Flow Behavior of Chain and Star Polymers and Their Mixtures



Abstract
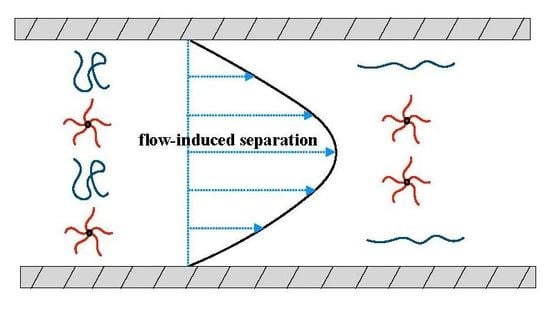
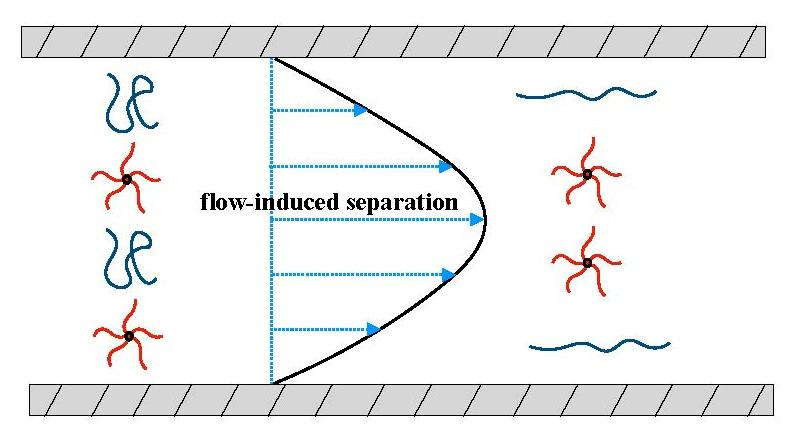{kind=link}
{kind=link}
{kind=link}
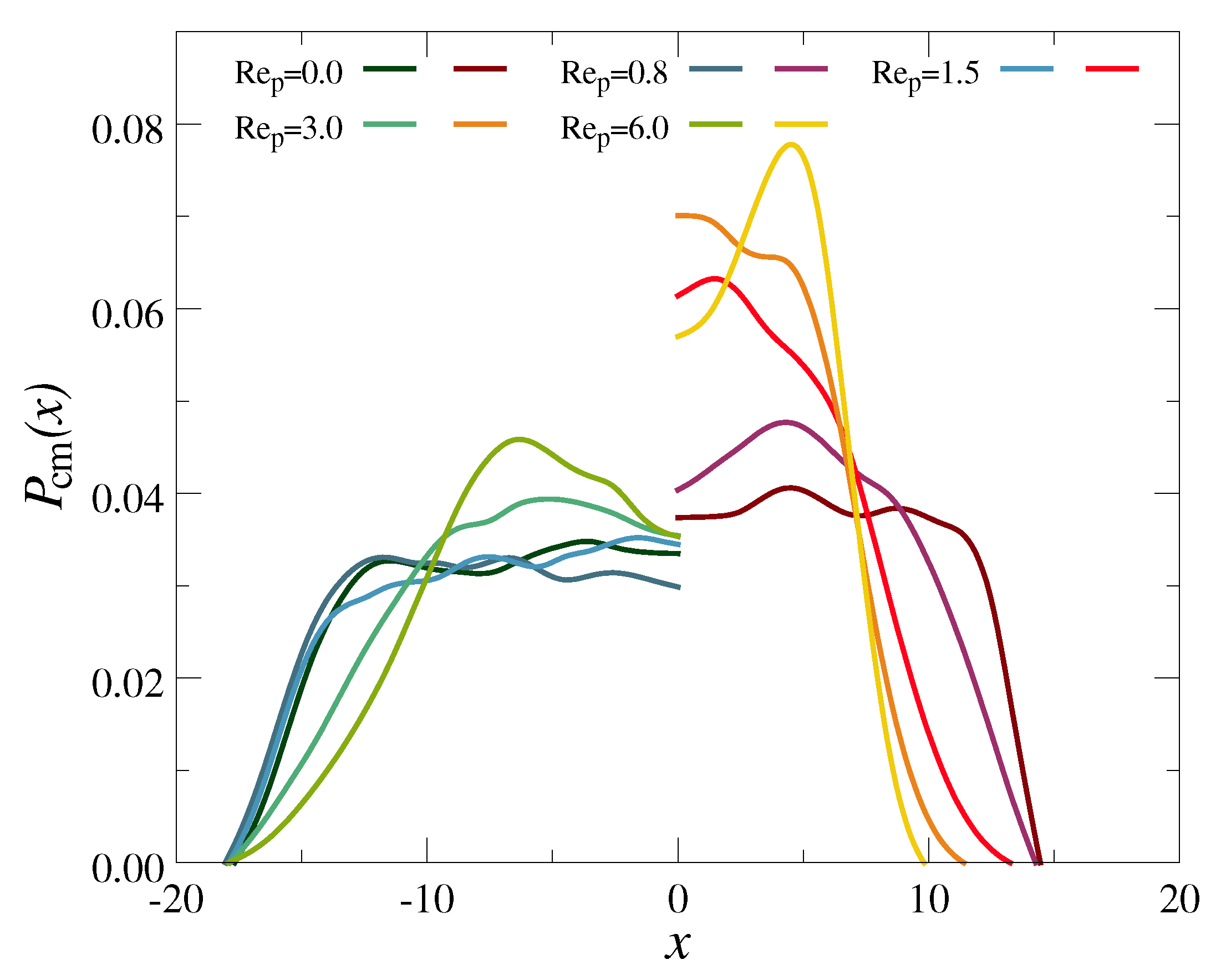{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Model and Simulation Method
3. Results and Discussion
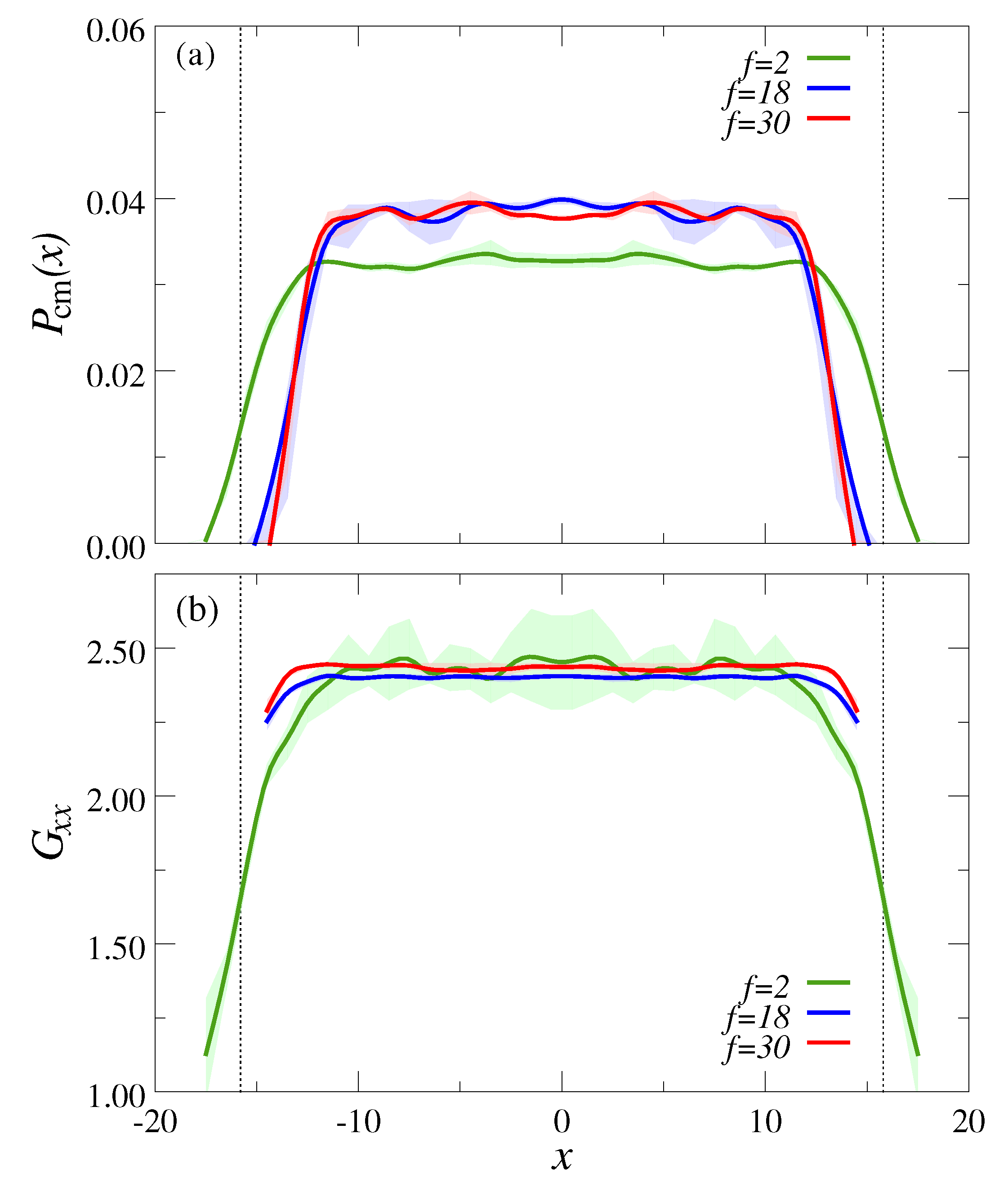
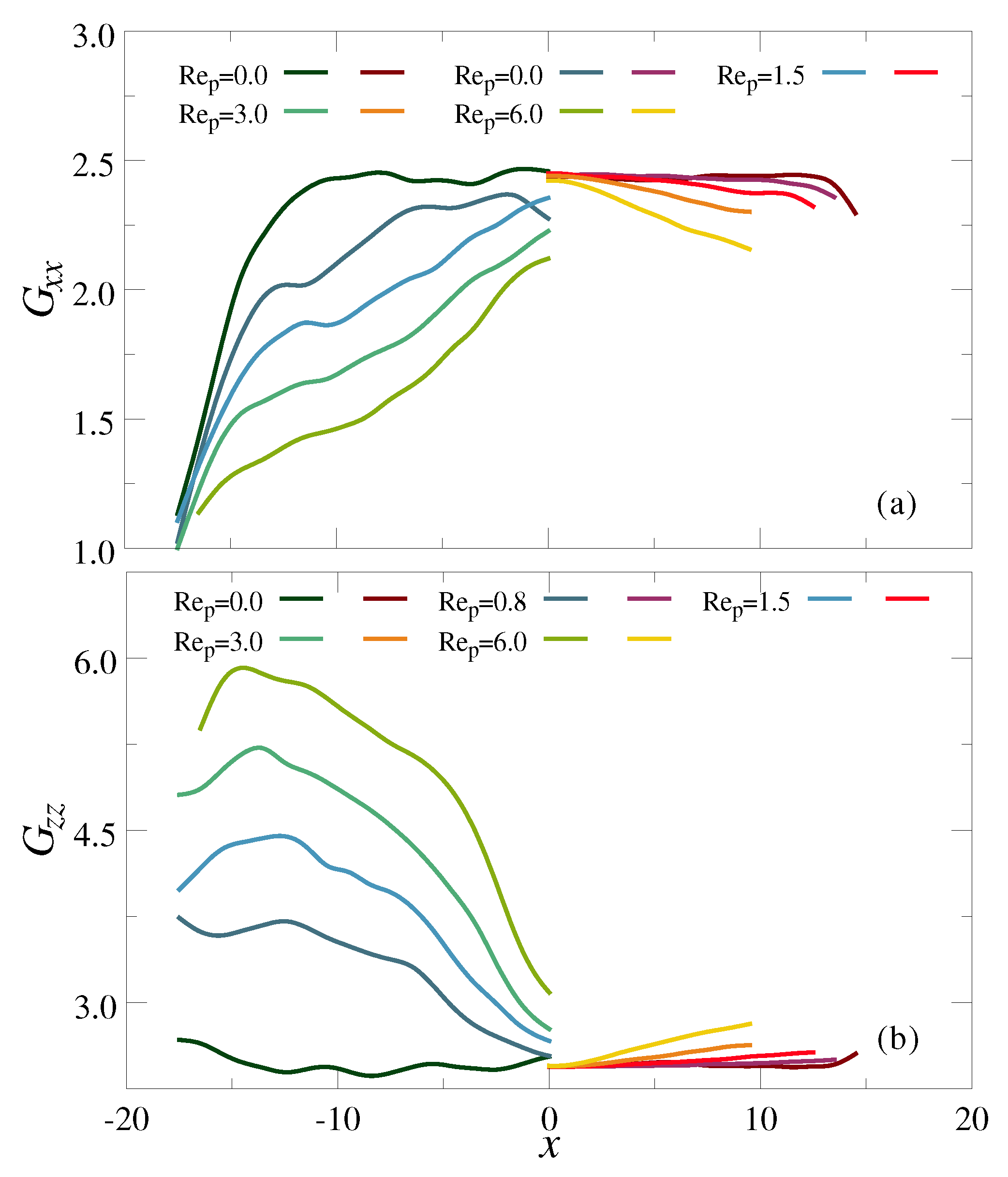
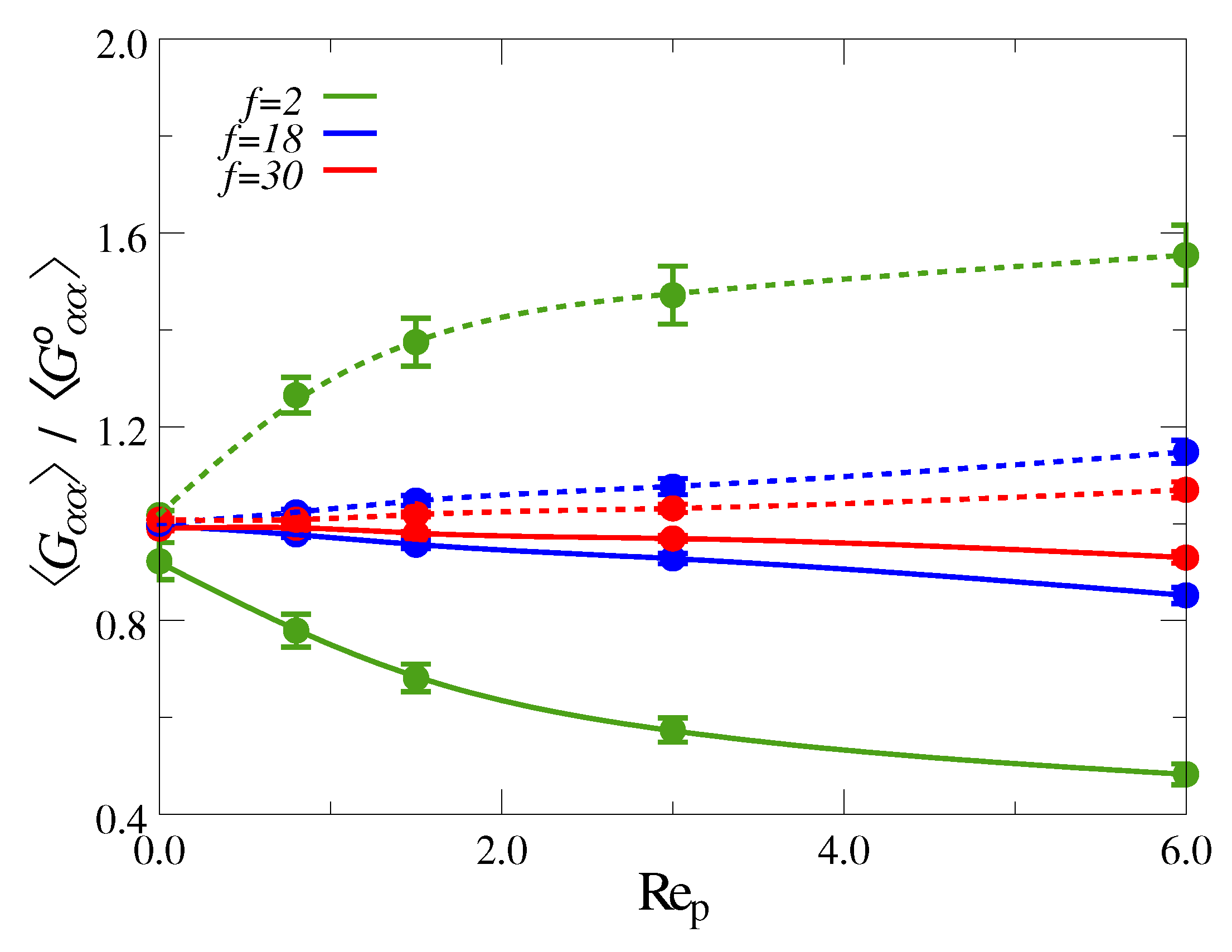
3.1. Ultradiulte Conditions
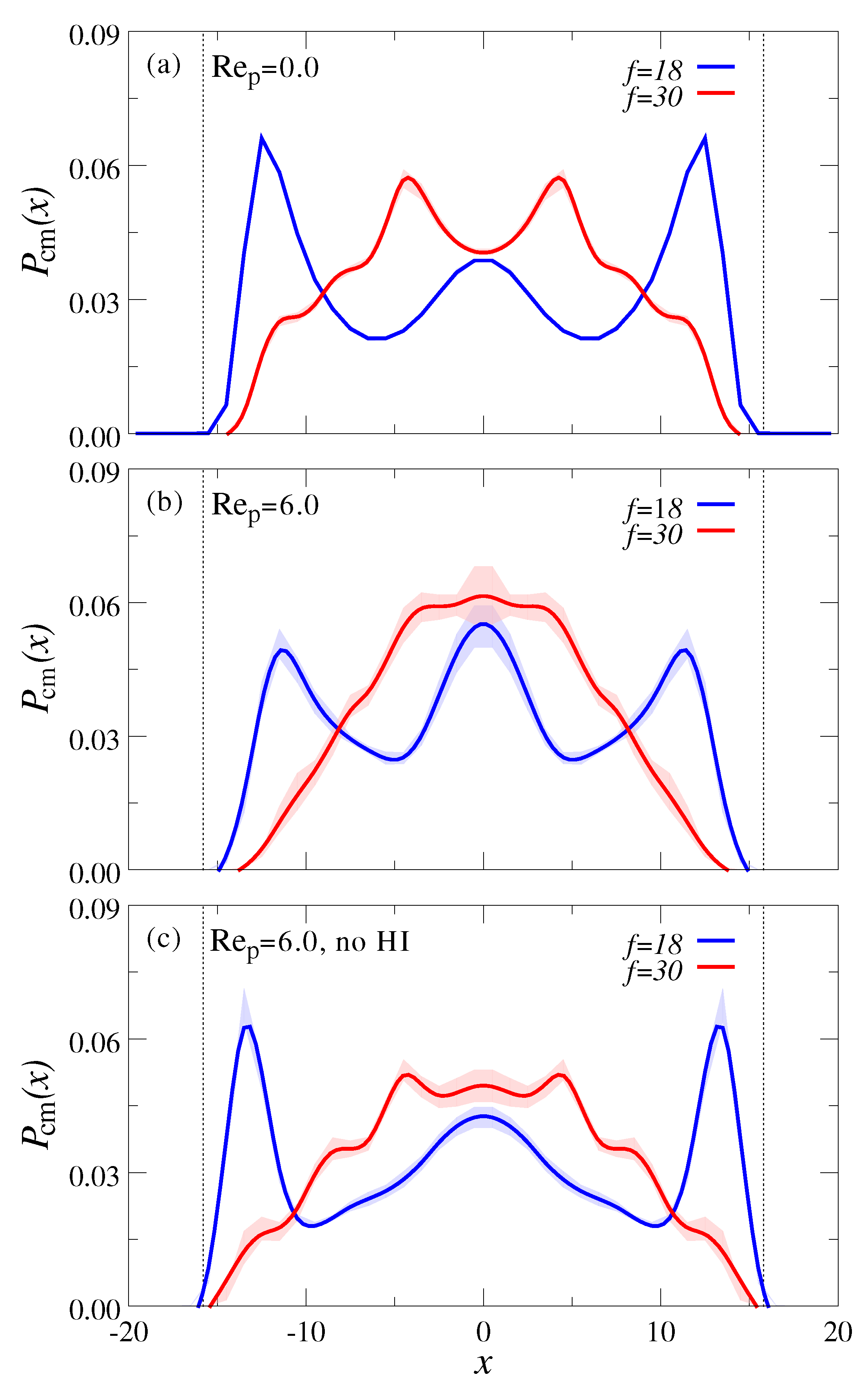
3.2. Polymer Mixtures
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CM | Center of mass |
| FENE | Finitely extensible nonlinear elastic |
| HI | Hydrodynamic interactions |
| MD | Molecular Dynamics |
| MPCD | Multi-particle collision dynamics |
| PDMS | Polydimethylsiloxane |
| PEO | Poly(ethylene oxide) |
| WCA | Weeks-Chandler-Andersen |
References
- Suresh, S. Biomechanics and biophysics of cancer cells. Acta Biomater. 2007, 3, 413–438. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K. Sickle cell anemia with few painful crises is characterized by decreased red cell deformability and increased number of dense cells. Am. J. Hematol. 1991, 36, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Squires, T.M.; Quake, S.R. Microfluidics: Fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005, 77, 977. [Google Scholar] [CrossRef]
- Chen, X.; Cui, D.F.; Liu, C.C.; Li, H. Microfluidic chip for blood cell separation and collection based on crossflow filtration. Sens. Actuators B Chem. 2008, 130, 216–221. [Google Scholar] [CrossRef]
- Fedosov, D.A.; Fornleitner, J.; Gompper, G. Margination of White Blood Cells in Microcapillary Flow. Phys. Rev. Lett. 2012, 108, 028104. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lee, S.S.; Ahn, S.W.; Kang, K.; Shim, W.; Lee, G.; Hyun, K.; Kim, J.M. Deformability-selective particle entrainment and separation in a rectangular microchannel using medium viscoelasticity. Soft Matter 2012, 8, 5011. [Google Scholar] [CrossRef]
