Surface Segregation of Cyclic Chains in Binary Melts of Thin Polymer Films: The Influence of Constituent Concentration


Abstract
:1. Introduction
2. Model and Simulation Details
2.1. The Bead-Spring Model
2.2. Simulation Details
3. Results and Discussions
3.1. Density and Radius of Gyration
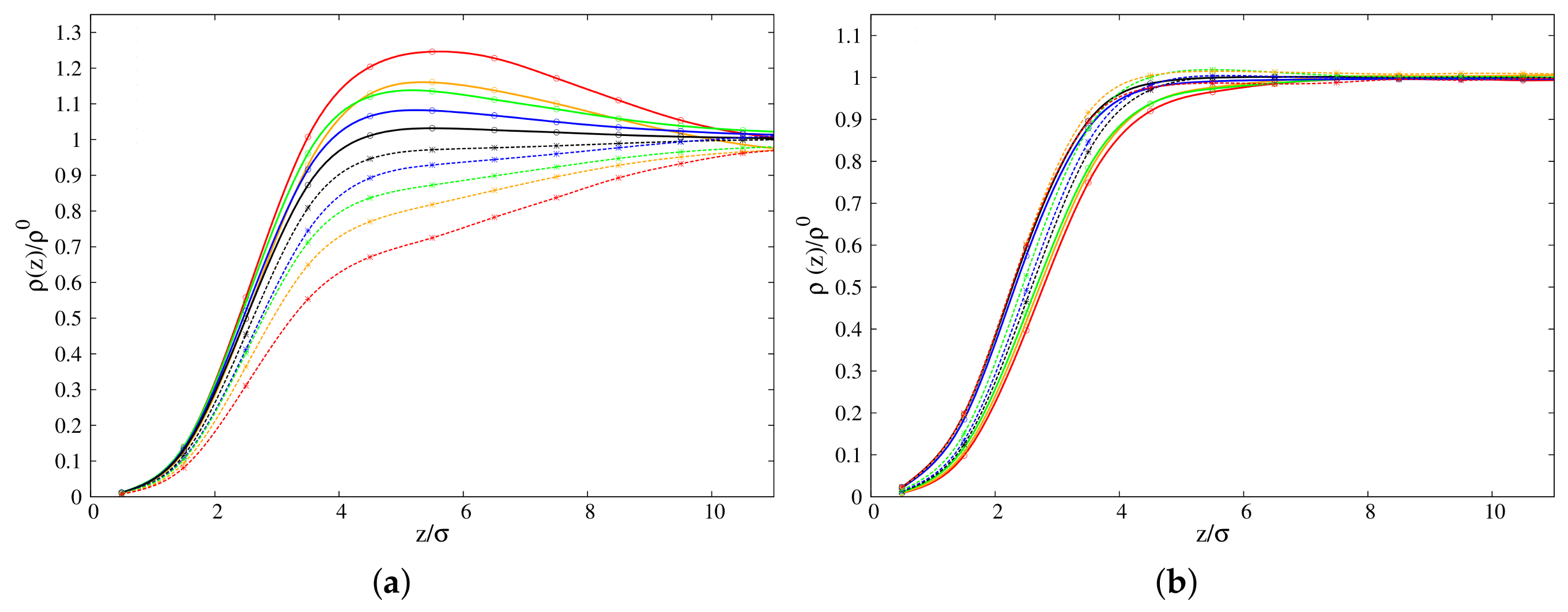
3.1.1. Density
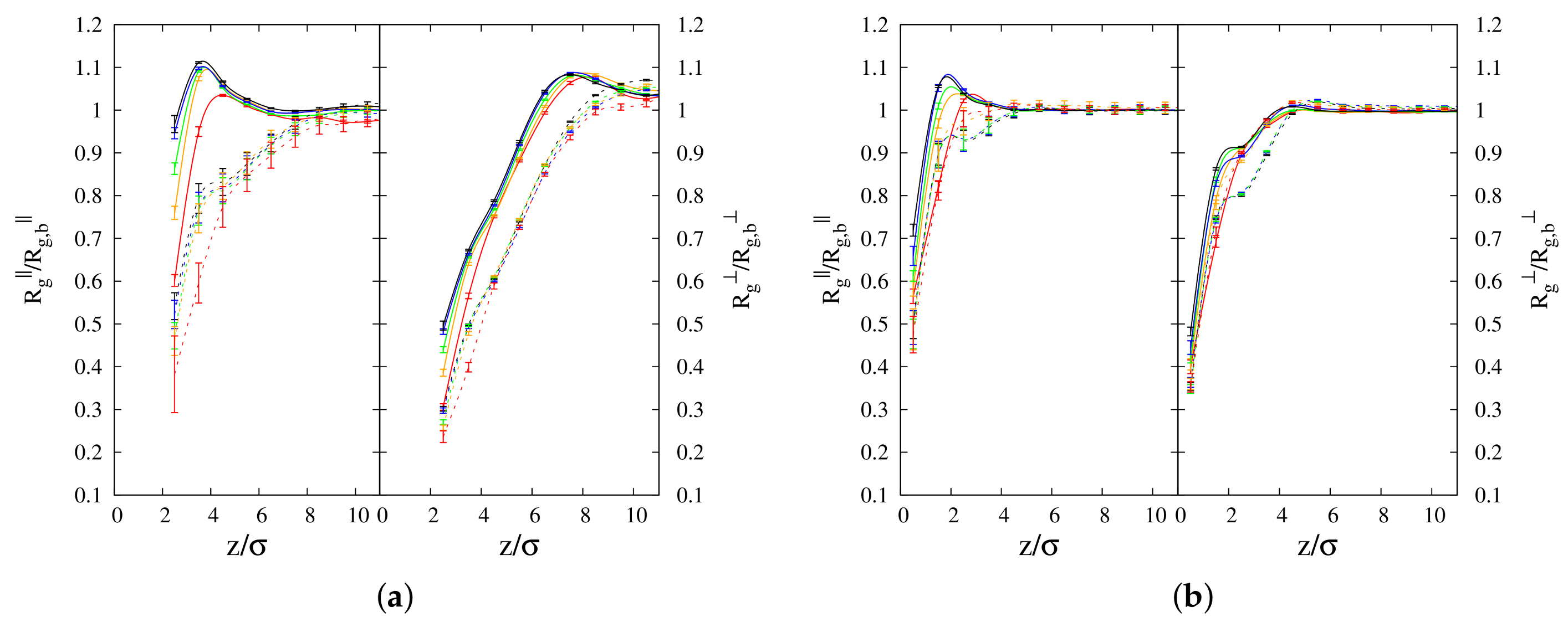
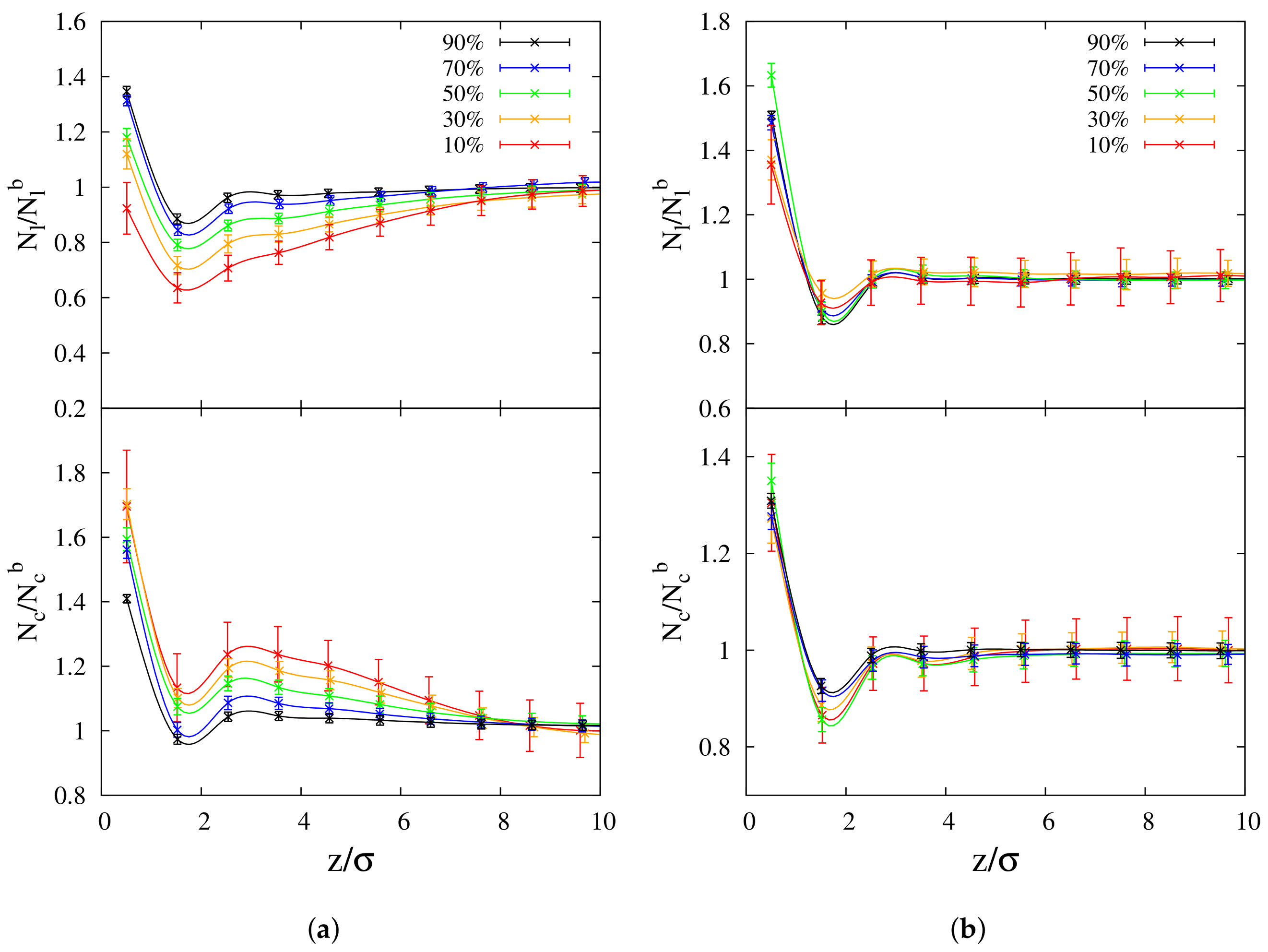
3.1.2. Radius of Gyration
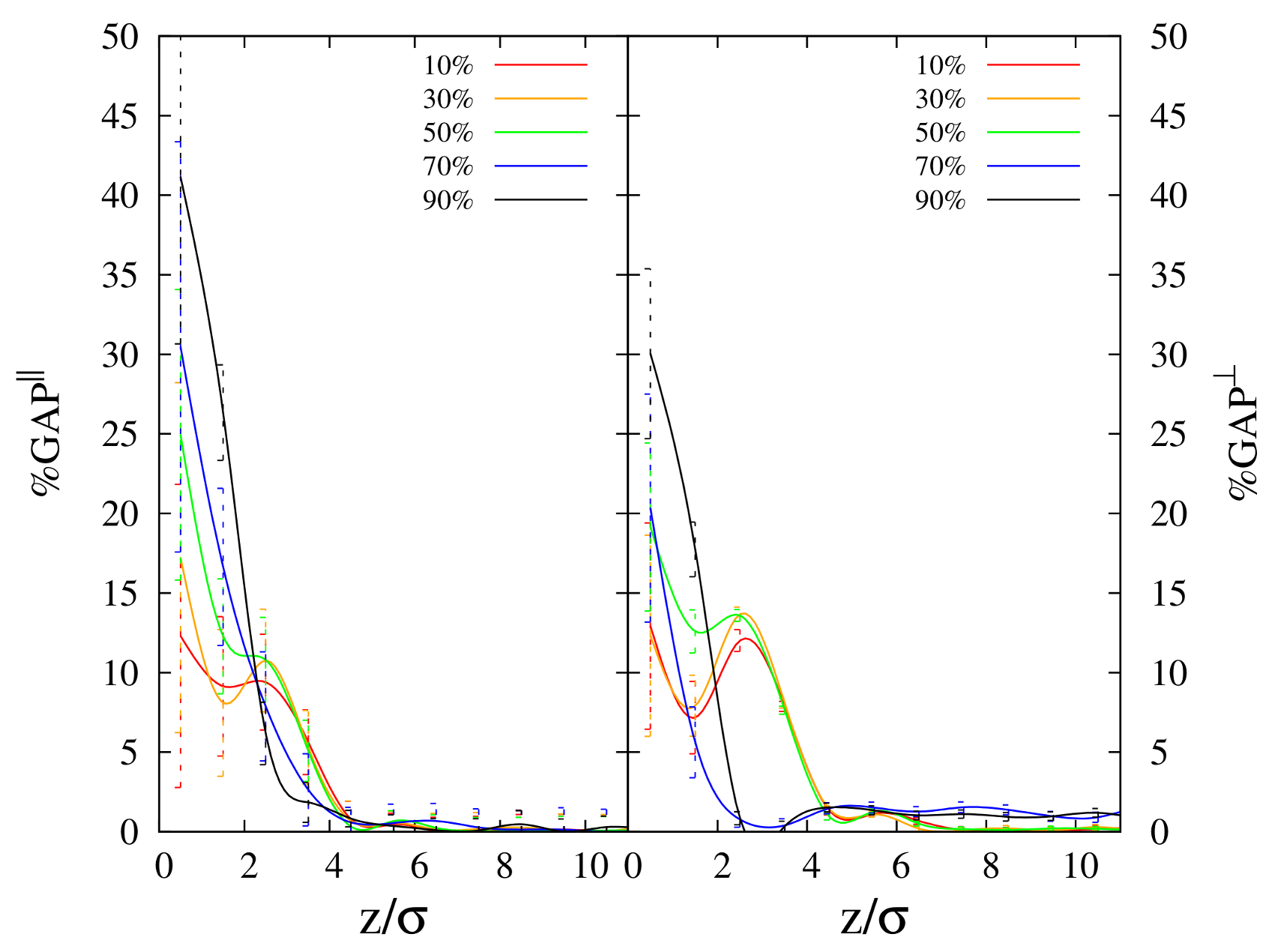
3.2. Instantaneous Surface Profile
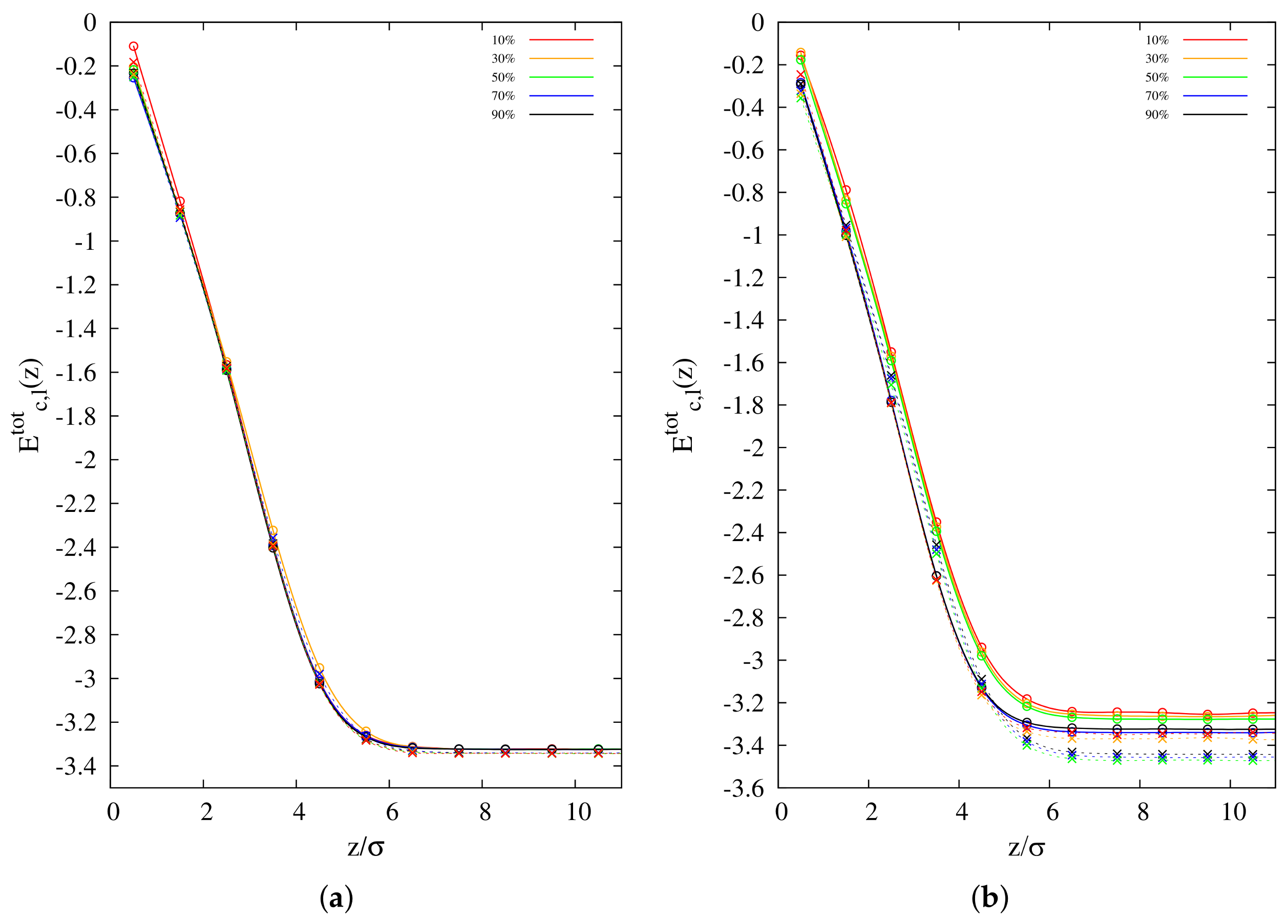
3.3. Surface Interaction Energy
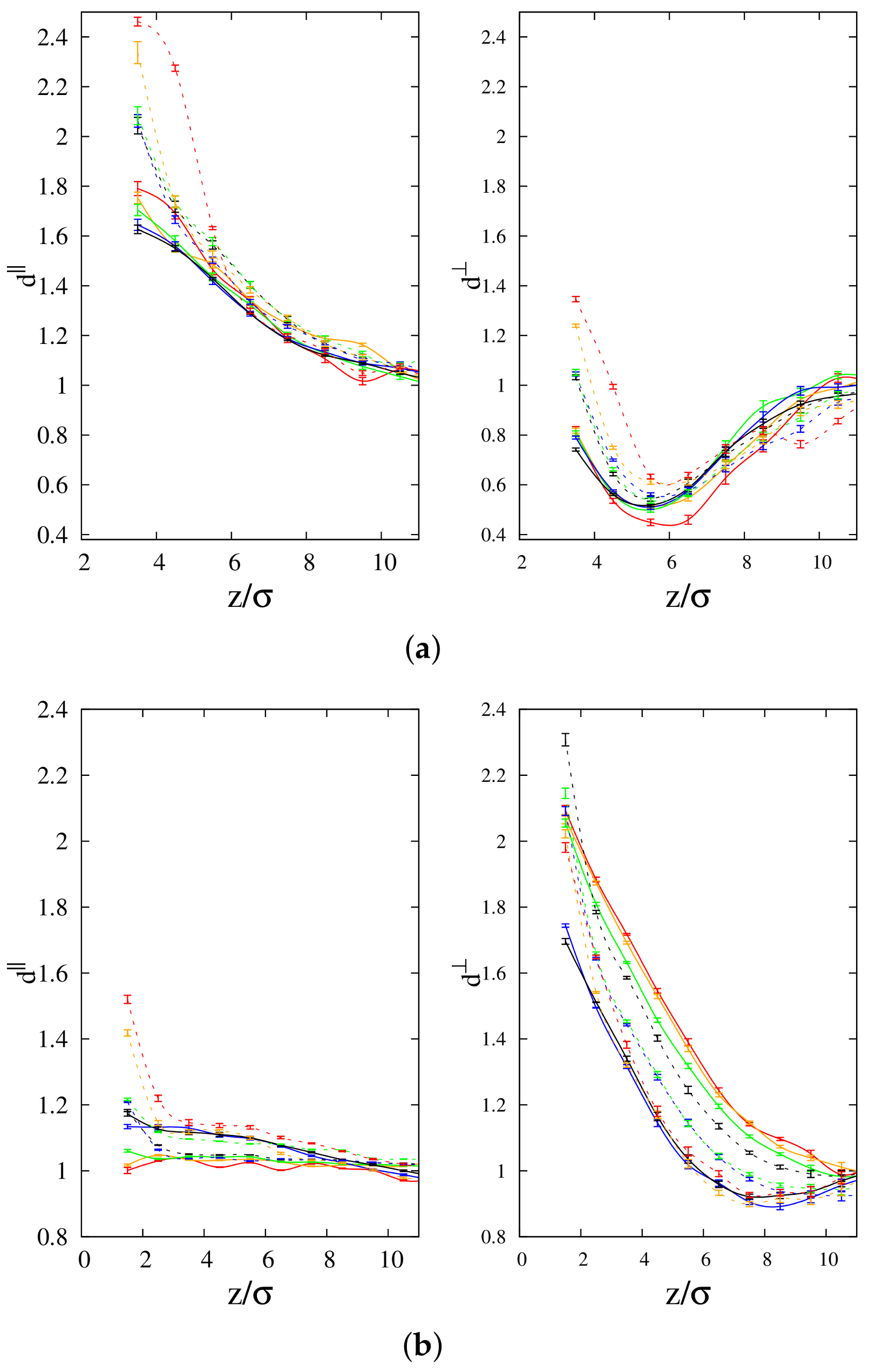
3.4. Diffusion Coefficient
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, D.; Nakahara, A.; Wei, D.; Nordlund, D.; Russell, T.P. P3HT/PCBM Bulk Heterojunction Organic Photovoltaics: Correlating Efficiency and Morphology. Nano Lett. 2011, 11, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Luo, Y.; Wei, H.; Luo, J.; Dong, J.; Lv, S.; Xiao, J.; Xu, Y.; Zhu, L.; Xu, X.; et al. Modified Two-Step Deposition Method for High-Efficiency TiO2/CH3NH3PbI3 Heterojunction Solar Cells. ACS Appl. Mater. Interfaces 2014, 6, 9711–9718. [Google Scholar] [CrossRef] [PubMed]
- Andrady, A.L.; Neal, M.A. Applications and societal benefits of plastics. Philos. Trans. R. Soc. B 2009, 364, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Drelich, J.; Payne, J.; Kim, T.; Miller, J. Selective froth flotation of PVC from PVC/PET mixtures for the plastics recycling industry. Polym. Eng. Sci. 1998, 38, 1378–1386. [Google Scholar] [CrossRef]
