Synthesis and Properties of Silk Fibroin/Konjac Glucomannan Blend Beads

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Beads Synthesis
2.1.1. Solutions Preparation
2.1.2. Beads Synthesis
2.2. Physicochemical Characterizations
2.2.1. Surface Tension and Size Beads
2.2.2. Morphological Characterization
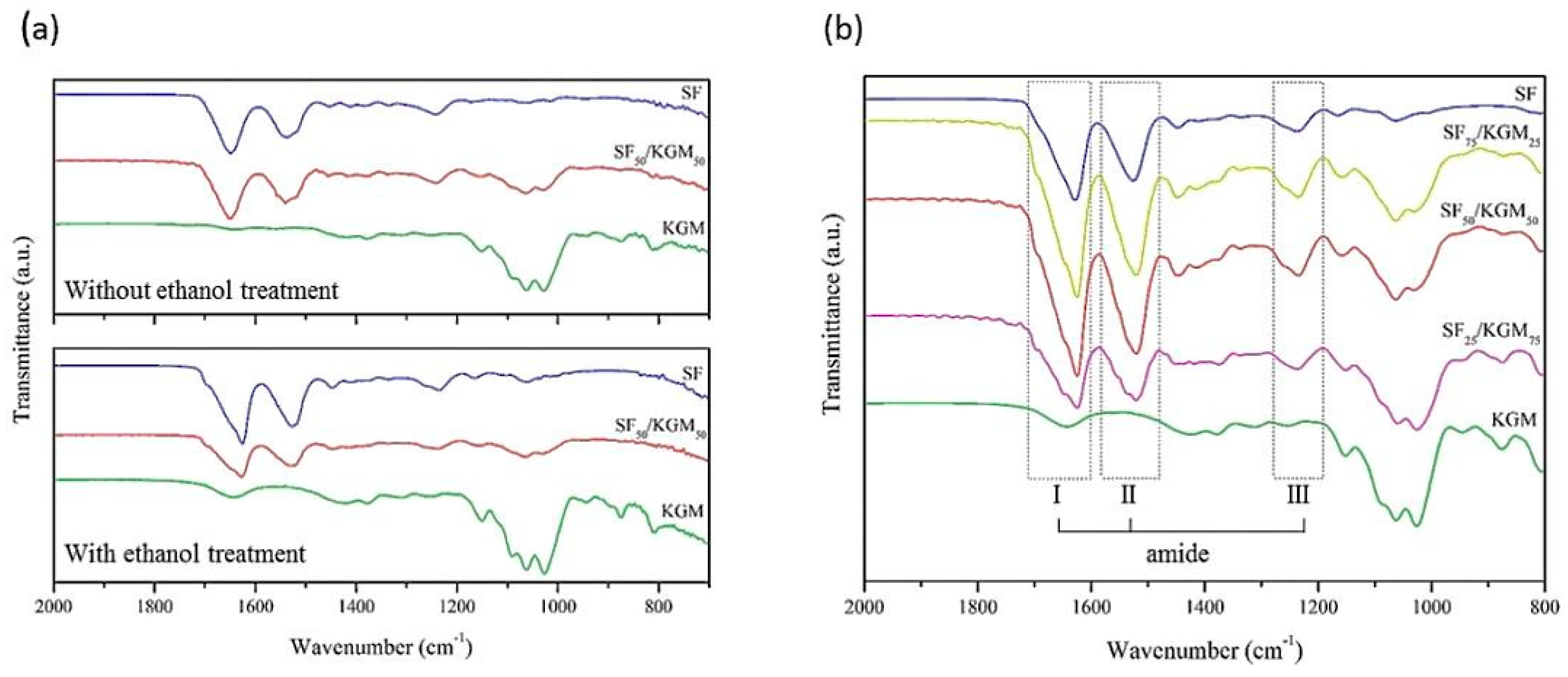
2.2.3. Chemical Characterization
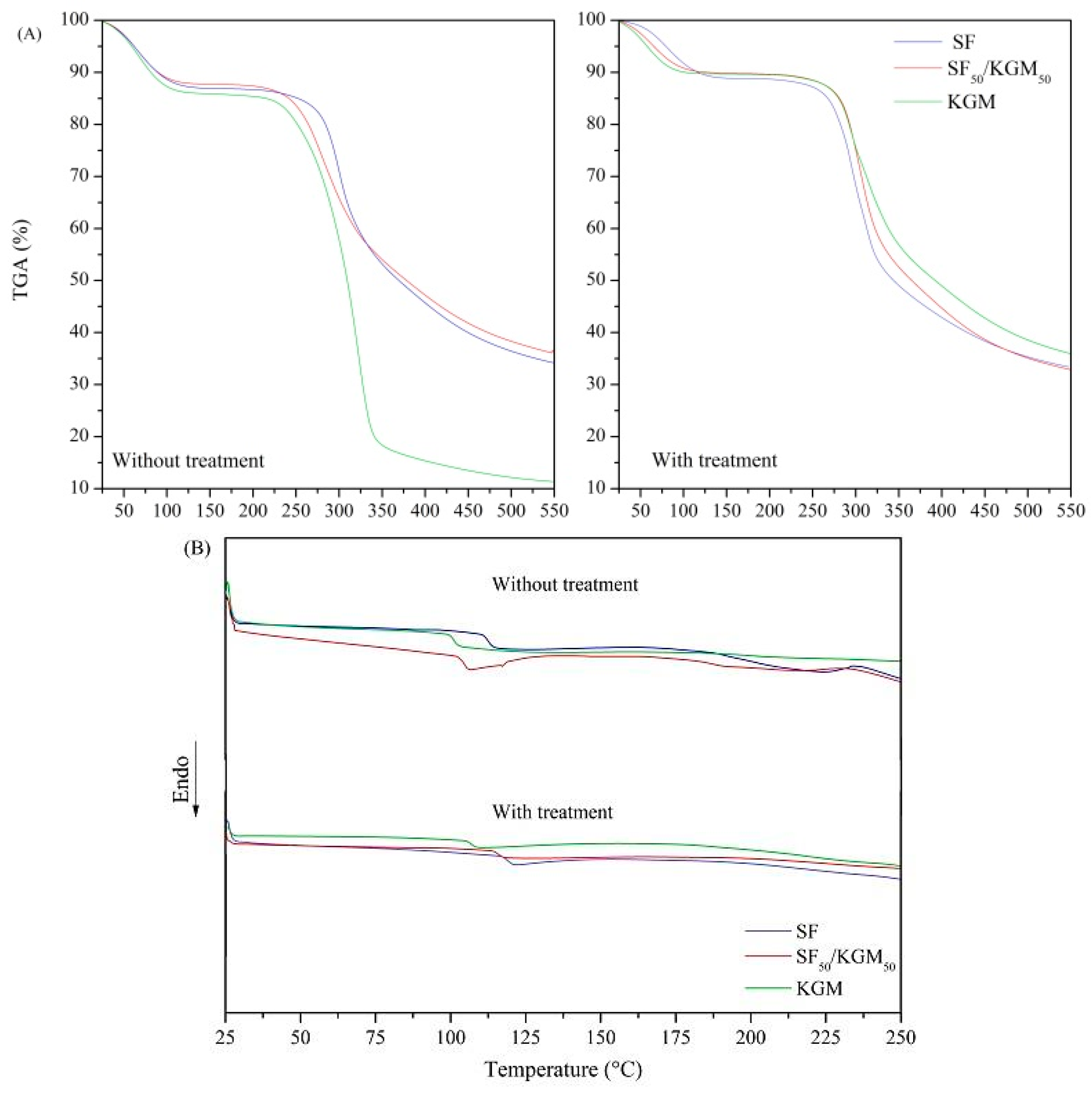
2.2.4. Thermogravimetric Characterization
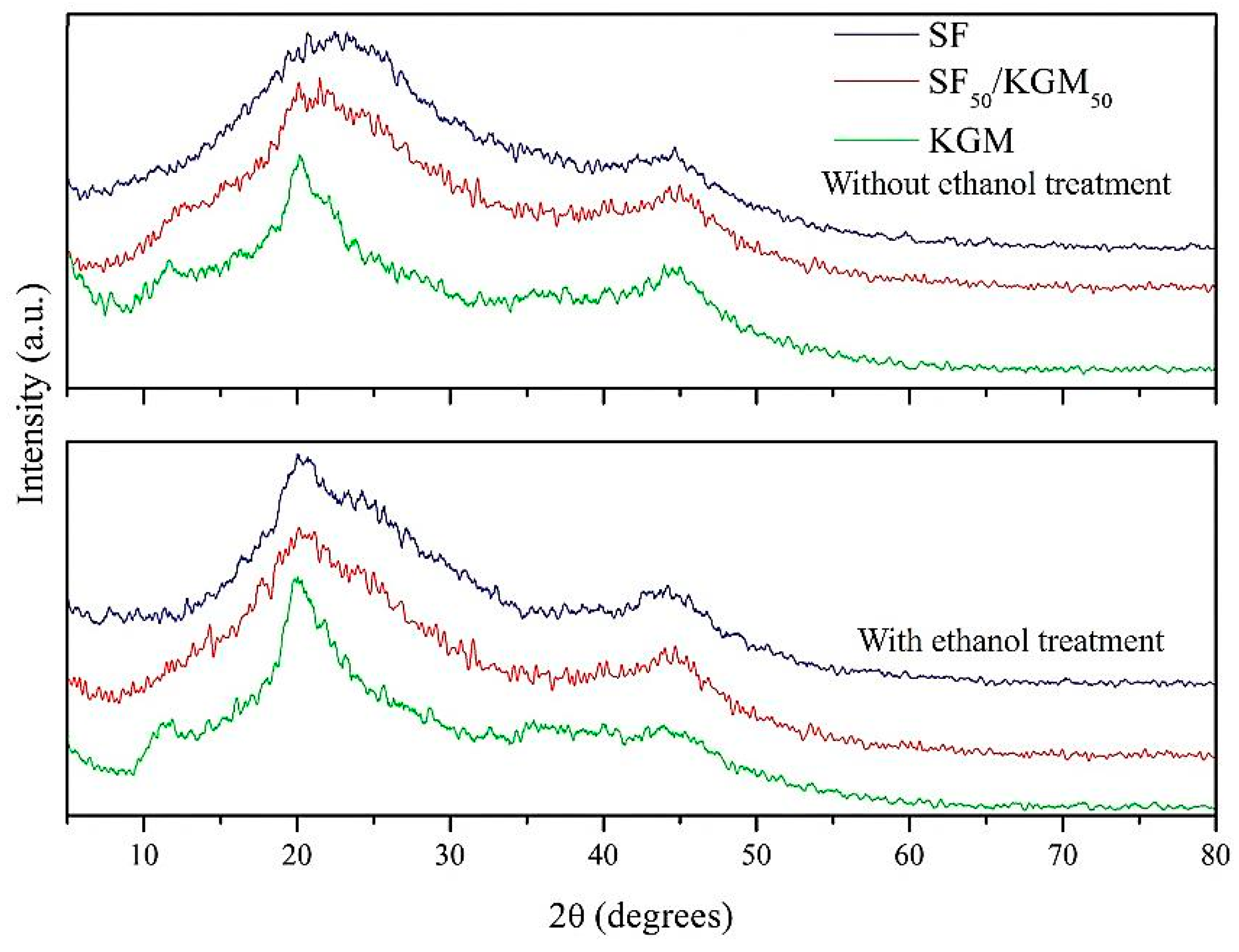
2.2.5. Crystallinity
2.3. Mechanical Assays
2.4. Cytotoxicity Assays
2.4.1. Cell Culture
2.4.2. MTT Reduction Assay
2.5. Statistical Analysis
3. Results and Discussions
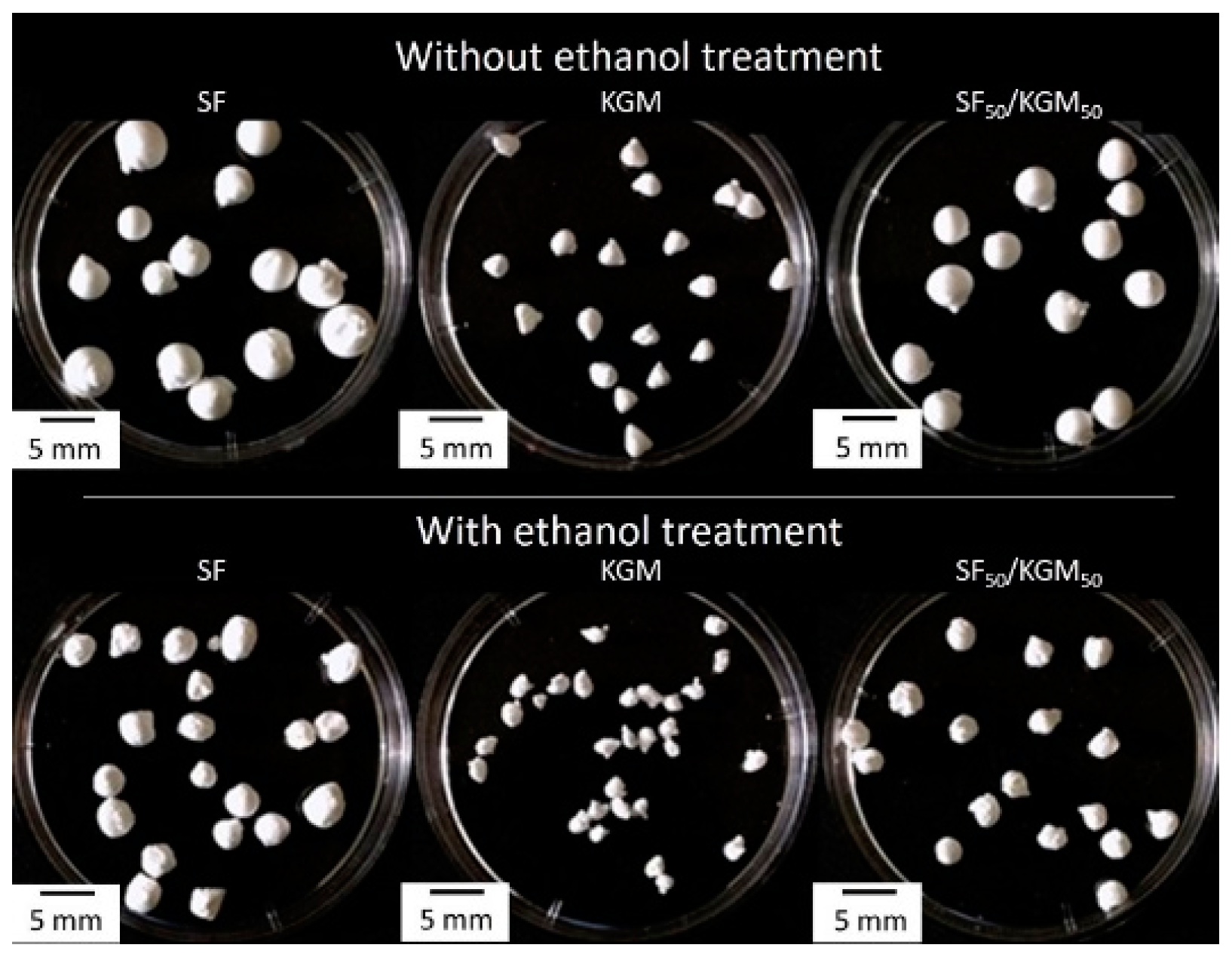
3.1. Size Beads and Surface Tension
3.2. Morphological Characterization
3.3. Chemical Characterization
3.4. Thermal Characterization
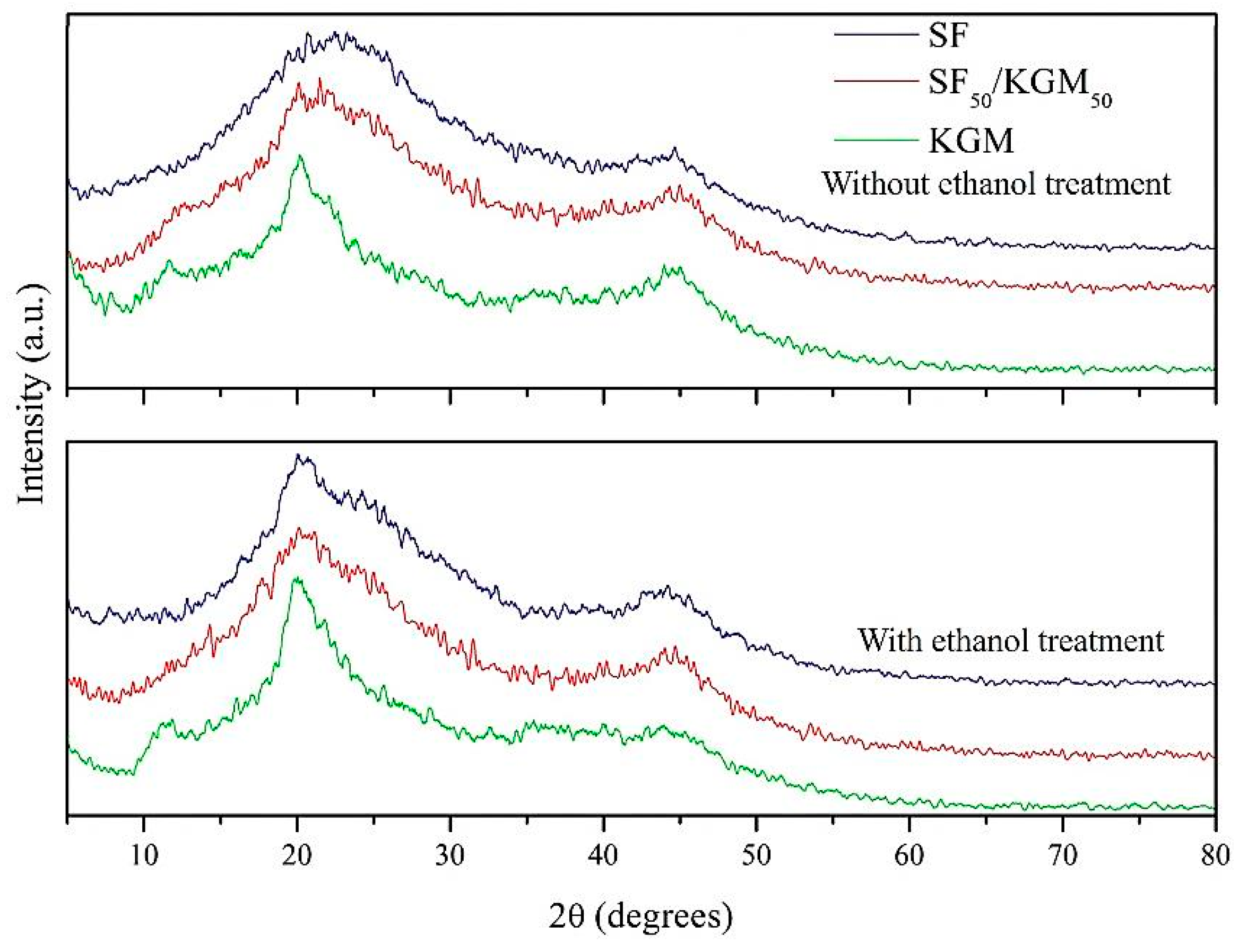
3.5. Crystallinity
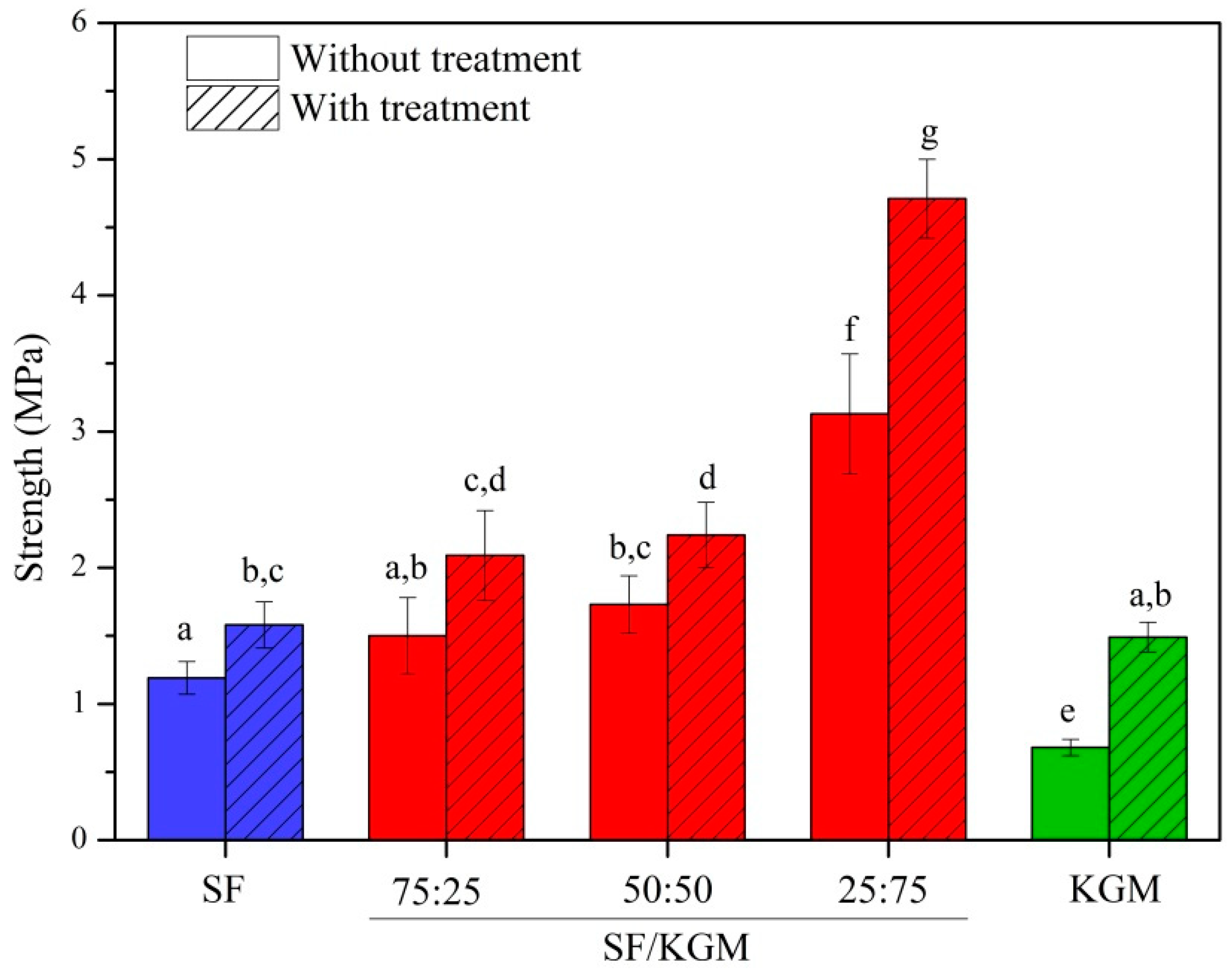
3.6. Mechanical Assays
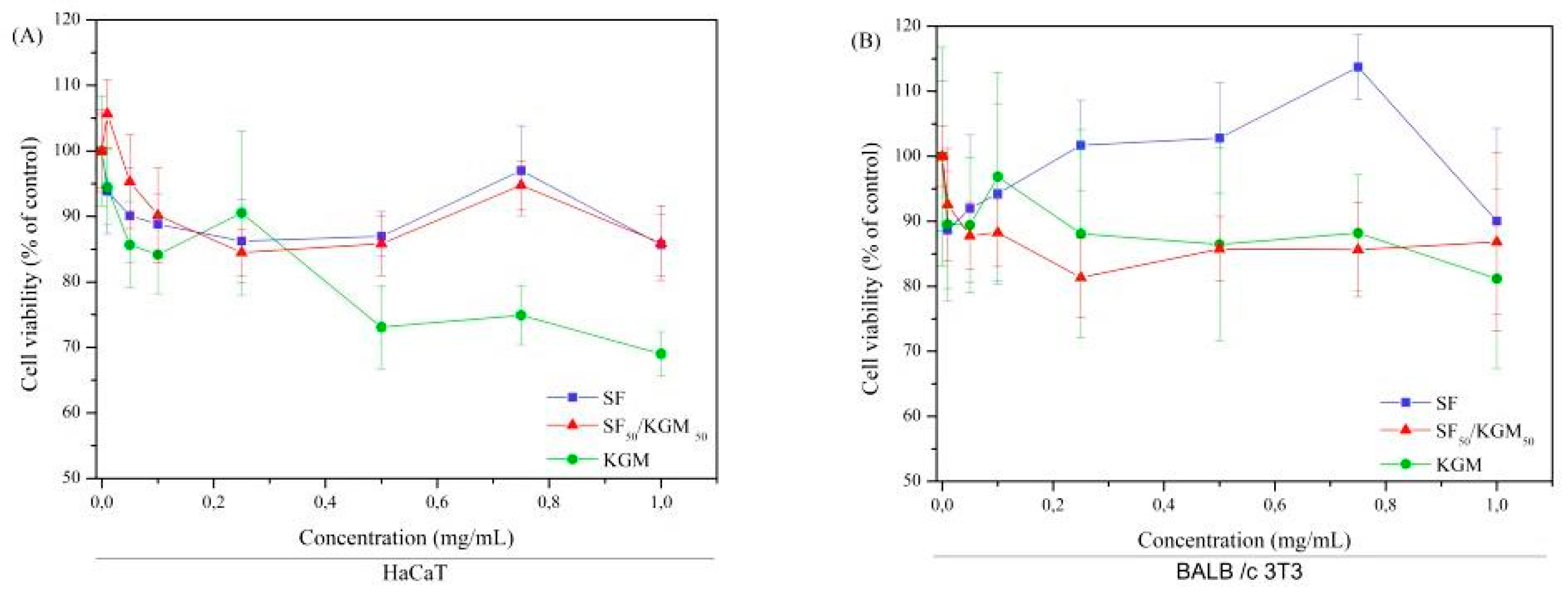
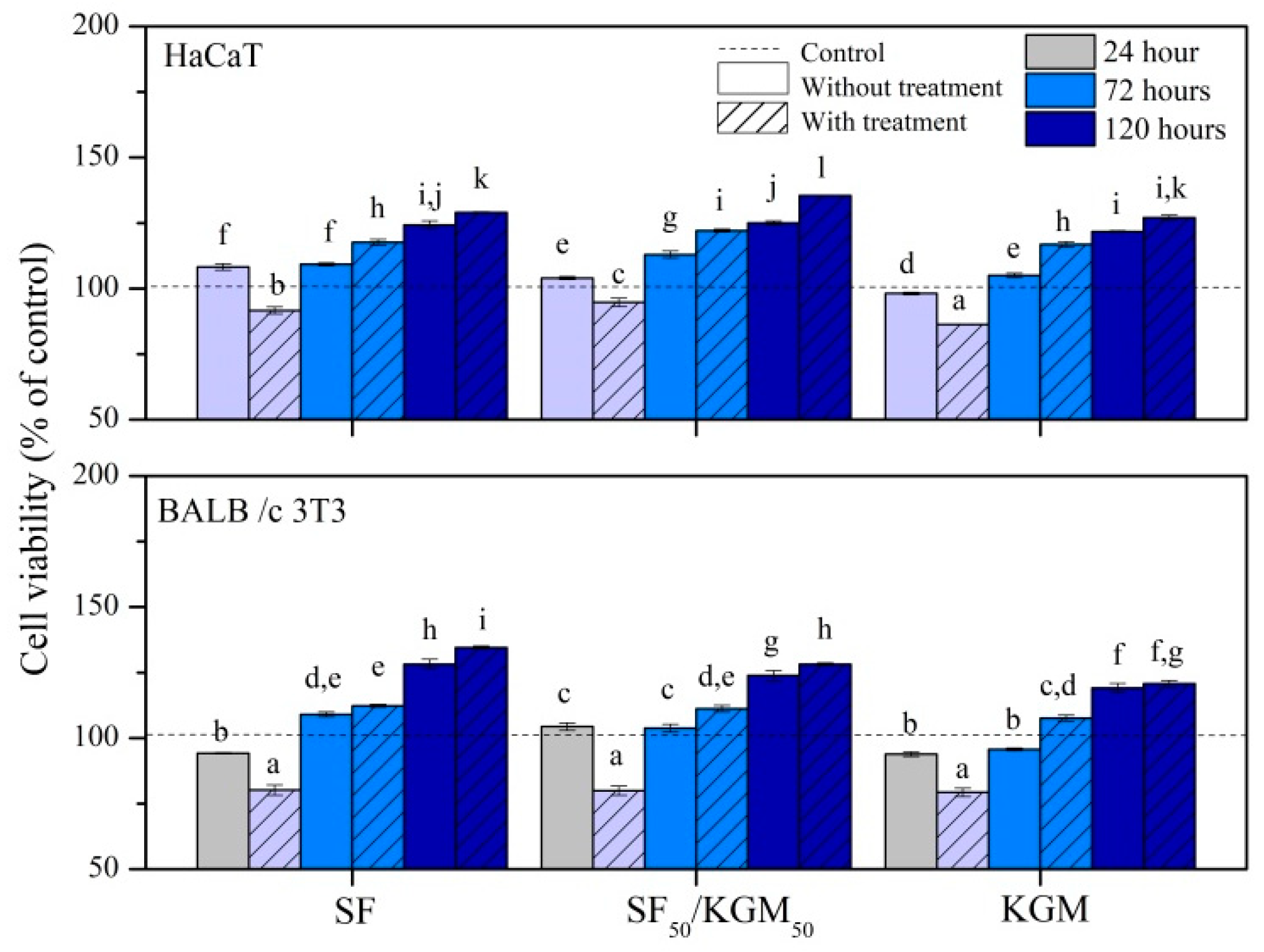
3.7. Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, X.; Wang, S.; Lu, M.; Chen, Y.; Zhao, L.; Li, W.; Yuan, Q.; Norde, W.; Li, Y. Formation and characterization of light-responsive TEMPO-oxidized konjac glucomannan microspheres. Biomacromolecules 2014, 15, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yucel, T.; Lu, Q.; Hu, X.; Kaplan, D.L. Silk nanospheres and microspheres from silk/pva blend films for drug delivery. Biomaterials 2010, 31, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, H.; Tsukada, M. New silk protein: Modification of silk protein by gene engineering for production of biomaterials. Rev. Mol. Biotecnol. 2000, 74, 95–103. [Google Scholar] [CrossRef]