- D’Avino, G.; Romeo, G.; Villone, M.M.; Greco, F.; Netti, P.A.; Maffettone, P.L. Single line particle focusing induced by viscoelasticity of the suspending liquid: Theory, experiments and simulations to design a micropipe flow-focuser. Lab Chip 2012, 12, 1638. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Yazdi, S.; Ardekani, A.M. Hydrodynamic mechanisms of cell and particle trapping in microfluidics. Biomicrofluidics 2013, 7, 021501. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Jimenez, M.; Birdle, H. Deterministic lateral displacement for particle separation: A review. Lab Chip 2014, 14, 4139. [Google Scholar] [CrossRef] [PubMed]
- Krüger, T.; Holmes, D.; Coveney, P.V. Deformability-based red blood cell separation in deterministic lateral displacement devices—A simulation study. Biomicrofluidics 2014, 8, 054114. [Google Scholar] [CrossRef] [PubMed]
- Zeming, K.K.; Salafi, T.; Chen, C.H.; Zhang, Y. Asymmetrical deterministic lateral displacement gaps for dual functions of enhanced separation and throughput of red blood cells. Sci. Rep. 2016, 6, 22934. [Google Scholar] [CrossRef] [PubMed]
- Sia, S.K.; Whitesides, G.M. Microfluidic devices fabricated in Poly(dimethylsiloxane) for biological studies. Electrophoresis 2003, 24, 3563–3576. [Google Scholar] [CrossRef] [PubMed]
- Segré, G.; Silberberg, A. Radial Particle Displacements in Poiseuille Flow of Suspensions. Nature 1961, 189, 209–210. [Google Scholar] [CrossRef]
- Karnis, K.; Mason, S.G. Particle motions in sheared suspensions. XIX. Viscoelastic media. Trans. Soc. Rheol. 1966, 10, 571. [Google Scholar] [CrossRef]
- Gauthier, F.; Goldsmith, H.L.; Mason, S.G. Particle motions in non-Newtonian media. I. Couette Flow. Rheol. Acta 1971, 10, 344–364. [Google Scholar] [CrossRef]
