Tunable Structure and Properties of Segmented Thermoplastic Polyurethanes as a Function of Flexible Segment




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Polyurethane Synthesis
2.3. Characterization Techniques
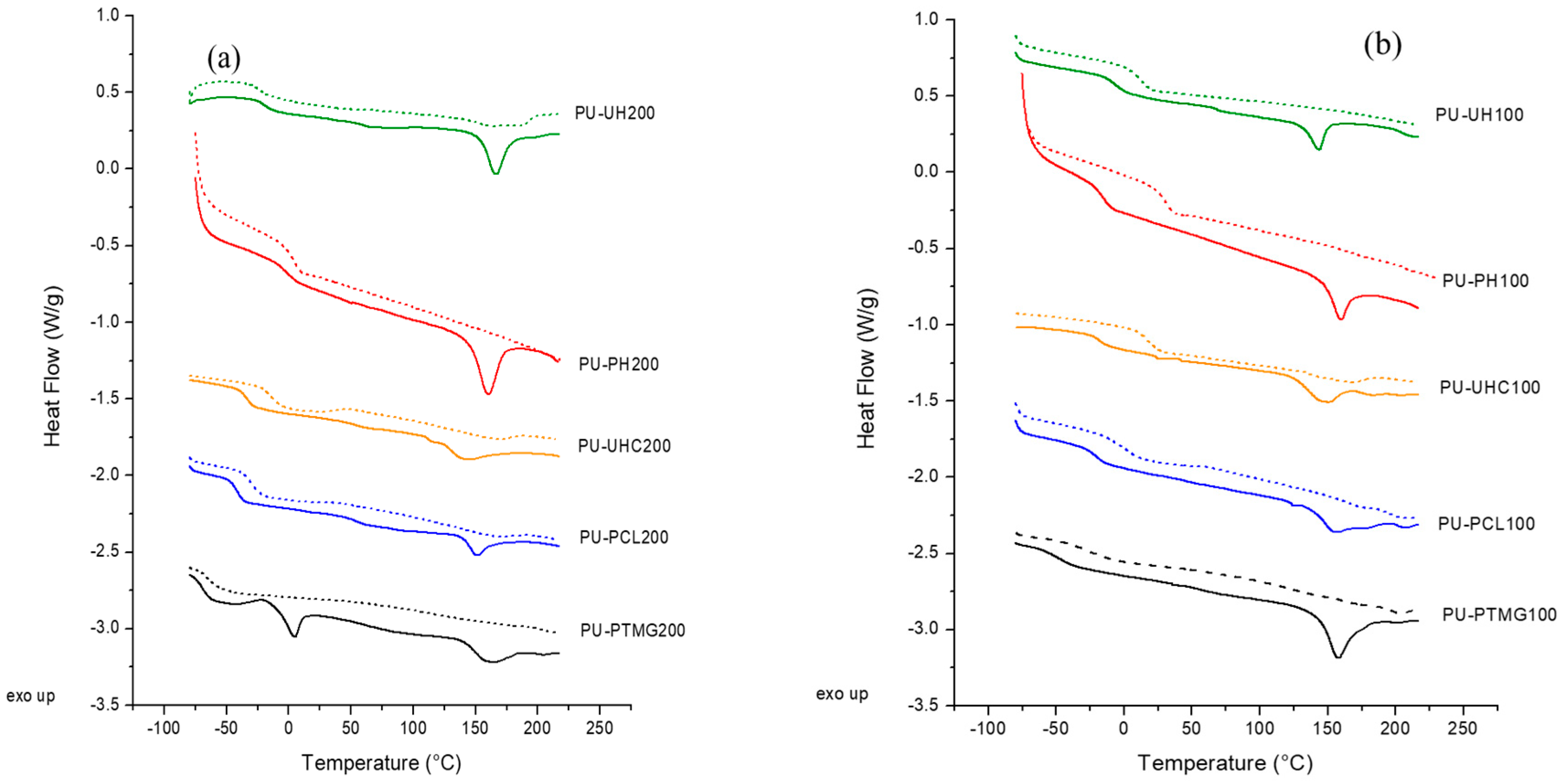
2.3.1. Differential Scanning Calorimetry (DSC)
2.3.2. Fourier Transform Infrared-Attenuated Total Reflection Spectroscopy (FTIR-ATR)
2.3.3. Dynamic Mechanical Analysis (DMA)
2.3.4. Synchrotron Small-Angle X-Ray Scattering (SAXS)
2.3.5. Shore D Hardness
2.3.6. Tensile Properties
2.3.7. Durability Tests
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brains, P.F. Polyurethanes Technology; John Wiley and Sons: New York, NY, USA, 1969. [Google Scholar]
- Hepburn, C. Polyurethane Elastomers, 2nd ed.; Elsevier Applied Science: London, UK, 1991. [Google Scholar]
- Wirpsza, Z. Polyurethane: Chemistry, Technology and Applications; Ellis Horwood: New York, NY, USA, 1993. [Google Scholar]
- Chattopadhyay, D.K.; Raju, K.V.S.N. Structural engineering of polyurethane coatings for high performance applications. Prog. Polym. Sci. 2007, 32, 352–418. [Google Scholar] [CrossRef]
- Król, P. Synthesis methods, chemical structures and phase structures of linear polyurethanes. Properties and applications of linear polyurethanes in polyurethane elastomers, copolymers and ionomers. Prog. Mater. Sci. 2007, 52, 915–1015. [Google Scholar] [CrossRef]
- Puska, A. Thermal and mechanical behavior of new transparent thermoplastic polyurethane elastomers derived from cycloaliphatic diisocyanate. Polymers 2018, 16, 537. [Google Scholar] [CrossRef] [PubMed]
- De Greef, T.F.A.; Smulders, M.M.J.; Wolffs, M.; Schenning, A.P.H.; Sijbesma, R.P.; Meijer, E.W. Supramolecular Polymerization. Chem. Rev. 2009, 109, 5687–5754. [Google Scholar] [CrossRef] [PubMed]
- Strawhecker, K.E.; Hsieh, A.J.; Chantawansri, T.L.; Kalciogl, Z.I.; Van Vliet, K.J. Influence of microstructure on micro-/nano-mechanical measurements of select model transparent poly(urethane urea) elastomers. Polymer 2013, 54, 901–908. [Google Scholar] [CrossRef]
- Lee, D.-K.; Tsai, H.-B.; Tsai, R.-S.; Chen, P.H. Preparation and properties of transparent thermoplastic segmented polyurethanes derived from different polyols. Polym. Eng. Sci. 2007, 47, 695–701. [Google Scholar] [CrossRef]
- Spírková, M.; Hodan, J.; Serkis-Rodze´n, M.; Kredatusová, J.; Zhigunov, A.; Kotek, J. The effect of pre-set extension on the degree of hydrolytic degradation in multicomponent polyurethane elastomers. Polym. Degrad. Stab. 2017, 142, 69–78. [Google Scholar] [CrossRef]
