Gellan Gum-Based Hydrogel for the Transdermal Delivery of Nebivolol: Optimization and Evaluation



Abstract
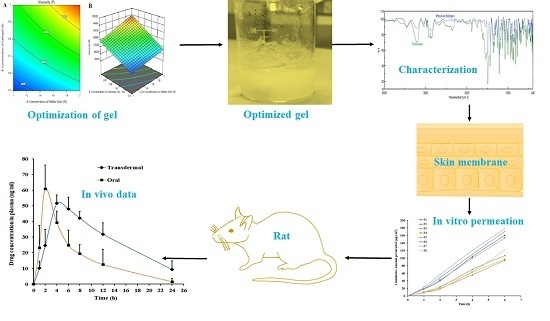
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HPLC Determination of Nebivolol
2.3. Fourier-Transform Infrared (FTIR) Spectroscopy
2.4. Formulation of Design Batches Using 23 Full Factorial
2.5. Evaluation of Gels
2.5.1. Differential Scanning Calorimetry (DSC)
2.5.2. Drug Content
2.6. Drug Release
2.7. Ex Vivo Permeation
2.8. In Vivo Studies
2.9. Stability Studies
2.10. Skin Irritation
2.11. Data Analysis
3. Result and Discussion
3.1. FTIR Spectroscopy
3.2. Physicochemical Properties of Gels
3.3. DSC
3.4. The Effect of Formulation Variables on Viscosity
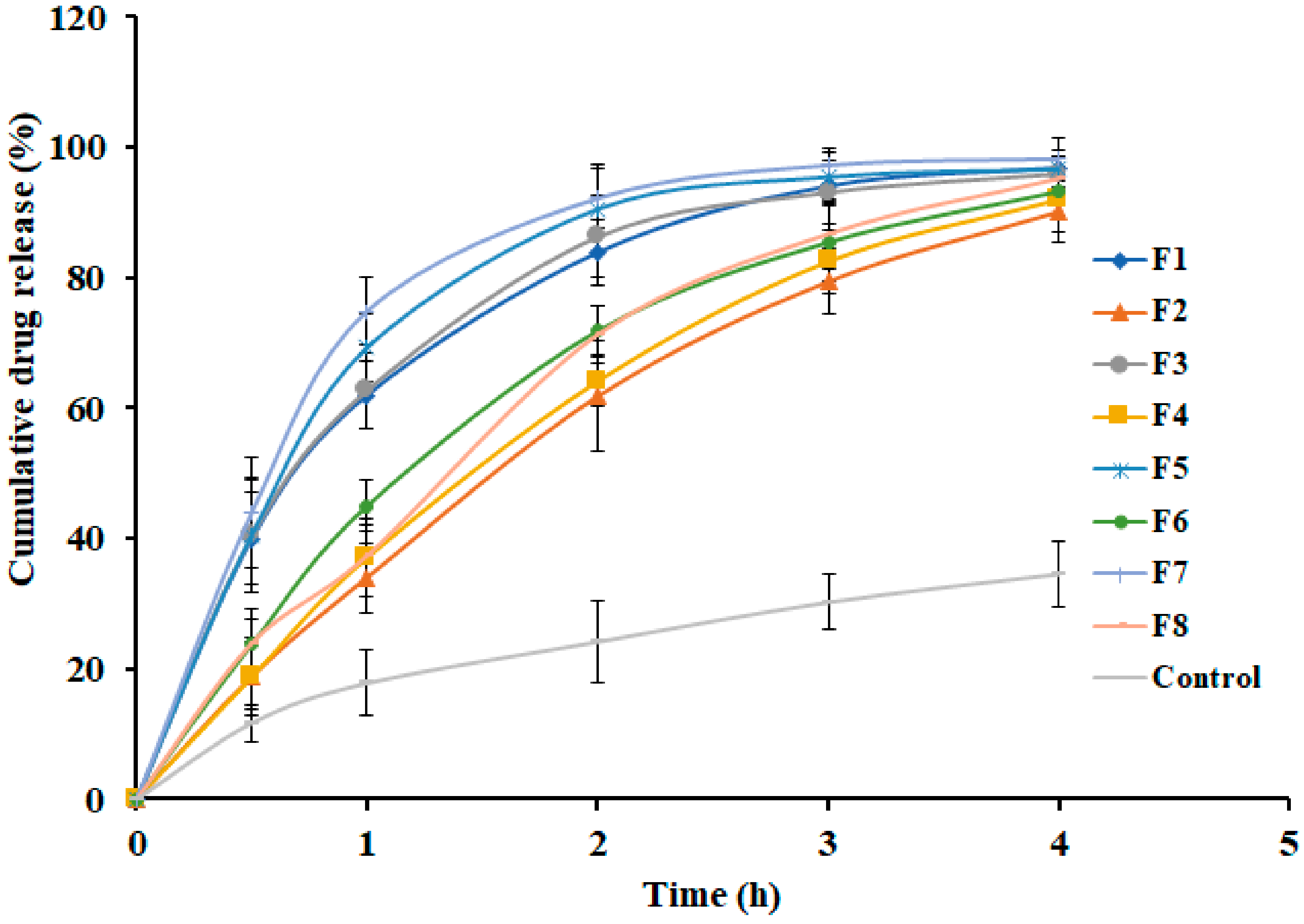
3.5. Effect of Formulation Variables on Drug Release
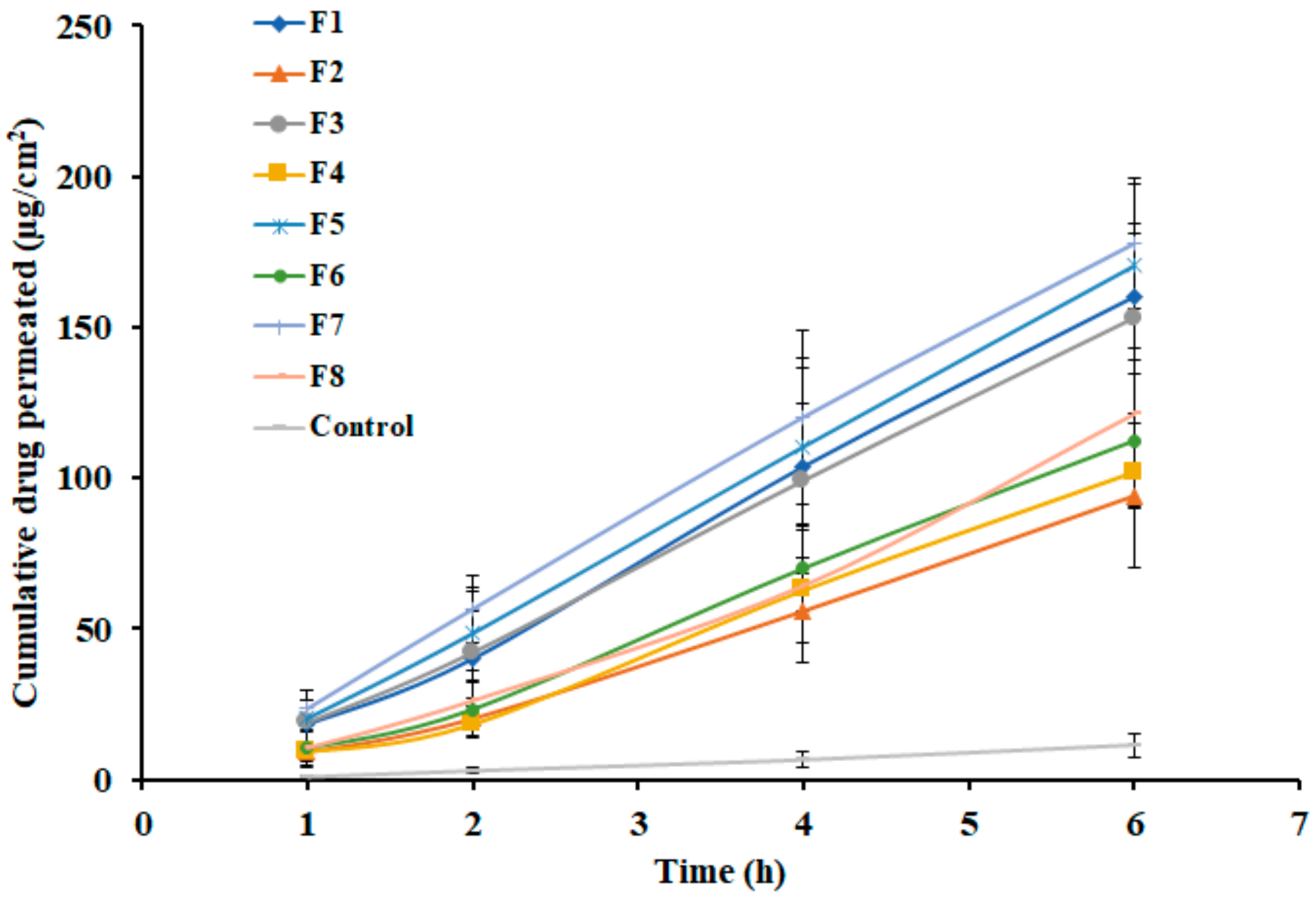
3.6. Effect of Formulation Variables on Ex Vivo Permeation
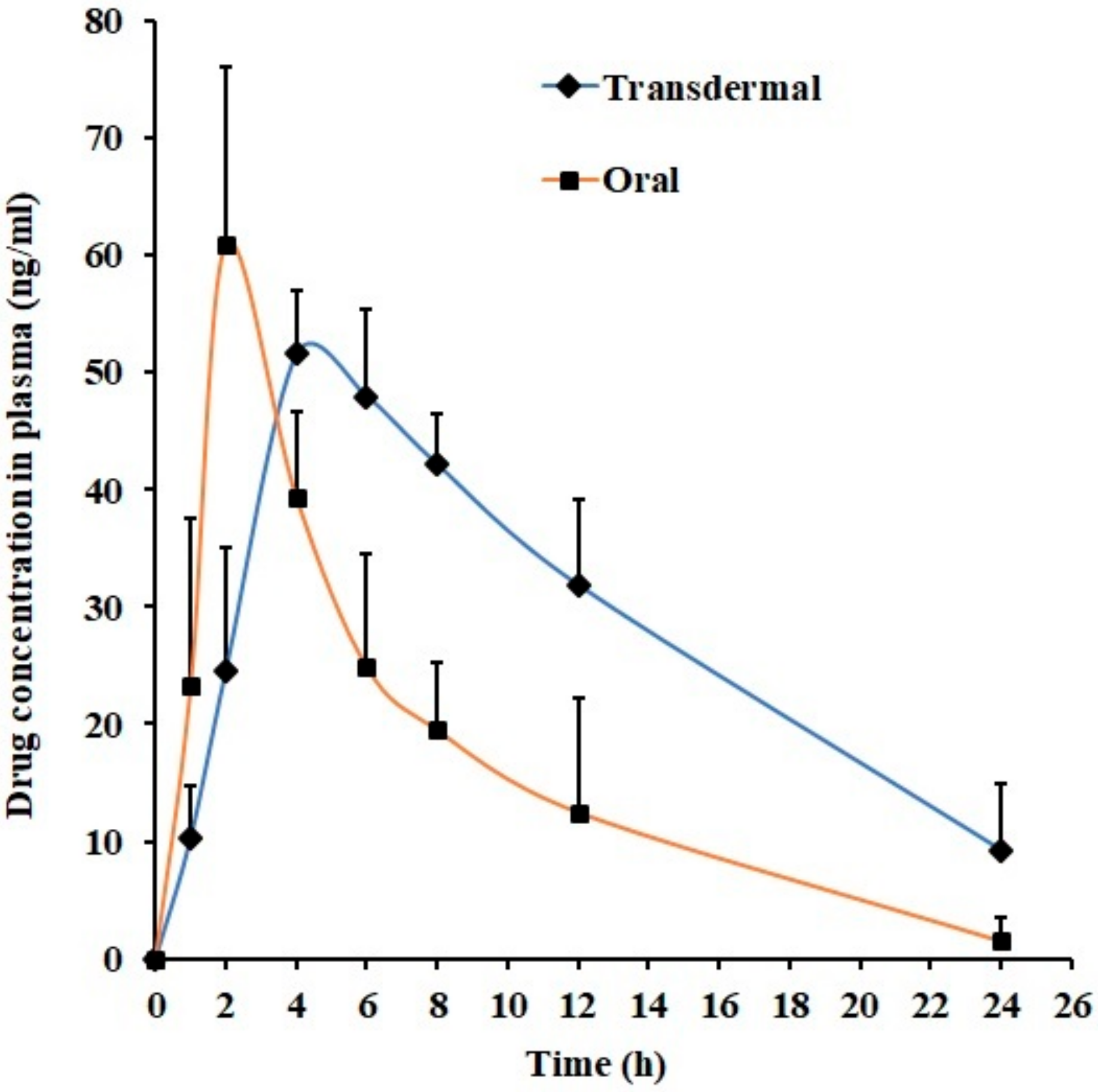
3.7. In Vivo Studies
3.8. Stability and Skin Irritation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. A Global Brief on Hypertension; WHO: Geneva, Switzerland, 2013. [Google Scholar]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; AlMazroa, M.A.; Amann, M.; Anderson, H.R.; Andrews, K.G.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- He, Y.M.; Yang, X.J.; Zhao, X.; Cheng, X.J.; Xu, H.F.; Qian, Y.X.; Li, X. β-Blockers in heart failure: Benefits of β-blockers according to varying male proportions of study patients. Clin. Cardiol. 2012, 35, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Boudonas, G.E. β-Blockers in coronary artery disease management. Hippokratia 2010, 14, 231–235. [Google Scholar] [PubMed]
- Rehsia, N.S.; Dhalla, N.S. Mechanisms of the beneficial effects of beta-adrenoceptor antagonists in congestive heart failure. Exp. Clin. Cardiol. 2010, 15, e86–e95. [Google Scholar] [PubMed]
- Ritter, J.M. Nebivolol: Endothelium-mediated vasodilating effect. J. Cardiovasc. Pharmacol. 2001, 38, S13–S16. [Google Scholar] [CrossRef]