- Park, S.M.; Liang, X.; Harteneck, B.D.; Pick, T.E.; Hiroshiba, N.; Wu, Y.; Helms, B.A.; Olynick, D.L. Sub-10 nm Nanofabrication via Nanoimprint Directed Self-Assembly of Block Copolymers. ACS Nano 2011, 5, 8523–8531. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Babcock, H.P.; Chu, S. Single-Polymer Dynamics in Steady Shear Flow. Science 1999, 283, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.M.; Teixeria, R.E.; Shaqfeh, E.S.G.; Chu, S. Characteristic Periodic Motion of Polymers in Shear Flow. Phys. Rev. Lett. 2005, 95, 018301. [Google Scholar] [CrossRef] [PubMed]
- Lagally, E.T.; Medintz, I.; Mathies, R.A. Single-Molecule DNA Amplification and Analysis in an Integrated Microfluidic Device. Anal. Chem. 2001, 73, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Nikoubashman, A.; Likos, C.N. Flow-induced polymer translocation through narrow and patterned channels. J. Chem. Phys. 2010, 133, 074901. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ahn, S.W.; Lee, S.S.; Kim, J.M. Lateral migration and focusing of colloidal particles and DNA molecules under viscoelastic flow. Lab Chip 2012, 12, 2807–2814. [Google Scholar] [PubMed]
- Ollila, S.T.T.; Denniston, C.; Karttunen, M.; Ala-Nissila, T. Biopolymer Filtration in Corrugated Nanochannels Santtu. Phys. Rev. Lett. 2014, 112, 118301. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.B.; Nikoubashman, A.; Likos, C.N. Topology-Sensitive Microfluidic Filter for Polymers of Varying Stiffness. ACS Macro Lett. 2017, 6, 1426–1431. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, MS, USA, 2003. [Google Scholar]
- Khare, R.; Graham, M.D.; de Pablo, J.J. Cross-stream migration of flexible molecules in a nanochannel. Phys. Rev. Lett. 2006, 96, 224505. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortiz, J.P.; Ma, H.; de Pablo, J.J.; Graham, M.D. Cross-stream-line migration in confined flowing polymer solutions: Theory and simulation. Phys. Fluids 2006, 18, 123101. [Google Scholar] [CrossRef]
- Cannavacciulo, L.; Winkler, R.G.; Gompper, G. Mesoscale simulations of polymer dynamics in microchannel flows. Europhys. Lett. 2008, 83, 34007. [Google Scholar] [CrossRef][Green Version]