- Kojio, K.; Nonaka, Y.; Masubucho, T.; Furukawa, M. Effect of the composition ratio of copolymerized poly(carbonate) glycol on the microphase-separated structures and mechanical properties of polyurethane elastomers. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4448–4450. [Google Scholar] [CrossRef]
- Wiggins, M.J.; MacEwan, M.; Anderson, J.M.; Hiltner, A. Effect of soft-segment chemistry on polyurethane biostability during in vitro fatigue loading. J. Biomed. Mater. Res. 2004, 68, 668–683. [Google Scholar] [CrossRef]
- Khan, I.; Smith, N.; Jones, E.; Finch, D.S.; Cameron, R.E. Analysis and evaluation of a biomedical polycarbonate urethane tested in a vitro study and an ovine arthroplasty model Part I: Materials selection and evaluation. Biomaterials 2005, 26, 621–631. [Google Scholar] [CrossRef]
- Geary, C.; Birkinshaw, C.; Jones, E. Characterization of Biomate polycarbonate polyurethanes for orthopaedic applications. J. Mater. Sci. Mater. Med. 2008, 19, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Fernando, T.L. Studies on ageing performance of some novel polyurethanes. J. Chem. Pharm. Res. 2011, 3, 848–862. [Google Scholar]
- Costa, V.; Nohales, A.; Felix, P.; Guillem, C.; Gómez, C.M. Enhanced polyurethanes based on different polycarbonatediols. J. Elastomers Plast. 2012, 45, 217–238. [Google Scholar] [CrossRef]
- Costa, V.; Nohales, A.; Félix, P.; Guillem, C.; Gutierrez, D.; Gómez, C.M. Structure–property relationships of polycarbonate diol-based polyurethanes as a function of soft segment content and molar mass. J. Appl. Polym. Sci. 2015, 132, 41704–41714. [Google Scholar] [CrossRef]
- Martins, J.N.; Bianchi, O.; Wanke, C.H.; Castel, C.D.; Oliveira, R.V.B. Effects of POSS addition on Non-isothermal crystallization and morphology of PVDF. J. Polym. Res. 2015, 22, 224. [Google Scholar] [CrossRef]
- Albrecht, T.; Strobl, G. Observation of the Early Stages of Crystallization in Polyethylene by Time-Dependent SAXS: Transition from Individual Crystals to Stacks of Lamellae. Macromolecules 1996, 29, 783–785. [Google Scholar] [CrossRef]
- Denchev, Z.; Nogales, A.; Ezquerra, T.A.; Fernandes-Nascimento, J.; Baltà-Calleja, F.J. On the origin of the multiple melting behavior in poly(ethylene naphthalene-2,6-dicarboxylate): Microstructural study as revealed by differential scanning calorimetry and X-ray scattering. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 1167–1182. [Google Scholar] [CrossRef]