- Kanbay, M.; Afsar, B. Are all β-blockers the same? Nebivolol vasodilator properties and evidence for relevance in treatment of hypertension. J. Clin. Hypertens. 2015, 17, 20–21. [Google Scholar] [CrossRef]
- Howlett, J.G. Nebivolol: Vasodilator properties and evidence for relevance in treatment of cardiovascular disease. Can. J. Cardiol. 2014, 30, S29–S37. [Google Scholar] [CrossRef]
- Hilas, O.; Ezzo, D. Nebivolol (bystolic), a novel beta blocker for hypertension. Pharm. Therap. 2009, 34, 188–192. [Google Scholar]
- Weiss, R. Nebivolol: A novel beta-blocker with nitric oxide-induced vasodilatation. Vasc. Health Risk Manag. 2006, 2, 303–308. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S.; Al-Dhubiab, B.E.; Alhumam, R.N. Influence of skin permeation enhancers on the transdermal delivery of palonosetron: An in vitro evaluation. J. App. Biomed. 2018, 16, 192–197. [Google Scholar] [CrossRef]
- Gupta, H.; Babu, R.J. Transdermal delivery: Product and patent update. Recent Pat. Drug Deliv. Formul. 2013, 7, 184–205. [Google Scholar] [CrossRef] [PubMed]
- Anroop, B.; Ghosh, B.; Parcha, V.; Khanam, J. Transdermal delivery of atenolol: Effect of prodrugs and iontophoresis. Curr. Drug Deliv. 2009, 6, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Haq, A.; Michniak-Kohn, B. Effects of solvents and penetration enhancers on transdermal delivery of thymoquinone: Permeability and skin deposition study. Drug Deliv. 2018, 25, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Marwah, H.; Garg, T.; Goyal, A.K.; Rath, G. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2016, 23, 564–578. [Google Scholar] [CrossRef]
- Jhawat, V.; Gupta, S.; Saini, V. Formulation and evaluation of novel controlled release of topical pluronic lecithin organogel of mefenamic acid. Drug Deliv. 2016, 23, 3573–3581. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Kiran Vaka, S.R.; Gupta, S.; Repka, M.A.; Murthy, S.N. In vitro and in vivo evaluation of a hydrogel-based prototype transdermal patch system of alfuzosin hydrochloride. Pharm. Dev. Technol. 2012, 17, 158–163. [Google Scholar] [CrossRef]