- Ripoll, M.; Winkler, R.G.; Gompper, G. Star polymers in shear flow. Phys. Rev. Lett. 2006, 96, 188302. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, M.; Winkler, R.G.; Gompper, G. Hydrodynamic screening of star polymers in shear flow. Eur. Phys. J. E 2007, 23, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Nikoubashman, A.; Likos, C.N. Branched polymers under shear. Macromolecules 2010, 43, 1610–1620. [Google Scholar] [CrossRef][Green Version]
- Vlassopoulos, D. Macromolecular topology and rheology: Beyond the tube model. Rheol. Acta 2016, 55, 613–632. [Google Scholar] [CrossRef]
- Winkler, R.G. Semiflexible polymers in shear flow. Phys. Rev. Lett. 2006, 97, 128301. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G. Conformational and Rheological properties of semiflexible polymers in shear flow. J. Chem. Phys. 2010, 133, 164905. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, R.; Winkler, R.G.; Gompper, G. Migration of semiflexible polymers in microcapillary flow. Europhys. Lett. 2010, 91, 14001. [Google Scholar] [CrossRef]
- Reddig, S.; Stark, H. Cross-streamline migration of a semiflexible polymer in a pressure driven flow. J. Chem. Phys. 2011, 135, 165101. [Google Scholar] [CrossRef] [PubMed]
- Nikoubashman, A.; Howard, M.P. Equilibrium Dynamics and Shear Rheology of Semiflexible Polymers in Solution. Macromolecules 2017, 50, 8279–8289. [Google Scholar] [CrossRef]
- Likos, C.N.; Löwen, H.; Watzlawek, M.; Abbas, B.; Jucknischke, O.; Allgaier, J.; Richter, D. Star polymers viewed as ultrasoft colloidal particles. Phys. Rev. Lett. 1998, 80, 4450–4453. [Google Scholar] [CrossRef]
- Likos, C.N. Effective interactions in soft condensed matter physics. Phys. Rep. 2001, 348, 267–439. [Google Scholar] [CrossRef]
- Vlassopoulos, D.; Fytas, G.; Pakula, T.; Roovers, J. Multiarm star polymers dynamics. J. Phys. Condens. Matter 2001, 13, R855. [Google Scholar] [CrossRef]
