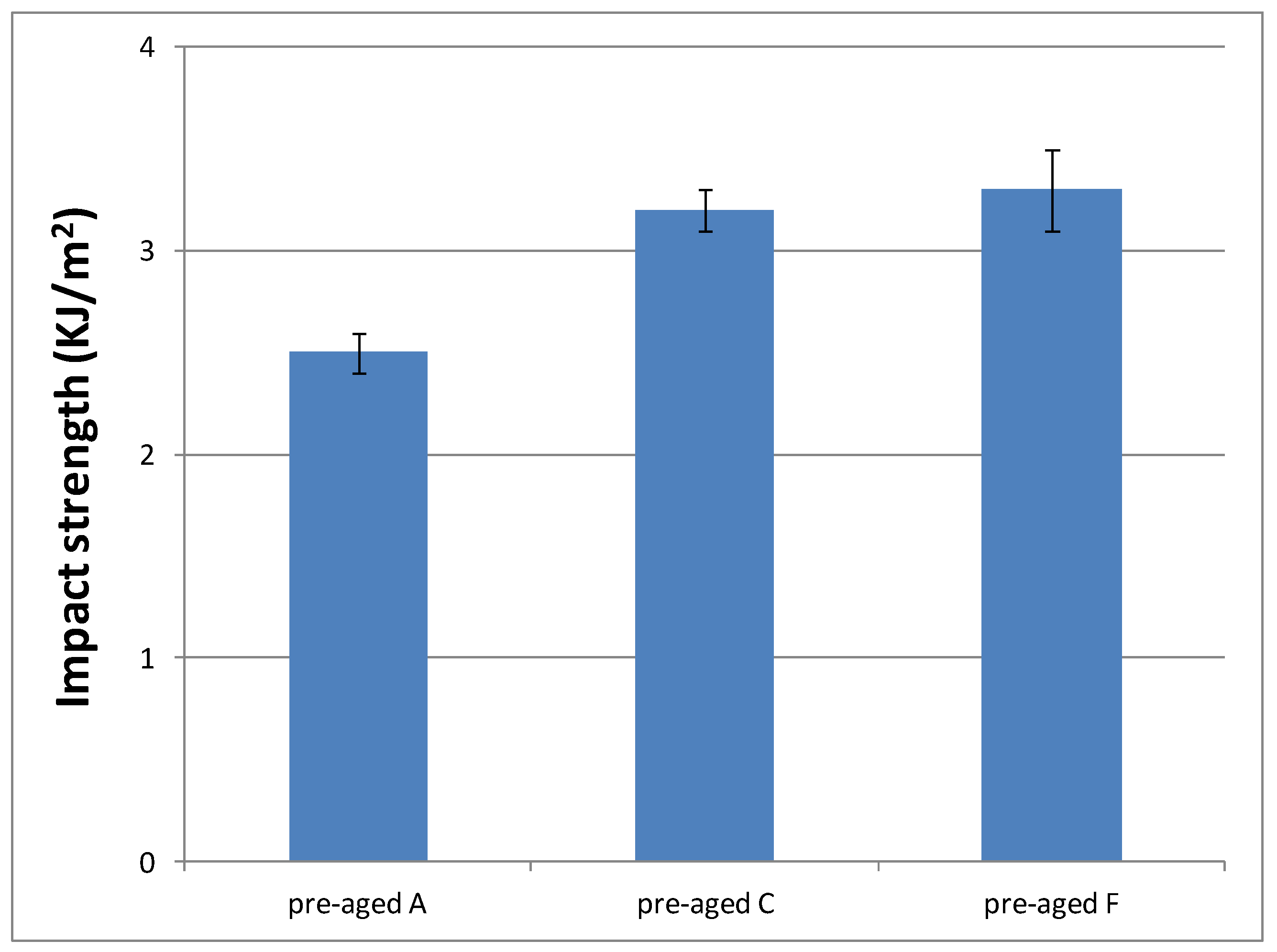
3.3.4. Impact Strength Characterization of the Cured Material
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Traxler, M.; Ackermann, J.; Juda, M.; Hirsch, D. Polymethyl Methacrylate (PMMA); Kunststoffe International; Carl Hanser Verlag: Munich, Germany, 2011; Volume 10. [Google Scholar]
- Decker, C. Photoinitiated crosslinking polymerization. Prog. Polym. Sci. 1996, 21, 593–650. [Google Scholar] [CrossRef]
- Lazzari, M.; Scalarone, D.; Malucelli, G.; Chiantore, O. Durability of acrylic films from commercial aqueous dispersion: Glass transition temperature and tensile behavior as indexes of photooxidative degradation. Prog. Org. Coat. 2011, 70, 116–121. [Google Scholar] [CrossRef]
- Wicks, Z.W., Jr.; Jones, F.N.; Pappas, S.P. Douglas A. Wicks. Wicks Organic Coatings: Science and Technology, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2007; p. 159. [Google Scholar]
- Bakioglu, L. Polymerization and Characterization of Poly(Ethyl Methacrylate). Master’s Thesis, The Middle East Technical University, Ankara, Turkey, 2003. [Google Scholar]