- Auvergne, R.S.; David, C.G.; Boutevin, B.; Pascault, J.P. Biobased Thermosetting Epoxy: Present and Future. Chem. Rev. 2014, 114, 1082–1115. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Scala, J.J.L.; Sadler, J.M.; Palmese, G.R. Synthesis and Characterization of Thermosetting Furan-Based Epoxy Systems. Macromolecules 2014, 47, 3332–3342. [Google Scholar] [CrossRef]
- Oseni, S.O.; Mola, G.T. Properties of functional layers in inverted thin film organic solar cells. Sol. Energy Mater. Sol Cells 2017, 160, 241–256. [Google Scholar] [CrossRef]
- Arbab, E.A.; Mola, G.T. V2O5 thin film deposition for application in organic solar cells. Appl. Phys. A 2016, 122, 405–412. [Google Scholar] [CrossRef]
- Costa, D.; Pellicane, G.; Caccamo, C.; Scholl-Paschinger, E.; Kahl, G. Theoretical description of phase coexistence in model. Phys. Rev. E 2003, 68, 021104. [Google Scholar] [CrossRef] [PubMed]
- Mano, J.F.; Koniarova, D.; Reis, R.L. Thermal properties if thermoplastic starch/synthetic polymer blends with potential biomedical applicability. J. Mater. Sci. Mater. Med. 2003, 14, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Nanda, M.; Tripatthy, D.K. Physico-mechanical and electrical properties of conductive carbon black reinforced chlorosulfonated polyethylene vulcanizates. Polym. Lett. 2008, 2, 855–865. [Google Scholar] [CrossRef]