- Sun, Y.S. Temperature-resolved SAXS studies of morphological changes in melt-crystallized poly(hexamethylene terephthalate) and its melting upon heating. Polymer 2006, 47, 8032–8043. [Google Scholar] [CrossRef]
- Carli, L.N.; Bianchi, O.; Machado, G.; Crespo, J.S.; Mauler, R.S. Morphological and structural characterization of PHBV/organoclay nanocomposites by small angle X-ray scattering. Mater. Sci. Eng. C. 2013, 33, 932–937. [Google Scholar] [CrossRef]
- Musselman, S.G.; Santosusso, T.M.; Barnes, J.D.; Sperling, L.H. Domain structure and interphase dimensions in poly(urethaneurea) elastomers using DSC and SAXS. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 2586–2600. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Leung, L.M. Compression-molded polyurethane block copolymers. 2.Evaluation of microphase compositions. Macromolecules 1992, 25, 6205–6213. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Stein, R.S. Small-angle x-ray scattering measurements of diffuse phase-boundary thicknesses in segmented polyurethane elastomers. J. Polym. Sci. 1983, 21, 2181–2200. [Google Scholar] [CrossRef]
- Eceiza, A.; Martin, M.D.; de la Caba, K.; Kortaberria, G.; Gabilondo, N.; Corcuera, M.A.; Mondragon, I. Thermoplastic polyurethane elastomers based on polycarbonate diols withdifferent soft segment molecular weight and chemical structure: mechanical and thermal properties. Polym. Eng. Sci. 2008, 48, 297–306. [Google Scholar] [CrossRef]
- Chen, P.H.; Yang, Y.F.; Lee, D.-K.; Lin, Y.-F.; Wang, H.-H.; Tsai, H.-B.; Tsai, R.-S. Synthesis and properties of transparent thermoplastic segmented polyurethanes. Adv. Polym. Technol. 2007, 26, 33–40. [Google Scholar] [CrossRef]
- Small, P.A. Some factors affecting the solubility of polymers. J. Appl. Chem. 1953, 3, 71–80. [Google Scholar] [CrossRef]
- Wang, C.B.; Cooper, S.L. Morphology and properties of segmented polyether polyurethaneureas. Macromolecules 1983, 16, 775–786. [Google Scholar] [CrossRef]
- Chang, A.L.; Briber, R.M.; Thomas, E.L.; Zdrahala, R.J.; Critchfield, F.E. Morphological study of the structure developed during the polymerization of a series of segmented polyurethanes. Polymer 1982, 23, 1060–1068. [Google Scholar] [CrossRef]
- Leung, L.M.; Koberstein, J.T. DSC annealing study of microphase separation and multiple endothermic behavior in polyether-based polyurethane block copolymers. Macromolecules 1986, 19, 706. [Google Scholar] [CrossRef]