- Vlassopoulos, D.; Cloitre, M. Tunable rheology of dense soft deformable colloids. Curr. Opin. Colloid Interface Sci. 2014, 19, 561–574. [Google Scholar] [CrossRef]
- Riest, J.; Athanasopoulou, L.; Egorov, S.A.; Likos, C.N.; Ziherl, P. Elasticity of polymeric nanocolloidal particles. Sci. Rep. 2015, 5, 15854. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.; Gao, H.; Scroggins, S.; Unruh, D.A.; Avestro, A.J.; Fréchet, J.M.J. Easy Access to a Family of Polymer Catalysts from Modular Star Polymers. J. Am. Chem. Soc. 2010, 132, 2570. [Google Scholar] [CrossRef] [PubMed]
- Kanibolotsky, A.L.; Perepichka, I.F.; Skabara, P.J. Star-shaped Pi-conjugated oligomers and their applications in organic electronics and photonics. Chem. Soc. Rev. 2010, 39, 2695. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Duong, M.R.; Whittaker, M.R.; Davis, T.P.; Boyer, C. Synthesis of Functional Core, Star Polymers via RAFT Polymerization for Drug Delivery Applications. Macromol. Rapid Commun. 2012, 33, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Chatterji, A.; Gompper, G.; Winkler, R.G. Dynamical and Rheological Properties of Ultrasoft Colloids under Shear Flow. Macromolecules 2013, 46, 8026–8036. [Google Scholar] [CrossRef]
- Ge, H.; Pispas, S.; Wu, C. How does a star chain (nanooctopus) crawl through a nanopore? Polym. Chem. 2011, 2, 1071–1076. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Xiao, M.; Wang, R.; Chen, Y.L. Conformation-dependent translocation of a star polymer through a nanochannel. Biomicrofluidics 2014, 8, 054107. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Gompper, G.; Winkler, R.G. Steady state sedimentation of ultrasoft colloids. J. Chem. Phys. 2018, 148, 084901. [Google Scholar] [CrossRef] [PubMed]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Bishop, M.; Kalos, M.H.; Frisch, H.L. Molecular dynamics of polymeric systems. J. Chem. Phys. 1979, 70, 1299–1304. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts—A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Malevanets, A.; Kapral, R. Mesoscopic model for solvent dynamics. J. Chem. Phys. 1999, 110, 8605–8613. [Google Scholar] [CrossRef]