- Hakimi, O.; Knight, D.P.; Vollrath, F.; Vadgama, P. Spider and mulberry silkworm silks as compatible biomaterials. Compos. Part. B Eng. 2007, 38, 324–337. [Google Scholar] [CrossRef]
- Li, M.Z.; Lu, S.Z.; Wu, Z.Y.; Tan, K.; Minoura, N.; Kuga, S. Structure and properties of silk fibroin-poly(vinyl alcohol) gel. Int. J. Biol. Macromol. 2002, 30, 89–94. [Google Scholar] [CrossRef]
- Altman, G.H.; Diaz, F.; Jakuba, C.; Calabro, T.; Horan, R.L.; Chen, J.S.; Lu, H.; Richmond, J.; Kaplan, D.L. Silk-based biomaterials. Biomaterials 2003, 24, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Sande, M.; Teijeiro-Osorio, D.; Remunan-Lopez, C.; Alonso, M.J. Glucomannan, a promising polysaccharide for biopharmaceutical purposes. Eur. J. Pharm. Biopharm. 2009, 72, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mancenido, F.; Landin, M.; Lacik, I.; Martinez-Pacheco, R. Konjac glucomannan and konjac glucomannan/xanthan gum mixtures as excipients for controlled drug delivery systems. Diffusion of small drugs. Int. J. Pharm. 2008, 349, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, K.Y.; Ha, W.S.; Park, S.Y. Structural changes and their effect on mechanical properties of silk fibroin/chitosan blends. J. Appl. Polym. Sci. 1999, 74, 2571–2575. [Google Scholar] [CrossRef]