- Coveney, S.; Clarke, N. Pattern Formation in Polymer Blend Thin Films: Surface Roughening Couples to Phase Separation. Phys. Rev. Lett. 2014, 113, 218301. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, N.H.; Peri, S.; Foster, M.D.; Majkrzak, C.F.; Hu, R.; Wu, D.T. Surface segregation driven by molecular architecture asymmetry in polymer blends. Phys. Rev. Lett. 2014, 113, 225702. [Google Scholar] [CrossRef] [PubMed]
- Pellicane, G.; Megnidio-Tchoukouegno, M.; Mola, G.T.; Tsige, M. Surface enrichment driven by polymer topology. Phys. Rev. E 2016, 93, 050501. [Google Scholar] [CrossRef] [PubMed]
- Linse, P. Effect of solvent quality on the polymer adsorption from bulk solution onto planar surfaces. Soft Matter 2012, 8, 5140–5150. [Google Scholar] [CrossRef]
- Lee, E.; Jung, Y.J. Segregated structures of ring polymer melts near. the surface: A molecular dynamics simulation study. Soft Matter 2015, 11, 6018–6028. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shentu, B.; Feller, R. Molecular dynamics of different polymer blend containing poly(2,6-dimethyl-1,4-phenylene ether. J. Chem. Phys. 2015, 17, 4714–4723. [Google Scholar] [CrossRef] [PubMed]
- Graessley, W.W.; Edwards, S.F. Entanglement interactions in polymers and the chain contour concentration. Polymer 1981, 22, 1329–1334. [Google Scholar] [CrossRef]
- Subramanian, G.; Shanbhang, S. Self-Diffusion in Binary Blends of Cyclic and Linear Polymers. Macromolecules 2008, 41, 7239–7242. [Google Scholar] [CrossRef]
- Minina, E.; Arnold, A. Induction of entropic segregation: The first step is the hardest. Soft Matter 2008, 10, 5836–5841. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Matsuno, H.; Kawaguchi, D.; Yamada, N.L.; Tanaka, M.; Tanaka, K. Construction of a blood-compatible interface based on surface segregation in a polymer blend. Polymer 2015, 78, 219–224. [Google Scholar] [CrossRef]