- Gompper, G.; Ihle, T.; Kroll, D.; Winkler, R.G. Multi-particle collision dynamics: A particle-based mesoscale simulation approach to the hydrodynamics of complex fluids. Adv. Polym. Sci. 2009, 221, 1–87. [Google Scholar]
- Huang, C.C.; Gompper, G.; Winkler, R.G. Hydrodynamic correlations in multiparticle collision dynamics fluids. Phys. Rev. E 2012, 86, 056711. [Google Scholar] [CrossRef] [PubMed]
- Allahyarov, E.; Gompper, G. Mesoscopic solvent simulations: Multiparticle-collision dynamics of three-dimensional flows. Phys. Rev. E 2002, 66, 036702. [Google Scholar] [CrossRef] [PubMed]
- Ihle, T.; Kroll, D.M. Stochastic rotation dynamics: A Galilean-invariant mesoscopic model for fluid flow. Phys. Rev. E 2001, 63, 020201. [Google Scholar] [CrossRef] [PubMed]
- Lamura, A.; Gompper, G.; Ihle, T.; Kroll, D.M. Multi-particle collision dynamics: Flow around a circular and a square cylinder. Europhys. Lett. 2001, 56, 319. [Google Scholar] [CrossRef]
- Nikoubashman, A.; Mahynski, N.A.; Pirayandeh, A.H.; Panagiotopoulos, A.Z. Flow-induced demixing of polymer-colloid mixtures in microfluidic channels. J. Chem. Phys. 2014, 140, 094903. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.P.; Panagiotopoulos, A.Z.; Nikoubashman, A. Inertial and viscoelastic forces on rigid colloids in microfluidic channels. J. Chem. Phys. 2015, 142, 224908. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Ripoll, M.; Mussawisade, K.; Winkler, R.G.; Gompper, G. Dynamic regimes of fluids simulated by multiparticle-collision dynamics. Phys. Rev. E 2005, 72, 016701. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Lorenz, C.D.; Travesset, A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 2008, 227, 5342–5359. [Google Scholar] [CrossRef]