- Hu, W.; Koberstein, J.T. The effect of thermal annealing on the thermal properties and molecular weight of a segmented polyurethane copolymer. Polym. Sci. Polym. Phys. 1994, 32, 437–446. [Google Scholar] [CrossRef]
- Chen, T.K.; Chui, J.Y.; Shieh, T.S.H. Glass transition behaviors of a polyurethane hard segment based on 4,4’-diisocyanatodiphenylmethane and 1,4-butanediol and the calculation of microdomain composition. Macromolecules 1997, 30, 5068–5074. [Google Scholar] [CrossRef]
- Korley, L.T.J.; Pate, B.D.; Thomas, E.L.; Hammond, P.T. Effect of the degree of soft and hard segment ordering on the morphology and mechanical behaviour of semicrystalline segmented polyurethanes. Polymer 2006, 47, 3073–3082. [Google Scholar] [CrossRef]
- Tanaka, H.; Kunimura, M. Mechanical properties of thermoplastic polyurethanes containing aliphatic polycarbonate soft segments with different chemical structures. Polym. Eng. Sci. 2002, 42, 1333–1349. [Google Scholar] [CrossRef]
- Alves, P.; Coelho, J.F.J.; Haack, J.; Rota, A.; Bruinink, A.; Gil, M.H. Surface modification and characterization of thermoplastic polyurethane. Eur. Polym. J. 2009, 45, 1412–1419. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Chou, N.-K.; Chen, K.-F.; Ho, G.-H.; Chang, C.-H.; Wang, S.-S.; Chu, S.-H.; K-Hsieh, H. Effect of soft segment length on properties of hydrophilic/hydrophobic polyurethanes. Polym. Int. 2007, 56, 1415–1422. [Google Scholar] [CrossRef]
- Harris, R.F.; Joseph, M.D.; Davidson, C.; Deporter, C.D.; Dais, V.A. Polyurethane elastomers based on molecular weight advanced poly(ethylene ether carbonate) diols. I. Comparison to commercial diols. J. Appl. Polym. Sci. 1990, 41, 487. [Google Scholar] [CrossRef]
- Son, T.W.; Lee, D.W.; Lim, S.K. Thermal and phase behavior of polyurethane based on chain extender 2,2-bis-[4-(2-hydroxyethoxy) phenyl]propane. Polym. J. 1999, 31, 563–568. [Google Scholar] [CrossRef]
- Barikani, M.; Barmar, M. Thermoplastic polyurethane elastomers: Synthesis and study of effective structural parameters. Iran. Polym. J. 1996, 5, 231–235. [Google Scholar]
- Kim, H.; Lee, T.J.; Huh, J.H.; Lee, D.J. Preparation and properties of segmented thermoplastic polyurethane elastomers with two different soft segments. J. Appl. Polym. Sci. 1999, 73, 345–352. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, C.P. Synthesis, characterization and mechanical properties of polyester-based aliphatic polyurethane elastomers containing hyperbranched polyester segments. Eur. Polym. J. 2008, 44, 3708–3714. [Google Scholar] [CrossRef]
