Trehalose-Rich, Degradable Hydrogels Designed for Trehalose Release under Physiologically Relevant Conditions



Abstract
:
1. Introduction
2. Experimental
2.1. Materials and General Methods
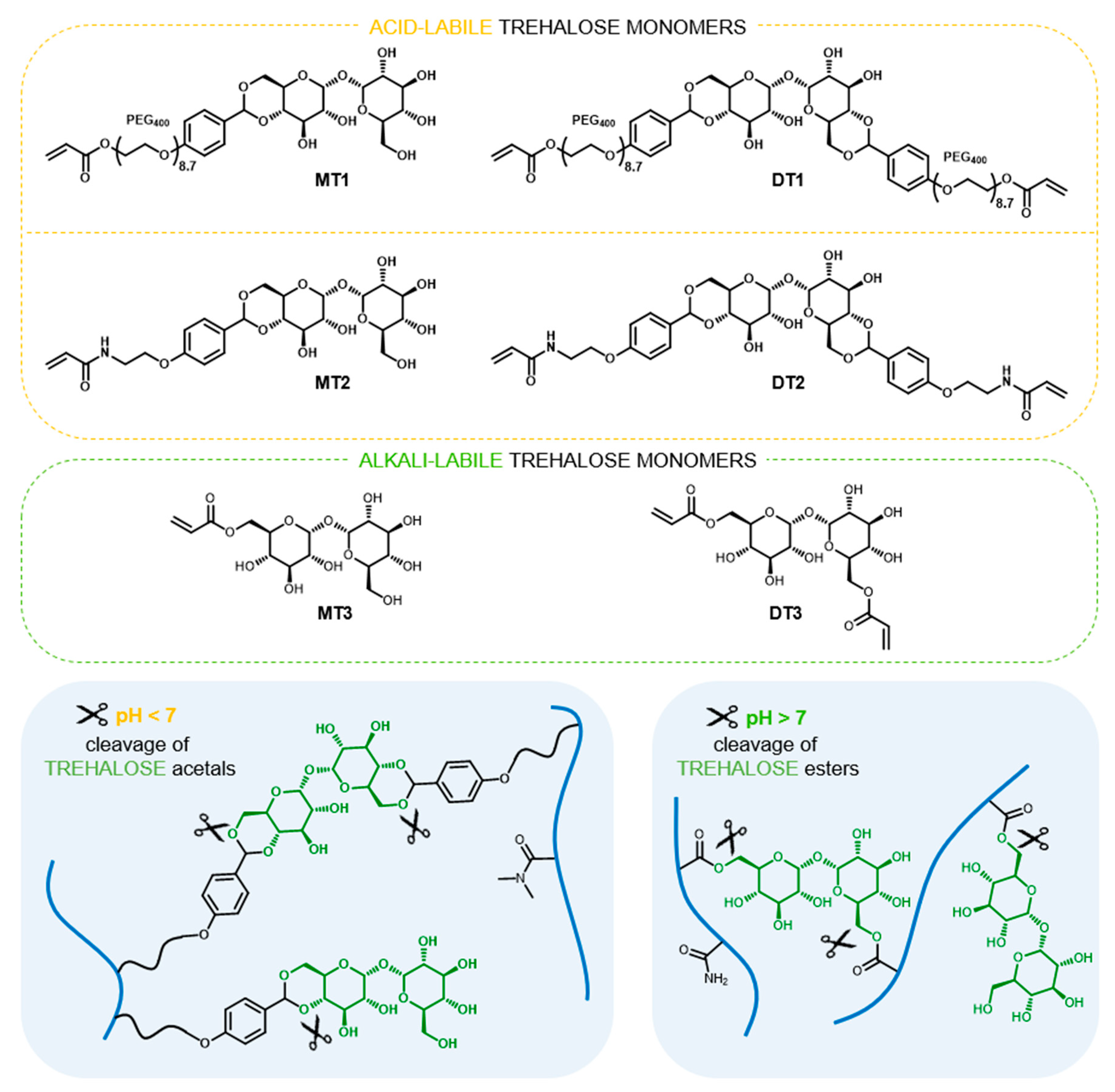
2.2. Synthesis and Characterization of Trehalose Monomers
2.3. Synthesis and Study on Hydrogels
2.3.1. Synthesis of Acid-Labile Hydrogels
2.3.2. Synthesis of Alkali-Labile Hydrogels