- He, J.; Liu, F.; Vallittu, P.K.; Lassila, L.V.J. Synthesis of dimethacrylates monomers with low polymerization shrinkage and its application in dental composites materials. J. Polym. Res. 2012, 19, 9932. [Google Scholar] [CrossRef]
- Lee, J.K.; Choi, J.Y.; Lim, B.S.; Lee, Y.K.; Sakaguchi, R.L. Change of properties during storage of a udma/tegdma dental resin. J. Biomed. Mater. Res. B Appl. Biomater. 2004, 68B, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Asmussen, E.; Peutzfeldt, A. Influence of uedma, bisgma and tegdma on selected mechanical properties of experimental resin composites. Dent. Mater. 1998, 14, 51–56. [Google Scholar] [CrossRef]
- Muhtaroǧullari, I.Y.; Doǧan, A.; Muhtaroǧullari, M.; Usanmaz, A. Thermal and mechanical properties of microwave and heat-cured poly(methyl methacrylate) used as dental base material. J. Appl. Polym. Sci. 2003, 90, 251–256. [Google Scholar] [CrossRef]
- Baki, G.; Alexander, K.S. Introduction to Cosmetic and Formulation Technology; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Moossavi, M.; Scher, R.K. Nail care products. Clin. Dermatol. 2001, 19, 445–448. [Google Scholar] [CrossRef]
- Marks, J.G.; Bishop, M.E.; Willis, W.F. Allergic contact dermatitis to sculptured nails. Arch. Dermatol. 1979, 115, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, L.; Henriks-Eckerman, M.L.; Jolanki, R.; Estalander, T. Plastics/acrylics: Material safety data sheet need to be improved. Clin. Dermatol. 1997, 15, 533–546. [Google Scholar] [CrossRef]
- Ramírez, F.; Félix, M.; Romero, A.; Guerrero, A. Mechanical properties of acrylic nails in the guitar playing. Afinidad LXXII 2015, 572, 249–255. [Google Scholar]
- Sideridou, I.D.; Achilias, D.S.; Karava, O. Reactivity of benzoyl peroxide/amine system as an initiator for the free radical polymerization of dental and orthopaedic dimethacrylate monomers: Effect of the amine and monomer chemical structure. Macromolecules 2004, 37, 4254–4265. [Google Scholar] [CrossRef]