- Pan, J.; Li, G.; Chen, Z.; Chen, X.; Zhu, W.; Xu, K. Alternative block polyurethanes based on poly(3-hydroxybutyrate-co-4-hydroxybutyrate) and poly(ethylene glycol). Biomaterials 2009, 30, 2975–2984. [Google Scholar] [CrossRef]
- Velayutham, T.S.; Abd Majid, W.H.; Ahmad, A.B.; Kang, G.Y.; Gan, S.N. Synthesis and characterization of polyurethane coatings derived from polyols synthesized with glycerol, phthalic anhydride and oleic acid. Prog. Org. Coat. 2009, 66, 367–371. [Google Scholar] [CrossRef]
- Zhao, Q.; Cheng, G.; Li, H.; Ma, X.; Zhang, L. Synthesis and characterization of biodegradable poly(3-hydroxybutyrate) and poly(ethylene glycol) multiblock copolymers. Polymer 2005, 46, 10561–10567. [Google Scholar] [CrossRef]
- Spirkova, M.; Pavlicevic, J.; Strachota, A.; Poreba, R.; Bera, O.; Kapralkova, L.; Baldrian, J.; Slouf, M.; Lazic, N.; Budinski-Simendic, J. Novel polycarbonate-based polyurethane elastomers: Composition–property relationship. Eur. Polym. J. 2011, 47, 959–972. [Google Scholar] [CrossRef]
- Fernández d’Arlas, B.; Rueda, L.; De la Caba, K.; Mondragon, I.; Eceiza, A. Microdomain composition and properties differences of biodegradable polyurethanes based on MDI and HDI. Polym. Eng. Sci. 2008, 48, 519–529. [Google Scholar] [CrossRef]
- Sung, C.H.S.P.; Schneider, N.S. Infrared studies of hydrogen bonding in toluene diisocyanate based polyurethanes. Macromolecules 1975, 8, 68–73. [Google Scholar] [CrossRef]
- Javni, I.; Bilić, O.; Bilić, N.; Petrović, Z.S.; Eastwood, E.A.; Zhang, F.; Ilavsk, J. Thermoplastic polyurethanes with controlled morphology based on methylenediphenyldiisocyanate/isosorbide/butanediol hard segments. Polym. Int. 2015, 64, 1607–1616. [Google Scholar] [CrossRef]
- Choi, T.; Masser, K.A.; Moore, E.; Weksler, J.; Padsalgikar, A.; Runt, J. Segmented polyurethanes derived from novel siloxane–carbonate soft segments for biomedical applications. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 865–872. [Google Scholar] [CrossRef]
- Leung, L.M.; Koberstein, J.T. Small-angle scattering analysis of hard-microdomain structure and microphase mixing in polyurethane elastomers. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 1883–1913. [Google Scholar] [CrossRef]
- Martin, D.J.; Meijs, G.; Renwick, G.M.; Gunatillake, P.A.; McCarthy, S.J. Effect of soft-segment CH2/O ratio on morphology and properties of a series of polyurethane elastomers. J. Appl. Polym. Sci. 1996, 60, 557. [Google Scholar] [CrossRef]