- Hirata, T.; Matsuno, H.; Tanaka, M.; Tanaka, K. Surface segregation of poly(2-methoxyethyl acrylate) in a mixture with poly(methyl methacrylate). Phys. Chem. Chem. Phys. 2015, 13, 4928–4934. [Google Scholar] [CrossRef] [PubMed]
- Hawks, S.A.; Aguirre, J.C.; Schelhas, L.T.; Thompson, R.J.; Huber, R.C.; Ferrreira, A.S.; Zang, G.; Herzing, A.A.; Tolbert, S.H.; Schwartz, B.J. Comparing Matched Polymer:Fullerene Solar Cells Made by Solution-Sequential Processing and Traditional Blend Casting: Nanoscale Structure and Device Performance. J. Phys. Chem. C 2014, 31, 17413–17425. [Google Scholar] [CrossRef]
- Gagliardi, S.; Arrighi, V.; Ferguson, R.; Dagger, A.C.; Semlyen, J.A.; Higgins, J.S. On the difference in scattering behavior of cyclic and linear polymers in bulk. J. Chem. Phys. 2005, 122, 064904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Chan, C.M.; Weng, L.T.; Jiang, M. Surface segregation in polymer blends and interpolymer complexes with increasing hydrogen bonding interactions. J. Polym. Sci. 2005, 43, 1924–1930. [Google Scholar] [CrossRef]
- Hariharan, A.; Kumar, S.K.; Russell, T.P. Reversal of the isotopic effect in the surface behavior of binary polymer blends. J. Chem. Phys. 1993, 98, 4163–4173. [Google Scholar] [CrossRef]