- Kweon, H.; Ha, H.C.; Um, I.C.; Park, Y.H. Physical properties of silk fibroin/chitosan blend films. J. Appl. Polym. Sci. 2001, 80, 928–934. [Google Scholar] [CrossRef]
- Lee, K.G.; Kweon, H.Y.; Yeo, J.H.; Woo, S.O.; Lee, J.H.; Park, Y.H. Structural and physical properties of silk fibroin/alginate blend sponges. J. Appl. Polym. Sci. 2004, 93, 2174–2179. [Google Scholar] [CrossRef]
- She, Z.D.; Zhang, B.F.; Jin, C.R.; Feng, Q.L.; Xu, Y.X. Preparation and in vitro degradation of porous three-dimensional silk fibroin/chitosan scaffold. Polym. Degrad. Stab. 2008, 93, 1316–1322. [Google Scholar] [CrossRef]
- De Alteriis, R.; Vecchione, R.; Attanasio, C.; De Gregorio, M.; Porzio, M.; Battista, E.; Netti, P.A. A method to tune the shape of protein-encapsulated polymeric microspheres. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Um, I.C.; Kweon, H.Y.; Park, Y.H.; Hudson, S. Structural characteristics and properties of the regenerated silk fibroin prepared from formic acid. Int. J. Biol. Macromol. 2001, 29, 91–97. [Google Scholar] [CrossRef]
- Qu, J.Q.; Wang, L.W.; Hu, Y.; Wang, L.; You, R.Y.; Li, M.L. Preparation of silk fibroin microspheres and its cytocompatibility. Biomat. Nanobiotechnol. 2013, 4, 84–90. [Google Scholar] [CrossRef]
- Li, J.; Ye, T.; Wu, X.; Chen, J.; Wang, S.; Lin, L.; Li, B. Preparation and characterization of heterogeneous deacetylated konjac glucomannan. Food Hydrocoll. 2014, 40, 9–15. [Google Scholar] [CrossRef]
- Sun, L.; Xiong, Z.; Zhou, W.; Liu, R.; Yan, X.; Li, J.; An, W.; Yuan, G.; Ma, G.; Su, Z. Novel konjac glucomannan microcarriers for anchorage-dependent animal cell culture. Biochem. Eng. J. 2015, 96, 46–54. [Google Scholar] [CrossRef]