- Kultys, A.; Rogulska, M.; Pikus, S. New thermoplastic segmented polyurethanes with hard segments derived from 4,4′-diphenylmethane diisocyanate and methylenebis(1,4-phenylenemethylenethio) dialcanols. J. Appl. Polym. Sci. 2012, 123, 331. [Google Scholar] [CrossRef]
- Gunatillake, P.A. Polyurethane elastomers based on novel polyether macrodiols and MDI: Synthesis, mechanical properties and resistance to hydrolysis and oxidation. J. Appl. Polym. Sci. 1992, 46, 319–328. [Google Scholar] [CrossRef]
- Harris, R.F.; Joseph, M.D.; Davidson, C. Polyurethane elastomers based on molecular weight advanced poly(ethylene ether carbonate) polyols. IV. Effects of poly(propylene glycol) modified diols. J. Appl. Polym. Sci. 1992, 46, 1843–1857. [Google Scholar] [CrossRef]
- Bajsic, E.G.; Rek, V.; Agic, A. Thermal degradation of polyurethane elastomers: Determination of kinetic parameters. J. Elast. Plast. 2003, 35, 311–323. [Google Scholar] [CrossRef]
- Clemitson, I.R. Castable Polyurethane Elastomers; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Structure | Molecular Weight (g·mol−1) | Tg (°C) | Tm (°C) |
|---|---|---|---|---|
| UH |  | 1000 2000 | −69.34 −52.30 | 42.8 50.4 |
| PH |  | 1000 2000 | −56.4 −52.1 | |
| UHC |  | 1000 2000 | −66.0 −59.1 | 11.3 14 |
| PCL |  | 1000 2000 | −71.1 −63.9 | 32 51.7 |
| PTMG |  | 1000 2000 | −75.8 −76.2 | 20.9 24.4 |
| BD |  | 90.12 | - | 20.1 |
| MDI |  | 250.12 | - | 40 |
| System | Tg (°C) | (Tg − Tg,S) (°C) | Tm (°C) | ΔH(J/g) | wH,DSC | Xb | WH,FTIR |
|---|---|---|---|---|---|---|---|
| PU-UH100 | −6.4 | 62.9 | 143.72 | 8.99 | 0.35 | 0.51 | 0.31 |
| PU-PH100 | −3.3 | 53.1 | 159.58 | 21.59 | 0.32 | 0.48 | 0.32 |
| PU-UHC100 | −17.1 | 48.9 | 148.83 | 17.11 | 0.28 | 0.54 | 0.30 |
| PU-PCL100 | −20.2 | 50.9 | 157.62 | 17.58 | 0.28 | 0.56 | 0.29 |
| PU-PTMG100 | −46.2 | 29.6 | 154.93 | 21.35 | 0.16 | 0.78 | 0.17 |
| PU-UH200 | −18.1 | 34.2 | 166.51 | 13.44 | 0.21 | 0.56 | 0.17 |
| PU-PH200 | −14.5 | 37.6 | 160.11 | 12.34 | 0.23 | 0.56 | 0.17 |
| PU-UHC200 | −30.2 | 28.9 | 148.28 | 10.39 | 0.17 | 0.57 | 0.17 |
| PU-PCL200 | −41.2 | 22.7 | 151.78 | 6.64 | 0.13 | 0.64 | 0.15 |
| PU-PTMG200 | −69.7 | 6.5 | 162.23 | 7.84 | 0.04 | 0.78 | 0.09 |
| Sample | aLp (nm) | bLp (nm) | H (nm) | S (nm) | P | THS | ||
|---|---|---|---|---|---|---|---|---|
| PU-UH100 | 10.41 | 9.78 | 2.71 | 7.07 | 0.21 | 0.88 | 2.01 | 2.19 |
| PU-PH100 | 10.46 | 10.00 | 2.86 | 7.14 | 0.23 | 0.94 | 1.99 | 2.41 |
| PU-UHC100 | 10.57 | 10.31 | 2.95 | 7.13 | 0.22 | 0.99 | 1.95 | 2.32 |
| PU-PCL100 | 10.29 | 9.88 | 3.02 | 6.85 | 0.33 | 1.09 | 1.96 | 3.40 |