- Ruocco, N.; Dahbi, L.; Driva, P.; Hadjichristidis, N.; Radulescu, A.; Sharp, M.; Lindner, P.; Straube, F.; Pyckhout-Hintzen, W.; Richter, D.; et al. Microscopic Relaxation Processes in Branched-Linear Polymer Blends by Rheo-SANS. Macromolecules 2013, 46, 9122–9133. [Google Scholar] [CrossRef]
- Read, D.J. Calculation of Scattering from Stretched Copolymers Using the Tube Model: Incorporation of the Effect of Elastic Inhomogeneities. Macromolecules 2004, 37, 5065–5092. [Google Scholar] [CrossRef]
- Mamun, C.K. Free Energy of Mixing of Cross-Linked Polymer Blends. Langmuir 2005, 21, 240–250. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, S.F.; Hu, R.; Akgun, B.; Tormey, C.; Peri, S.; Wu, D.T.; Foster, M.D. Evidence and Limits of Universal Topological Surface Segregation of Cyclic Polymers. Phys. Rev. Lett. 2005, 118, 167801–167805. [Google Scholar] [CrossRef] [PubMed]
- Quirk, R.P.; Wang, S.; Foster, M.D.; Wesdemiotis, C.; Yol, A.M. Synthesis of Cyclic Polystyrenes Using Living Anionic Polymerization and Metathesis Ring-Closure. Macromolecules 2011, 44, 7537–7545. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Agapov, R.L.; Wesdemiotis, C.; Foster, M.D. Probing Surface Concentration of Cyclic/Linear Blend Films Using Surface Layer MALDI-TOF Mass Spectrometry. ACS Macro. Lett. 2012, 1, 1024–1027. [Google Scholar] [CrossRef]
- Megnidio-Tchoukouegno, M.; Gaitho, F.M.; Mola, G.T.; Tsige, M.; Pellicane, G. Unravelling the surface composition of symmetric linear-cyclic polymer blends. Fluid Phase Equilib. 2017, 441, 33–42. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5060. [Google Scholar] [CrossRef]