- Wang, L.; Xie, H.G.; Qiao, X.Y.; Goffin, A.; Hodgkinson, T.; Yuan, X.F.; Sun, K.; Fuller, G.G. Interfacial rheology of natural silk fibroin at air/water and oil/water interfaces. Langmuir 2012, 28, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and s urvival: Application to proliferation and c ytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lautrup, B. Physics of Continuous Matter: Exotic and Everyday Phenomena in the Macroscopic World; CRC Press: Boca Raton, FL, USA, 2011; p. 696. [Google Scholar]
- Gong, Z.; Huang, L.; Yang, Y.; Chen, X.; Shao, Z. Two distinct beta-sheet fibrils from silk protein. Chem. Commun. 2009, 7506–7508. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.; Chan, K.; Hocking, T.J.; Williams, P.A.; Perry, C.J.; Baldwin, T.C. Methodologies for the extraction and analysis of konjac glucomannan from corms of Amorphophallus konjac K. Koch. Carbohydr. Polym. 2012, 87, 2202–2210. [Google Scholar] [CrossRef]
- Teimouri, A.; Ebrahimi, R.; Chermahini, A.N.; Emadi, R. Fabrication and characterization of silk fibroin/chitosan/nano gamma-alumina composite scaffolds for tissue engineering applications. RSC Adv. 2015, 5, 27558–27570. [Google Scholar] [CrossRef]
- Sepulveda, P.; Binner, J.G.P.; Rogero, S.O.; Higa, O.Z.; Bressiani, J.C. Production of porous hydroxyapatite by the gel-casting of foams and cytotoxic evaluation. J. Biomed. Mater. Res. 2000, 50, 27–34. [Google Scholar] [CrossRef]