- Rehman, K.; Zulfakar, M.H. Recent advances in gel technologies for topical and transdermal drug delivery. Drug Dev. Ind. Pharm. 2014, 40, 433–440. [Google Scholar] [CrossRef]
- Osmałek, T.; Froelich, A.; Tasarek, S. Application of gellan gum in pharmacy and medicine. Int. J. Pharm. 2014, 466, 328–340. [Google Scholar] [CrossRef]
- Carmona-Moran, C.A.; Zavgorodnya, O.; Penman, A.D.; Kharlampieva, E.; Bridges, S.L., Jr.; Hergenrother, R.W.; Singh, J.A.; Wick, T.M. Development of gellan gum containing formulations for transdermal drug delivery: Component evaluation and controlled drug release using temperature responsive nanogels. Int. J. Pharm. 2016, 509, 465–476. [Google Scholar] [CrossRef]
- Mahdi, M.H.; Conway, B.R.; Mills, T.; Smith, A.M. Gellan gum fluid gels for topical administration of diclofenac. Int. J. Pharm. 2016, 515, 535–542. [Google Scholar] [CrossRef]
- Picone, C.S.; Cunha, R.L. Influence of pH on formation and properties of gellan gels. Carbohyd. Polym. 2011, 84, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.R.; Nishinari, K.; Rinaudo, M. Gelation of gellan–a review. Food Hydrocoll. 2012, 28, 373–411. [Google Scholar] [CrossRef]
- Kathe, K.; Kathpalia, H. Film forming systems for topical and transdermal drug delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Baek, J.S.; Cho, C.W. Transdermal delivery of tadalafil using a novel formulation. Drug Deliv. 2016, 23, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Galgatte, U.C.; Kumbhar, A.B.; Chaudhari, P.D. Development of in situ gel for nasal delivery: Design, optimization, in vitro and in vivo evaluation. Drug Deliv. 2014, 21, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Ueda, C.T.; Shah, V.P.; Derdzinski, K.; Ewing, G.; Flynn, G.; Maibach, H.; Marques, M.; Rytting, H.; Shaw, S.; Thakker, K.; et al. Topical and transdermal drug products. Pharmacopeial Forum 2009, 35, 750–764. [Google Scholar] [CrossRef]