- Kroger, M.; de Angelis, E. An extended FENE dumbbell model theory for concentration dependent shear-induced anisotropy in dilute polymer solutions. J. Non-Newton. Fluid Mech. 1990, 125, 87–90. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Halverson, J.D.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Rheology of Ring Polymer Melts: From Linear Contaminants to Ring-Linear Blends. Phys. Rev. Lett. 2012, 108, 038301. [Google Scholar] [CrossRef] [PubMed]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. I. Statics. J. Chem. Phys. 2011, 134, 204904. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, H.S.; Zhang, B.K.; Li, J.; de Tian, W. A New Self-Consistent Field Model of Polymer/Nanoparticle Mixture. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yethiraj, A.; Hall, C.K. Monte Carlo simulations and integral equation theory for microscopic correlations in polymeric fluids. Macromolecules 1990, 23, 1865–1872. [Google Scholar] [CrossRef]
- Wang, J.; Kontopoulou, M.; Yea, Z.; Subramanian, R.; Zhu, S. Chain-topology-controlled hyperbranched polyethylene as effective polymer processing aid (PPA) for extrusion of a metallocene linear-low-density polyethylene (mLLDPE). J. Rheol. 2008, 52, 243–260. [Google Scholar] [CrossRef]
- Takeshita, H.; Shiomi, T.; Takenaka, K.; Arai, F. Crystallization and higher-order structure of multicomponent polymeric systems. Polymer 2013, 54, 4776–4789. [Google Scholar] [CrossRef]
- Wu, D.T.; Fredrickson, G.H. Effect of Architecture in the Surface Segregation of Polymer Blends. Macromolecules 1996, 29, 7919–7930. [Google Scholar] [CrossRef]