- Rusa, C.C.; Bridges, C.; Ha, S.W.; Tonelli, A.E. Conformational changes induced in Bombyx mori silk fibroin by cyclodextrin inclusion complexation. Macromolecules 2005, 38, 5640–5646. [Google Scholar] [CrossRef]
- Lv, Q.; Cao, C.B.; Zhang, Y.; Ma, X.L.; Zhu, H.S. Preparation of insoluble fibroin films without methanol treatment. J. Appl. Polym. Sci. 2005, 96, 2168–2173. [Google Scholar] [CrossRef]
- Zhang, H.; Yoshimura, M.; Nishinari, K.; Williams, M.A.K.; Foster, T.J.; Norton, I.T. Gelation behaviour of konjac glucomannan with different molecular weights. Biopolymers 2001, 59, 38–50. [Google Scholar] [CrossRef]
- Jin, W.; Song, R.; Xu, W.; Wang, Y.; Li, J.; Shah, B.R.; Li, Y.; Li, B. Analysis of deacetylated konjac glucomannan and xanthan gum phase separation by film forming. Food Hydrocoll. 2015, 48, 320–326. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, L.N.; Liu, Y.G. Structure and microporous formation of cellulose/silk fibroin blend membranes I. Effect of coagulants. J. Membr. Sci. 2000, 177, 153–161. [Google Scholar] [CrossRef]
- Shang, S.; Zhu, L.; Fan, J. Intermolecular interactions between natural polysaccharides and silk fibroin protein. Carbohydr. Polym. 2013, 93, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.X.; Hirabeyashi, K. Improvemets of the physical properties of fibroin membranes with sodium alginate. J. Appl. Polym. Sci. 1992, 45, 1937–1943. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.J.; Yu, T.Y. Conformation transition of silk fibroin induced by blending chitosan. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 2293–2296. [Google Scholar] [CrossRef]