- Zoller, A.; Gigmesa, D.; Guillaneuf, Y. Simulation of radical polymerization of methyl methacrylate at room temperature using a tertiary amine/BPO initiating system. Polym. Chem. 2015, 6, 5719–5727. [Google Scholar] [CrossRef]
- Vazquez, B.; Levenfeld, B.; Roman, J.S. Role of amine activators on the curing parameters, properties and toxicity of acrylic bone cements. Polym. Int. 1998, 46, 241–250. [Google Scholar] [CrossRef]
- Vazquez, B.; San Roman, J.; Deb, S.; Bonfield, W. Application of long chain amine activator in conventional acrylic bone cement. J. Biomed. Mater. Res. 1998, 43, 131–139. [Google Scholar] [CrossRef]
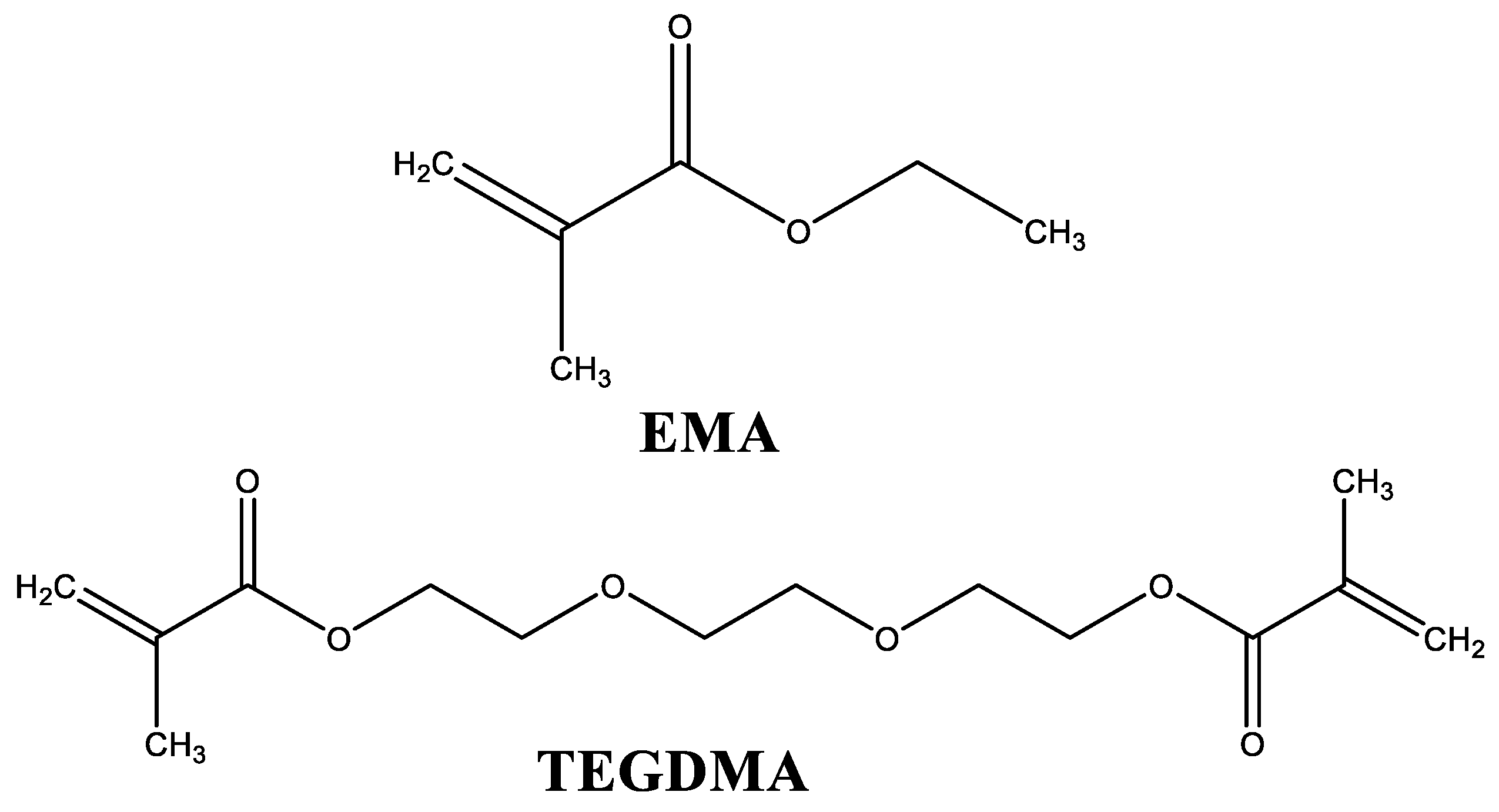
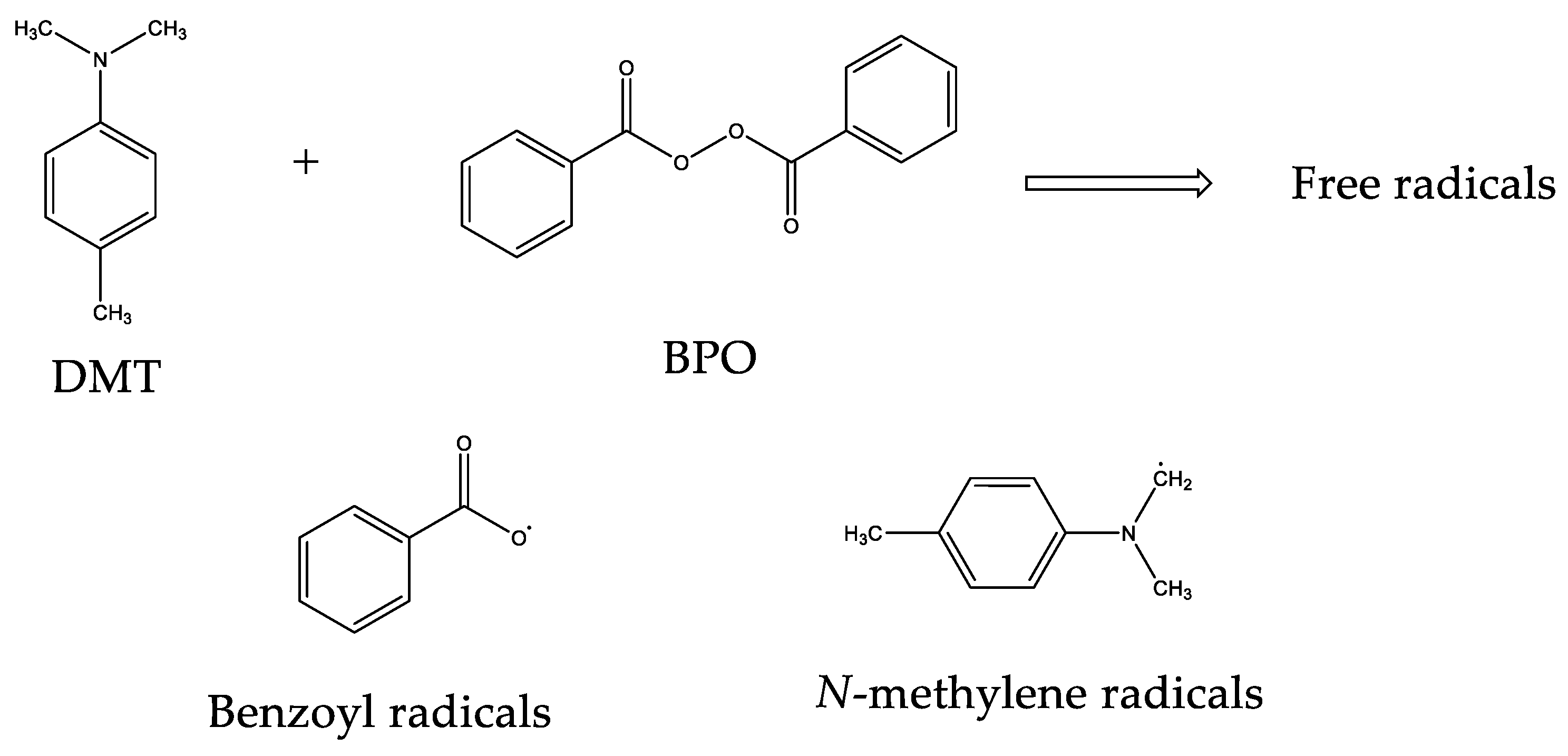
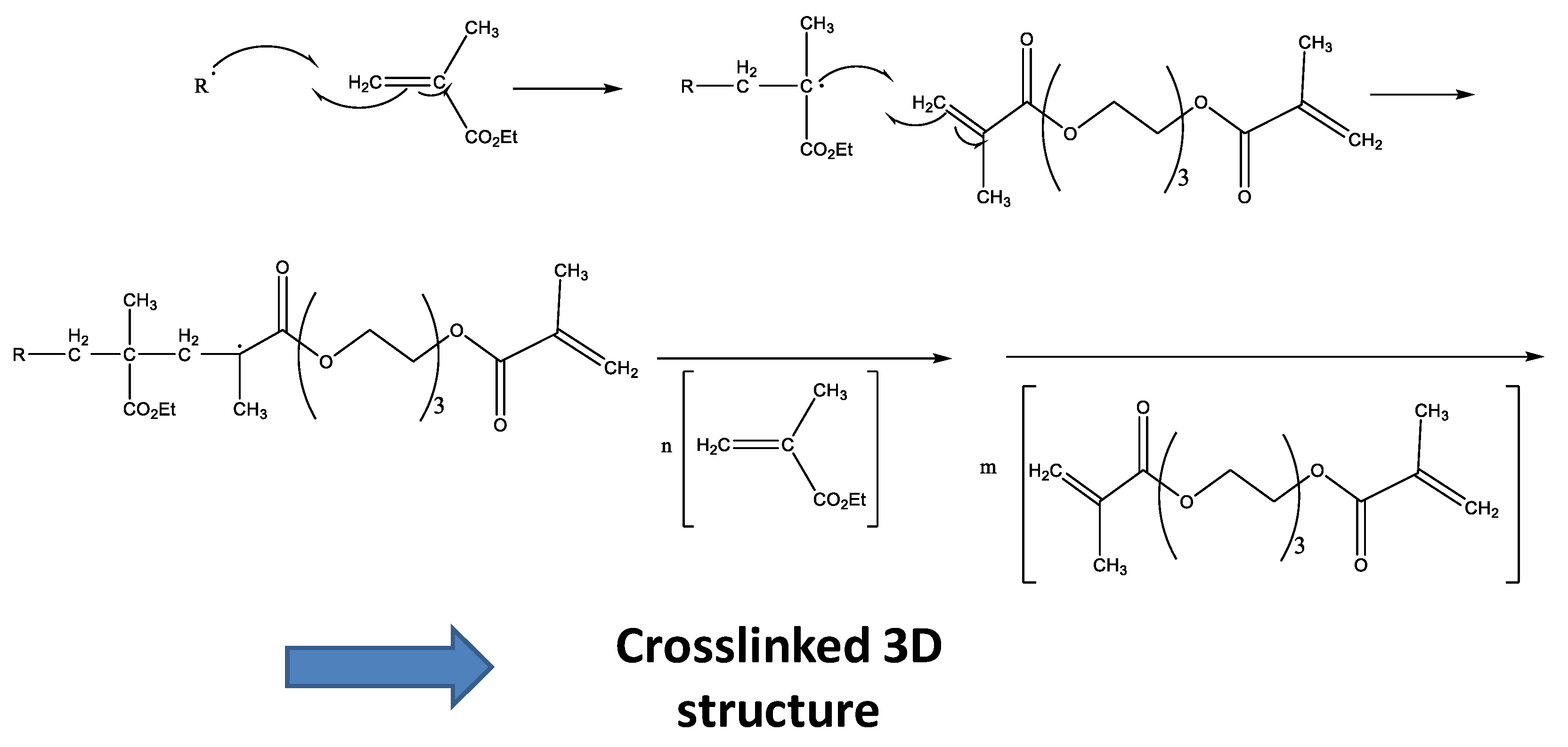


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | EMA | TEGDMA | DMT | B-3 |
|---|---|---|---|---|
| A | 83.5 | 15 | 1 | 0.50 |
| B | 88.5 | 10 | 1 | 0.50 |
| C | 93.5 | 5 | 1 | 0.50 |
| D | 83.63 | 15.13 | 0.75 | 0.50 |
| E | 88.63 | 10.13 | 0.75 | 0.50 |
| F | 93.63 | 5.13 | 0.75 | 0.50 |
| G | 83.75 | 15.25 | 0.50 | 0.50 |
| H | 88.75 | 10.25 | 0.50 | 0.50 |
| I | 93.75 | 5.25 | 0.50 | 0.50 |