- Rodenhausen, H. Einstein’s relation between diffusion constant and mobility for a diffusion model. J. Stat. Phys. 1989, 55, 1065–1088. [Google Scholar] [CrossRef]
- Alatas, P.V.; Tsalikis, D.G.; Mavrantzas, V.G. Detailed Molecular Dynamics Simulation of the Structure and Self-Diffusion of Linear and Cyclic n-Alkanes in Melt and Blends. Macromol. Theory Simul. 2016, 26, 1600049–1600066. [Google Scholar] [CrossRef]
- Von Meerwall, E.; Ozisik, R.; Mattice, W.L.; Pfister, P.M. Self-diffusion of linear and cyclic alkanes, measured with pulsed-gradient spin-echo nuclear magnetic resonance. J. Chem. Phys. 2003, 118, 3867–3873. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % Cyclic concentration | 10-mers | 100-mers | ||
|---|---|---|---|---|
| 10 | 3.13 | 0.35 | 3.23 | 0.33 |
| 30 | 2.44 | 1.06 | 2.54 | 1.02 |
| 50 | 1.74 | 1.78 | 1.87 | 1.69 |
| 70 | 1.05 | 2.48 | 1.15 | 2.41 |
| 90 | 0.35 | 3.20 | 0.40 | 3.17 |
| % Cyclic concentration | 100-mers | 10-mers | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Linear chain | Cyclic chain | Linear chain | Cyclic chain | |||||||||
| 10 | 5.06 | 2.84 | 2.79 | 3.77 | 2.18 | 2.08 | 1.45 | 0.81 | 0.79 | 1.14 | 0.66 | 0.64 |
| 30 | 5.10 | 2.86 | 2.82 | 3.68 | 2.11 | 2.07 | 1.44 | 0.81 | 0.79 | 1.14 | 0.65 | 0.64 |
| 50 | 5.09 | 2.87 | 2.78 | 3.63 | 2.08 | 2.04 | 1.44 | 0.81 | 0.79 | 1.14 | 0.65 | 0.64 |
| 70 | 5.01 | 2.82 | 2.76 | 3.57 | 2.05 | 2.00 | 1.26 | 0.71 | 0.70 | 1.22 | 0.70 | 0.68 |
| 90 | 5.04 | 2.84 | 2.81 | 3.51 | 2.01 | 1.98 | 1.20 | 0.68 | 0.67 | 1.17 | 0.67 | 0.65 |