- Hu, X.; Kaplan, D.; Cebe, P. Determining beta-sheet crystallinity in fibrous proteins by thermal analysis and infrared spectroscopy. Macromolecules 2006, 39, 6161–6170. [Google Scholar] [CrossRef]
- Matsumoto, A.; Chen, J.; Collette, A.L.; Kim, U.-J.; Altman, G.H.; Cebe, P.; Kaplan, D.L. Mechanisms of silk fibroin sol-gel transitions. J. Phys. Chem. B 2006, 110, 21630–21638. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Qi, Z.; Knight, D.P.; Shao, Z.; Chen, X. Synchrotron FTIR microspectroscopy of single natural silk fibers. Biomacromolecules 2011, 12, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Fowler, B.O.; Tung, M.S. Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, Y.; Ying, H.; Xiao, C. Preparation and characterization of a quaternary ammonium derivative of konjac glucomannan. Carbohydr. Polym. 2007, 69, 29–40. [Google Scholar] [CrossRef]
- Enomoto-Rogers, Y.; Ohmomo, Y.; Iwata, T. Syntheses and characterization of konjac glucomannan acetate and their thermal and mechanical properties. Carbohydr. Polym. 2013, 92, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Mei, T.; Wang, Y.; Xu, W.; Li, J.; Zhou, B.; Li, B. Synergistic degradation of konjac glucomannan by alkaline and thermal method. Carbohydr. Polym. 2014, 99, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Canevarolo, S.V., Jr. Técnicas de Caracterização de Polímeros; Artliber: São Paulo, Brazil, 2003; 448p. [Google Scholar]