| Formulation | Polymerization ΔH at 25 °C (10−2) (J/g) | Residual ΔH (10−2) (J/g) | t at α95 (min) |
|---|---|---|---|
| Commercial | 2.4 ± 0.1 | 0.21 ± 0.01 | 31 ± 1 |
| A | 2.3 ± 0.1 | 0.23 ± 0.01 | 32 ± 1 |
| B | 2.2 ± 0.2 | 0.24 ± 0.02 | 43 ± 2 |
| C | 2.3 ± 0.1 | 0.26 ± 0.05 | 66 ± 2 |
| D | 2.3 ± 0.1 | 0.27 ± 0.02 | 34 ± 1 |
| E | 2.2 ± 0.1 | 0.28 ± 0.02 | 46 ± 1 |
| F | 2.0 ± 0.3 | 0.22 ± 0.02 | 73 ± 1 |
| G | 2.4 ± 0.1 | 0.30 ± 0.01 | 37 ± 1 |
| H | 2.3 ± 0.1 | 0.25 ± 0.02 | 54 ± 1 |
| I | 1.7 ± 0.3 | 0.24 ± 0.01 | 80 ± 1 |
| Curing time at 25 °C (min) | Residual ΔH (J/g) |
|---|---|
| 30 | 11 ± 1 |
| 60 | 9 ± 1 |
| 90 | 6 ± 2 |
| 180 | 2 ± 1 |
| 300 | 0.5 ± 0.2 |
| Formulation | Onset TGA curve (5% Mass Loss) (°C) | Tmax (°C) |
|---|---|---|
| A | 273 | 390 |
| B | 251 | 389 |
| C | 256 | 389 |
| D | 267 | 388 |
| E | 236 | 388 |
| F | 245 | 386 |
| Formulation | Tg (°C) | E (MPa) |
|---|---|---|
| A | 95 ± 1 | 900 ± 34 |
| B | 95 ± 1 | 743 ± 37 |
| C | 94 ± 1 | 564 ± 29 |
| D | 91 ± 1 | 436 ± 27 |
| E | 91 ± 1 | 329 ± 22 |