| % Cyclic concentration | 100-mers | 10-mers | ||||||
|---|---|---|---|---|---|---|---|---|
| Linear chain | Cyclic chain | Linear chain | Cyclic chain | |||||
| 10 | 0.10 | 0.11 | 0.05 | 0.05 | 6.00 | 1.19 | 3.11 | 0.63 |
| 30 | 0.11 | 0.12 | 0.06 | 0.06 | 5.89 | 1.26 | 3.10 | 0.64 |
| 50 | 0.12 | 0.12 | 0.06 | 0.06 | 5.80 | 1.27 | 3.09 | 0.70 |
| 70 | 0.12 | 0.12 | 0.07 | 0.07 | 2.25 | 1.28 | 1.09 | 0.80 |
| 90 | 0.15 | 0.15 | 0.08 | 0.08 | 2.18 | 1.28 | 1.07 | 1.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaitho, F.M.; Tsige, M.; Mola, G.T.; Pellicane, G. Surface Segregation of Cyclic Chains in Binary Melts of Thin Polymer Films: The Influence of Constituent Concentration. Polymers 2018, 10, 324. https://doi.org/10.3390/polym10030324
Gaitho FM, Tsige M, Mola GT, Pellicane G. Surface Segregation of Cyclic Chains in Binary Melts of Thin Polymer Films: The Influence of Constituent Concentration. Polymers. 2018; 10(3):324. https://doi.org/10.3390/polym10030324
Chicago/Turabian StyleGaitho, Francis M., Mesfin Tsige, Genene T. Mola, and Giuseppe Pellicane. 2018. "Surface Segregation of Cyclic Chains in Binary Melts of Thin Polymer Films: The Influence of Constituent Concentration" Polymers 10, no. 3: 324. https://doi.org/10.3390/polym10030324
APA StyleGaitho, F. M., Tsige, M., Mola, G. T., & Pellicane, G. (2018). Surface Segregation of Cyclic Chains in Binary Melts of Thin Polymer Films: The Influence of Constituent Concentration. Polymers, 10(3), 324. https://doi.org/10.3390/polym10030324