- Teimouri, A.; Ebrahimi, R.; Emadi, R.; Beni, B.H.; Chermahini, A.N. Nano-composite of silk fibroin-chitosan/nano ZrO2 for tissue engineering applications: Fabrication and morphology. Int. J. Biol. Macromol. 2015, 76, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, B.; Kennedy, J.F.; Xie, B.J.; Huang, M. Characterization of konjac glucomannan-gellan gum blend films and their suitability for release of nisin incorporated therein. Carbohydr. Polym. 2007, 7, 192–197. [Google Scholar] [CrossRef]
- Lawrence, B.D.; Omenetto, F.; Chui, K.; Kaplan, D.L. Processing methods to control silk fibroin film biomaterial features. J. Mater. Sci. 2008, 43, 6967–6985. [Google Scholar] [CrossRef]
- Guang, S.; An, Y.; Ke, F.; Zhao, D.; Shen, Y.; Xu, H. Chitosan/silk fibroin composite scaffolds for wound dressing. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Yin, A.L.; Zhang, K.H.; McClure, M.J.; Huang, C.; Wu, J.L.; Fang, J.; Mo, X.M.; Bowlin, G.L.; Al-Deyab, S.S.; El-Newehy, M. Electrospinning collagen/chitosan/poly(l-lactic acid-co-epsilon-caprolactone) to form a vascular graft: Mechanical and biological characterization. J. Biomed. Mater. Res. Part A 2013, 101, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. Biological Evaluation of Medical Devices—Part 5 Tests for In Vitro Cytotoxicity; ISO 10993-5; ISO: Geneva, Switzerland, 2009. [Google Scholar]