| F | 90 ± 1 | 259 ± 21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donoso, R.; Reina, J.A.; Giamberini, M.; De La Flor, S.; Ferrando, F.; Cerruti, P. Thermal and Mechanical Characterization of EMA-TEGDMA Mixtures for Cosmetic Applications. Polymers 2018, 10, 256. https://doi.org/10.3390/polym10030256
Donoso R, Reina JA, Giamberini M, De La Flor S, Ferrando F, Cerruti P. Thermal and Mechanical Characterization of EMA-TEGDMA Mixtures for Cosmetic Applications. Polymers. 2018; 10(3):256. https://doi.org/10.3390/polym10030256
Chicago/Turabian StyleDonoso, Ruben, Jose Antonio Reina, Marta Giamberini, Silvia De La Flor, Francesc Ferrando, and Pierfrancesco Cerruti. 2018. "Thermal and Mechanical Characterization of EMA-TEGDMA Mixtures for Cosmetic Applications" Polymers 10, no. 3: 256. https://doi.org/10.3390/polym10030256
APA StyleDonoso, R., Reina, J. A., Giamberini, M., De La Flor, S., Ferrando, F., & Cerruti, P. (2018). Thermal and Mechanical Characterization of EMA-TEGDMA Mixtures for Cosmetic Applications. Polymers, 10(3), 256. https://doi.org/10.3390/polym10030256