- Kamil, N.; Nair, A.B.; Attimarad, M. Development of transdermal delivery system of dexamethasone, palonosetron and aprepitant for combination antiemetic therapy. Indian, J. Pharm. Educ. Res. 2016, 50, 472–481. [Google Scholar] [CrossRef]
- Prima, G.; Conigliaro, A.; De Caro, V. Mucoadhesive polymeric films to enhance barbaloin penetration into buccal mucosa: A novel approach to chemoprevention. AAPS PharmSciTech 2019, 20, 18. [Google Scholar] [CrossRef]
- Shah, J.; Nair, A.B.; Jacob, S.; Patel, R.K.; Shah, H.; Shehata, T.M.; Morsy, M.A. Nanoemulsion based vehicle for effective ocular delivery of moxifloxacin using experimental design and pharmacokinetic study in rabbits. Pharmaceutics 2019, 11, 230. [Google Scholar] [CrossRef]
- Nair, A.; Reddy, C.; Jacob, S. Delivery of a classical antihypertensive agent through the skin by chemical enhancers and iontophoresis. Skin Res. Technol. 2009, 15, 187–194. [Google Scholar] [CrossRef]
- Aziz, D.E.; Abdelbary, A.A.; Elassasy, A.I. Fabrication of novel elastosomes for boosting the transdermal delivery of diacerein: Statistical optimization, ex-vivo permeation, in-vivo skin deposition and pharmacokinetic assessment compared to oral formulation. Drug Deliv. 2018, 25, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Morsy, M.A.; Jacob, S. Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Dev. Res. 2018, 79, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Nair, A.B.; Al-Dhubiab, B.E. Preparation and evaluation of niosome gel containing acyclovir for enhanced dermal deposition. J. Liposome Res. 2017, 27, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Draize, J.H.; Woodard, G.; Calvery, H.O. Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J. Pharmacol. Exp. Ther. 1944, 82, 377–390. [Google Scholar]