- Glaser, J.; Nguyen, T.D.; Anderson, J.A.; Liu, P.; Spiga, F.; Millan, J.A.; Morse, D.C.; Glotzer, S.C. Strong scaling of general-purpose molecular dynamics simulations on GPUs. Comput. Phys. Commun. 2015, 192, 97–107. [Google Scholar] [CrossRef]
- Howard, M.P.; Anderson, J.A.; Nikoubashman, A.; Glotzer, S.C.; Panagiotopoulos, A.Z. Efficient neighbor list calculation for molecular simulation of colloidal systems using graphics processing units. Comput. Phys. Commun. 2016, 203, 45–52. [Google Scholar] [CrossRef]
- Jusufi, A.; Dzubiella, J.; Likos, C.N.; von Ferber, C.; Löwen, H. Effective interactions between star polymers and colloidal particles. J. Phys. Condens. Matter 2001, 13, 6177–6194. [Google Scholar] [CrossRef]
- Marzi, D.; Likos, C.N.; Capone, B. Coarse graining of star-polymer–colloid nanocomposites. J. Chem. Phys. 2012, 137, 014902. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Winkler, R.G.; Stutmann, G.; Gompper, G. Semidilute Polymer Solutions at Equilibrium and under Shear Flow. Macromolecules 2010, 43, 10107. [Google Scholar] [CrossRef]
- Eslami, H.; Müller-Plathe, F. Viscosity of Nanoconfined Polyamide-6,6 Oligomers: Atomistic Reverse Nonequilibrium Molecular Dynamics Simulation. J. Phys. Chem. B 2010, 114, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Zimm, B.H. Dynamics of Polymer Molecules in Dilute Solution: Viscoelasticity, Flow Birefringence and Dielectric Loss. J. Chem. Phys. 1956, 24, 269–278. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K.; Milner, S.T.; Witten, T.A. Relaxation of self-entangled many-arm star polymers. Macromolecules 1989, 22, 1904–1910. [Google Scholar] [CrossRef]
- Wohl, R.P.; Rubinow, S.I. The transverse force on a drop in an unbounded parabolic flow. J. Fluid Mech. 1974, 62, 185–207. [Google Scholar] [CrossRef]
- Chan, P.C.H.; Leal, L.G. The motion of a deformable drop in a second-order fluid. J. Fluid Mech. 1979, 92, 131–170. [Google Scholar] [CrossRef]