| PU-PTMG100 | 10.41 | 10.37 | 3.16 | 7.20 | 0.30 | 1.13 | 1.91 | 3.12 |
| PU-UH200 | 11.37 | 10.89 | 2.95 | 7.89 | 0.22 | 0.95 | 2.15 | 2.50 |
| PU-PH200 | 11.69 | 11.42 | 3.05 | 8.34 | 0.21 | 0.96 | 2.14 | 2.45 |
| PU-UHC200 | 12.05 | 11.70 | 3.20 | 8.55 | 0.25 | 1.07 | 2.14 | 3.01 |
| PU-PCL200 | 11.84 | 11.47 | 3.42 | 8.04 | 0.30 | 1.24 | 2.12 | 3.55 |
| PU-PTMG200 | 13.65 | 13.70 | 3.92 | 9.77 | 0.31 | 1.42 | 2.10 | 4.23 |
| System | Tensile Stress at 100% (MPa) | Tensile Stress at 200% (MPa) | Tensile Stress at 300% (MPa) | Tensile Strength at Break (MPa) | Elongation at Break (%) | Hardness (Shore A) |
|---|---|---|---|---|---|---|
| PU-UH100 | 30.7 ± 0.1 | 52.7 ± 0.9 | - | 50 ± 6 | 210 ± 10 | 95.8 ± 0.2 |
| PU-PH100 | 28.1 ± 0.1 | 49.8 ± 0.4 | - | 56 ± 3 | 235 + 16 | 95.0 |
| PU-UHC100 | 20.4 ± 0.3 | 35.7 ± 0.7 | - | 45 ± 4 | 270 ± 30 | 94.3 ± 0.3 |
| PU-PCL100 | 14.9 ± 0.2 | 25.8 ± 0.4 | 38.5 ± 0.7 | 46 ± 2 | 362 ± 7 | 93.0 |
| PU-PTMG100 | 14.39 ± 0.02 | 21.8 ± 0.2 | 32.7 ± 0.6 | 47 ± 2 | 420 + 20 | 92.5 ± 0.5 |
| PU-UH200 | 11.7 ± 0.5 | 24 ± 1 | 42 ± 3 | 49.1 ± 8.2 | 340 ± 20 | 87.3 ± 0.3 |
| PU-PH200 | 11.7 ± 0.2 | 24 ± 0.3 | 40.6 ± 0.2 | 55 ± 2 | 374 ± 7 | 86.3 ± 0.3 |
| PU-UHC200 | 8.24 ± 0.05 | 13.9 ± 0.2 | 22.9 ± 0.7 | 43 ± 4 | 480 ± 20 | 85.2 ± 0.2 |
| PU-PCL200 | 6.107 ± 0.004 | 10.53 ± 0.06 | 17.2 ± 0.2 | 52 ± 4 | 530 + 20 | 82.5 ± 0.2 |
| PU-PTMG200 | 6.8 ± 0.1 | 10.8 ± 0.2 | 16.4 ± 0.5 | 39 ± 2 | 600 ± 30 | 85.7 ± 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asensio, M.; Costa, V.; Nohales, A.; Bianchi, O.; Gómez, C.M. Tunable Structure and Properties of Segmented Thermoplastic Polyurethanes as a Function of Flexible Segment. Polymers 2019, 11, 1910. https://doi.org/10.3390/polym11121910
Asensio M, Costa V, Nohales A, Bianchi O, Gómez CM. Tunable Structure and Properties of Segmented Thermoplastic Polyurethanes as a Function of Flexible Segment. Polymers. 2019; 11(12):1910. https://doi.org/10.3390/polym11121910
Chicago/Turabian StyleAsensio, Manuel, Victor Costa, Andrés Nohales, Otávio Bianchi, and Clara M Gómez. 2019. "Tunable Structure and Properties of Segmented Thermoplastic Polyurethanes as a Function of Flexible Segment" Polymers 11, no. 12: 1910. https://doi.org/10.3390/polym11121910
APA StyleAsensio, M., Costa, V., Nohales, A., Bianchi, O., & Gómez, C. M. (2019). Tunable Structure and Properties of Segmented Thermoplastic Polyurethanes as a Function of Flexible Segment. Polymers, 11(12), 1910. https://doi.org/10.3390/polym11121910