- Bharate, S.S.; Bharate, S.B.; Bajaj, A.N. Interactions and incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: A comprehensive review. J. Excip. Food Chem. 2016, 1, 3–26. [Google Scholar]
- Gannu, R.; Yamsani, V.V.; Yamsani, S.K.; Palem, C.R.; Yamsani, M.R. Optimization of hydrogels for transdermal delivery of lisinopril by box - Behnken statistical design. AAPS PharmSciTech 2009, 10, 505–514. [Google Scholar] [CrossRef]
- Joshi, S.A.; Chavhan, S.S.; Sawant, K.K. Rivastigmine-loaded PLGA and PBCA nanoparticles: Preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur. J. Pharm. Biopharm. 2010, 76, 189–199. [Google Scholar] [CrossRef]
- Nayak, A.; Laha, B.; Sen, K. Development of hydroxyapatite-ciprofloxacin bone-implants using quality by design. Acta Pharm. 2011, 61, 25–36. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Arafa, M.G.; Ayoub, B.M. DOE optimization of nano-based carrier of pregabalin as hydrogel: New therapeutic & chemometric approaches for controlled drug delivery systems. Sci. Rep. 2017, 7, 41503. [Google Scholar]
- Alkilani, A.; McCrudden, M.T.; Donnelly, R. Transdermal drug delivery: Innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutics 2015, 7, 438. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Jacob, S.; Al-Dhubiab, B.; Attimarad, M.; Harsha, S. Basic considerations in the dermatokinetics of topical formulations. Braz. J. Pharm. Sci. 2013, 49, 423–434. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Formulations | |||||||
|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | |
| Nebivolol HCl (mg) | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 |
| Gellan gum (mg) | 300 | 600 | 300 | 600 | 300 | 600 | 300 | 600 |
| Carbopol (mg) | 300 | 300 | 300 | 300 | 150 | 150 | 150 | 150 |
| Polyethylene glycol 400 (µL) | 15 | 20 | 25 | 40 | 10 | 35 | 20 | 35 |