- Hernandez-Montelongo, J.; Lucchesi, E.G.; Nascimento, V.F.; Franca, C.G.; Gonzalez, I.; Macedo, W.A.A.; Machado, D.; Lancellotti, M.; Moraes, A.M.; et al. Antibacterial and non-cytotoxic ultra-thin polyethylenimine film. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 71, 718–724. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Surface Tension of Solutions (mPa) * | The Diameter of Beads without Ethanol Treatment (mm) * | The Diameter of Beads with Ethanol Treatment (mm) * |
|---|---|---|---|
| SF | 45.2 ± 0.2 a | 3.4 ± 0.1 d | 3.1 ± 0.1 h |
| KGM | 63.7 ± 0.8 b | 2.0 ± 0.2 f | 1.8 ± 0.1 e |
| SF50/KGM50 | 50.7 ± 0.7 c | 3.2 ± 0.15 d | 2.5 ± 0.1 g |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
França, C.G.; Nascimento, V.F.; Hernandez-Montelongo, J.; Machado, D.; Lancellotti, M.; Beppu, M.M. Synthesis and Properties of Silk Fibroin/Konjac Glucomannan Blend Beads. Polymers 2018, 10, 923. https://doi.org/10.3390/polym10080923
França CG, Nascimento VF, Hernandez-Montelongo J, Machado D, Lancellotti M, Beppu MM. Synthesis and Properties of Silk Fibroin/Konjac Glucomannan Blend Beads. Polymers. 2018; 10(8):923. https://doi.org/10.3390/polym10080923
Chicago/Turabian StyleFrança, Carla Giometti, Vicente Franco Nascimento, Jacobo Hernandez-Montelongo, Daisy Machado, Marcelo Lancellotti, and Marisa Masumi Beppu. 2018. "Synthesis and Properties of Silk Fibroin/Konjac Glucomannan Blend Beads" Polymers 10, no. 8: 923. https://doi.org/10.3390/polym10080923
APA StyleFrança, C. G., Nascimento, V. F., Hernandez-Montelongo, J., Machado, D., Lancellotti, M., & Beppu, M. M. (2018). Synthesis and Properties of Silk Fibroin/Konjac Glucomannan Blend Beads. Polymers, 10(8), 923. https://doi.org/10.3390/polym10080923