- Marson, R.; Huang, Y.; Huang, M.; Fu, T.; Larson, R.G. Inertio-Capillary Cross-Streamline Drift of Droplets in Poiseuille Flow using Dissipative Particle Dynamics Simulations. Soft Matter 2018, 14, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.; Stark, H. Inertial migration and axial control of deformable capsules. Soft Matter 2017, 13, 3544–3555. [Google Scholar] [CrossRef] [PubMed]
- Perram, J.W.; White, L.R. Structure of the liquid/vapour and liquid/solid interfaces. Faraday Discuss. Chem. Soc. 1975, 59, 29–37. [Google Scholar] [CrossRef]
- Henderson, D.; Abraham, F.F.; Barker, J.A. The Ornstein-Zernike equation for a fluid in contact with a surface. Mol. Phys. 1976, 31, 1291–1295. [Google Scholar] [CrossRef]
- Snook, I.K.; Henderson, D. Monte Carlo study of a hard-sphere fluid near a hard wall. J. Chem. Phys. 1978, 68, 2134. [Google Scholar] [CrossRef]
- Howard, M.P.; Gautam, A.; Panagiotopoulos, A.Z.; Nikoubashman, A. Axial dispersion of Brownian colloids in microfluidic channels. Phys. Rev. Fluids 2016, 1, 044203. [Google Scholar] [CrossRef]
- Devanand, K.; Selser, J.C. Asymptotic behavior and long-range interactions in aqueous solutions of poly(ethylene oxide). Macromolecules 1991, 24, 5943–5947. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastva, D.; Nikoubashman, A. Flow Behavior of Chain and Star Polymers and Their Mixtures. Polymers 2018, 10, 599. https://doi.org/10.3390/polym10060599
Srivastva D, Nikoubashman A. Flow Behavior of Chain and Star Polymers and Their Mixtures. Polymers. 2018; 10(6):599. https://doi.org/10.3390/polym10060599
Chicago/Turabian StyleSrivastva, Deepika, and Arash Nikoubashman. 2018. "Flow Behavior of Chain and Star Polymers and Their Mixtures" Polymers 10, no. 6: 599. https://doi.org/10.3390/polym10060599
APA StyleSrivastva, D., & Nikoubashman, A. (2018). Flow Behavior of Chain and Star Polymers and Their Mixtures. Polymers, 10(6), 599. https://doi.org/10.3390/polym10060599