| Tween 80 (mL) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Ethanol (mL) | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Water up to (mL) | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| Evaluation Parameters | Formulations | |||||||
|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | |
| pH | 5.69 ± 0.28 | 5.58 ± 0.35 | 5.78 ± 0.41 | 6.12 ± 0.25 | 6.9 ± 0.34 | 7.02 ± 0.29 | 7.1 ± 0.15 | 7.14 ± 0.22 |
| Viscosity (centi poise) | 14,281 ± 189 | 18,374 ± 224 | 12,993 ± 175 | 17,451 ± 152 | 9872 ± 80 | 11,221 ± 94 | 8943 ± 116 | 11,351 ± 127 |
| Drug content (mg/g) * | 2.41 ± 0.04 | 2.47 ± 0.06 | 2.49 ± 0.09 | 2.43 ± 0.05 | 2.45 ± 0.02 | 2.42 ± 0.04 | 2.47 ± 0.05 | 2.46 ± 0.06 |
| Formulations | Factors | Model Name | ||||
|---|---|---|---|---|---|---|
| Zero order | First order | Higuchi | Korsmeyer – Peppas | Weibull Model | ||
| F1 | r2 | 0.8080 | 0.9963 | 0.9703 | 0.9601 | 0.9989 |
| SSR | 1353.74 | 39.66 | 209.26 | 130.11 | 0.88 | |
| FR | 338.44 | 9.91 | 52.32 | 32.53 | 0.22 | |
| F2 | r2 | 0.9633 | 0.9909 | 0.9723 | 0.9915 | 0.9975 |
| SSR | 231.14 | 135.18 | 174.73 | 76.12 | 6.13 | |
| FR | 57.79 | 33.79 | 43.68 | 19.03 | 1.53 | |
| F3 | r2 | 0.7887 | 0.9855 | 0.9608 | 0.9469 | 0.9951 |
| SSR | 1481.09 | 183.99 | 274.79 | 171.99 | 6.36 | |
| FR | 370.27 | 45.99 | 68.70 | 43.00 | 1.59 | |
| F4 | r2 | 0.9559 | 0.9905 | 0.9733 | 0.9849 | 0.9992 |
| SSR | 293.39 | 173.67 | 177.92 | 117.83 | 3.02 | |
| FR | 73.35 | 43.42 | 44.48 | 29.46 | 0.75 | |
| F5 | r2 | 0.7481 | 0.9600 | 0.9374 | 0.8979 | 0.9778 |
| SSR | 1868.63 | 413.18 | 464.57 | 313.73 | 30.92 | |
| FR | 467.16 | 103.29 | 116.14 | 78.43 | 7.73 | |
| F6 | r2 | 0.9163 | 0.9984 | 0.9828 | 0.9721 | 0.9998 |
| SSR | 565.37 | 32.12 | 116.34 | 158.36 | 0.7015 | |
| FR | 141.34 | 8.03 | 29.08 | 39.59 | 0.1754 | |
| F7 | r2 | 0.7174 | 0.9753 | 0.9228 | 0.8733 | 0.9828 |
| SSR | 2147.59 | 365.13 | 586.63 | 319.66 | 30.66 | |
| FR | 536.90 | 91.28 | 146.66 | 79.90 | 7.67 | |
| F8 | r2 | 0.9401 | 0.9863 | 0.9751 | 0.9841 | 0.9886 |
| SSR | 428.25 | 350.90 | 177.81 | 131.89 | 32.94 | |
| FR | 107.06 | 87.72 | 44.45 | 32.97 | 8.24 | |
| Formulations | Lag time (h) | Flux (µg/cm2/h) | Cumulative Amount Permeated (12 h) (µg/cm2) | Permeability Coefficient (cm/h ×10−3) |
|---|---|---|---|---|
| F1 | 0.46 ± 0.21 | 28.94 ± 2.66 | 160.15 ± 20.91 | 2.67 ± 0.39 |
| F2 | 0.66 ± 0.29 | 17.30 ± 1.46 | 94.11 ± 24.02 | 1.57 ± 0.25 |
| F3 | 0.36 ± 0.12 | 27.14 ± 2.52 | 153.14 ± 35.51 | 2.55 ± 0.32 |
| F4 | 0.75 ± 0.22 | 19.25 ± 2.13 | 101.97 ± 29.75 | 1.70 ± 0.21 |
| F5 | 0.35 ± 0.16 | 30.17 ± 3.56 | 170.37 ± 27.02 | 2.84 ± 0.45 |
| F6 | 0.67 ± 0.30 | 20.95 ± 2.42 | 112.38 ± 22.09 | 1.87 ± 0.29 |
| F7 | 0.19 ± 0.05 | 30.86 ± 4.08 | 177.76 ± 21.76 | 2.96 ± 0.22 |
| F8 | 0.73 ± 0.25 | 22.05 ± 2.48 | 121.20 ± 31.51 | 2.02 ± 0.19 |
| Parameter | Transdermal Gel (F7) | Oral Suspension |
|---|---|---|
| Tmax (h) | 4 | 2 |
| Cmax (ng/mL) | 51.56 ± 5.41 | 60.95 ± 15.06 |
| AUC0-α (ng.h/mL) | 986.52 ± 382.63* | 422.90 ± 192.64 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.B.; Shah, J.; Aljaeid, B.M.; Al-Dhubiab, B.E.; Jacob, S. Gellan Gum-Based Hydrogel for the Transdermal Delivery of Nebivolol: Optimization and Evaluation. Polymers 2019, 11, 1699. https://doi.org/10.3390/polym11101699
Nair AB, Shah J, Aljaeid BM, Al-Dhubiab BE, Jacob S. Gellan Gum-Based Hydrogel for the Transdermal Delivery of Nebivolol: Optimization and Evaluation. Polymers. 2019; 11(10):1699. https://doi.org/10.3390/polym11101699
Chicago/Turabian StyleNair, Anroop B., Jigar Shah, Bader M. Aljaeid, Bandar E. Al-Dhubiab, and Shery Jacob. 2019. "Gellan Gum-Based Hydrogel for the Transdermal Delivery of Nebivolol: Optimization and Evaluation" Polymers 11, no. 10: 1699. https://doi.org/10.3390/polym11101699
APA StyleNair, A. B., Shah, J., Aljaeid, B. M., Al-Dhubiab, B. E., & Jacob, S. (2019). Gellan Gum-Based Hydrogel for the Transdermal Delivery of Nebivolol: Optimization and Evaluation. Polymers, 11(10), 1699. https://doi.org/10.3390/polym11101699