2.4. Hydrogels Characterization
2.4.1. Determination of Trehalose Content
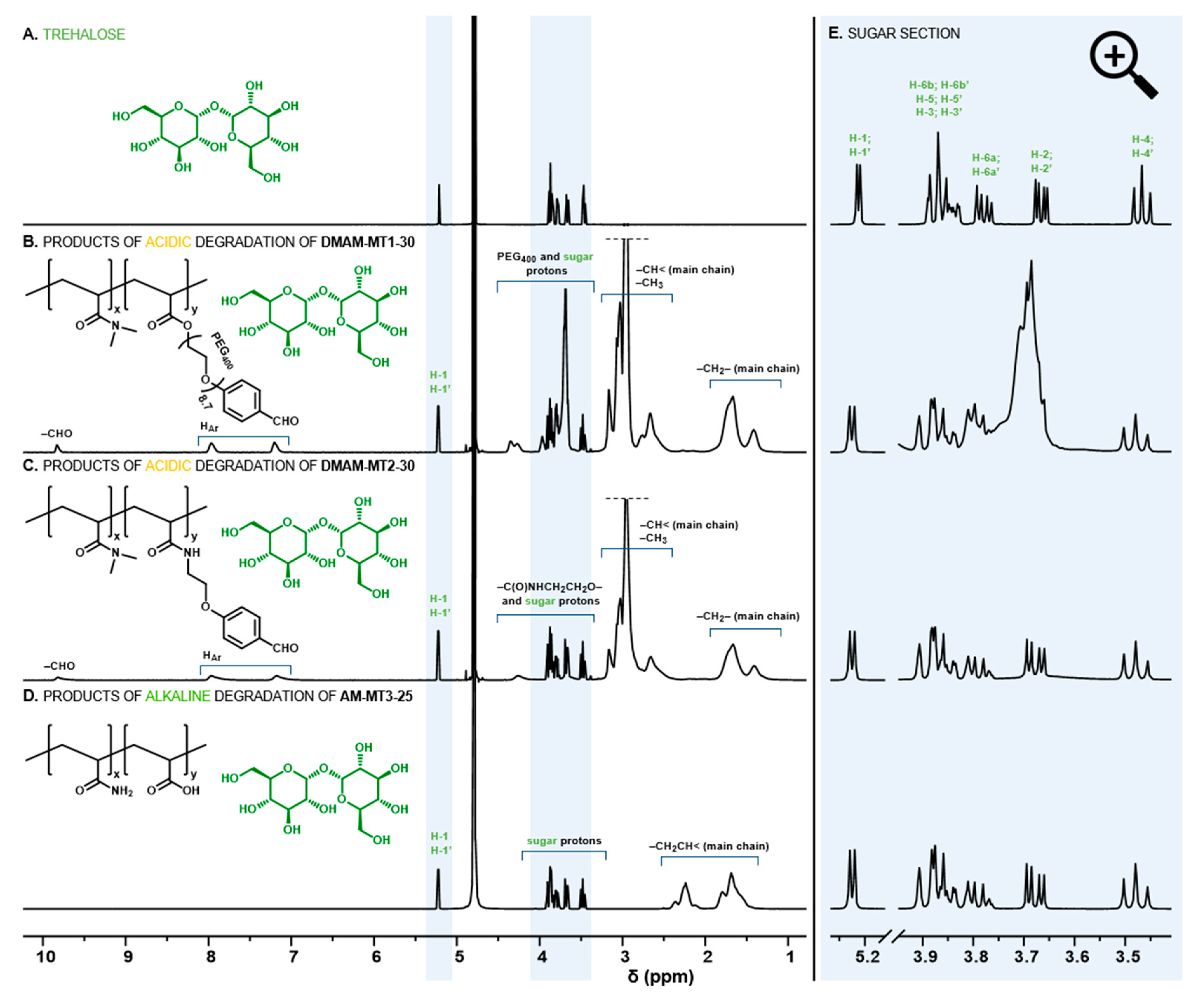
2.4.2. Characterization of Degradation Products by 1H NMR Spectroscopy
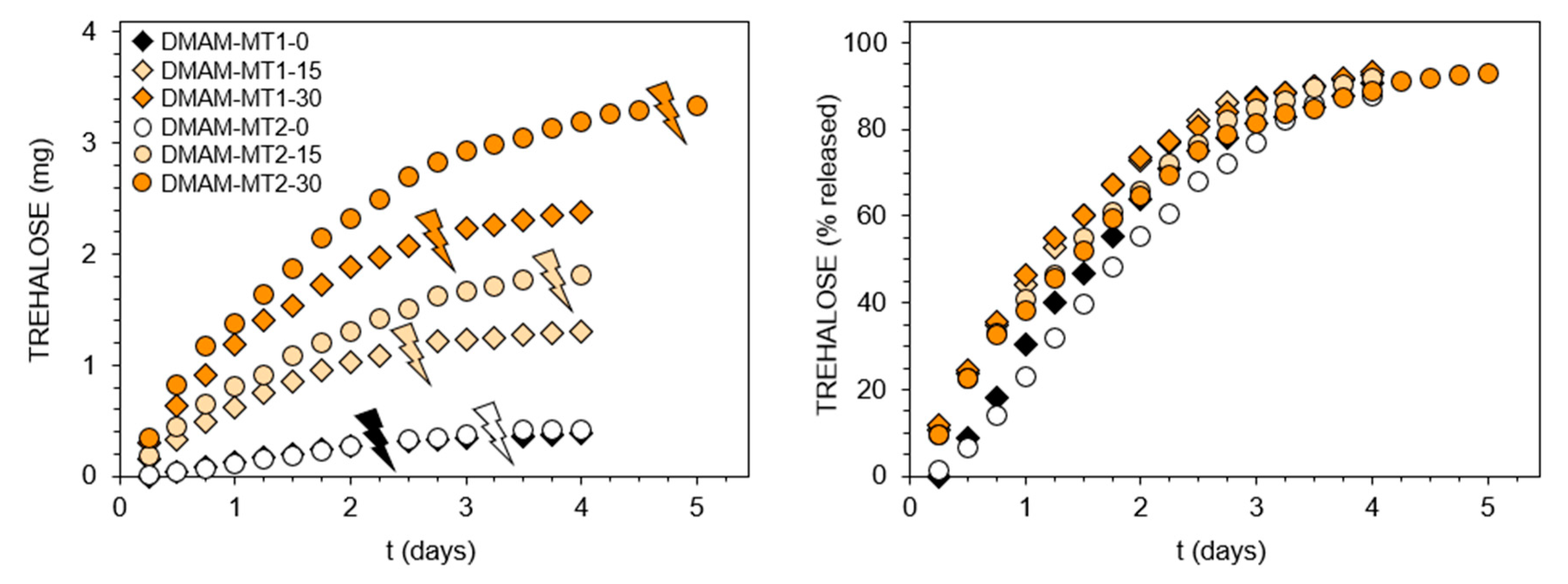
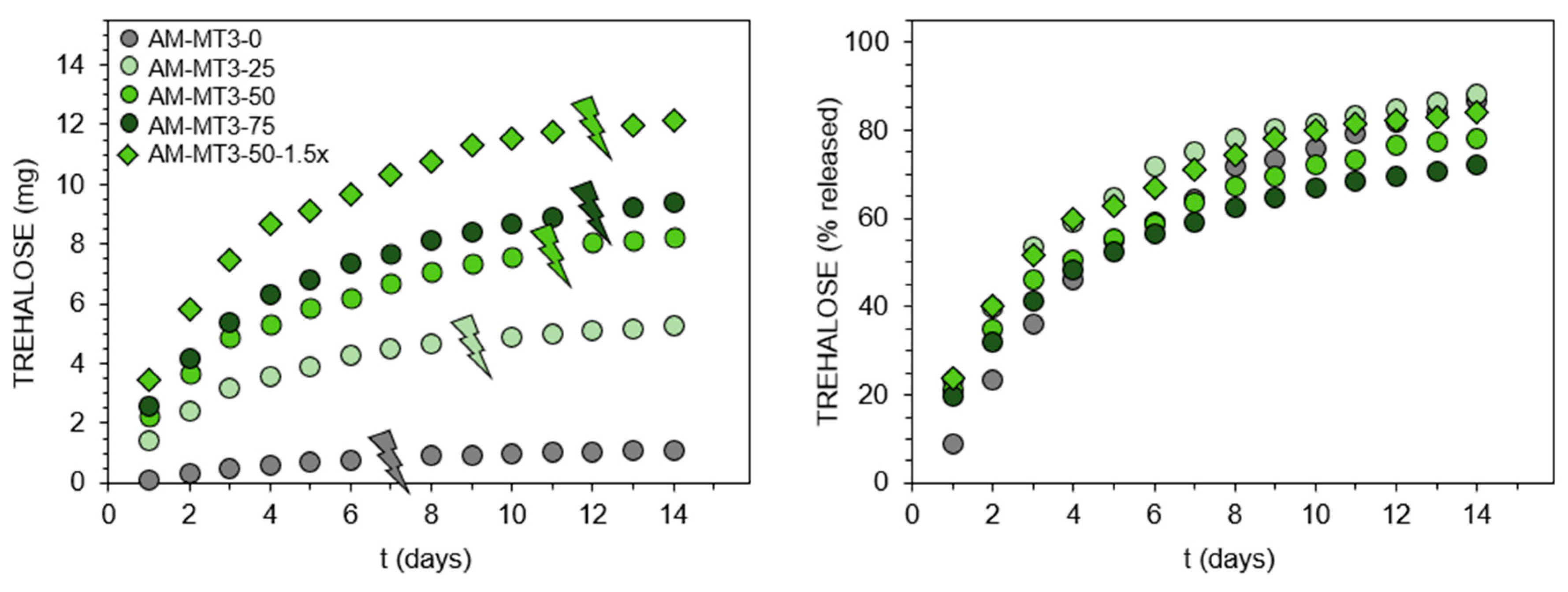
2.4.3. Trehalose Release Study
2.4.4. Equilibrium Swelling Ratio (ESR)
3. Results and Discussion
3.1. Hydrogels Capable of Trehalose Release under Mildly Acidic Conditions
3.2. Hydrogels Capable of Trehalose Release under Slightly Alkaline Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jain, N.K.; Roy, I. Effect of trehalose on protein structure. Protein Sci. 2009, 18, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour-Moghaddam, K.; Caraglia, M.; Sahebkar, A. Autophagy induction by trehalose: Molecular mechanisms and therapeutic impacts. J. Cell. Physiol. 2018, 233, 6524–6543. [Google Scholar] [CrossRef] [PubMed]
- Pierzynowska, K.; Gaffke, L.; Cyske, Z.; Puchalski, M.; Rintz, E.; Bartkowski, M.; Osiadły, M.; Pierzynowski, M.; Mantej, J.; Piotrowska, E.; et al. Autophagy stimulation as a promising approach in treatment of neurodegenerative diseases. Metab. Brain Dis. 2018, 33, 989–1008. [Google Scholar] [CrossRef]
- Zhao, J.; Zhi, X.; Pan, L.; Zhou, P. Trehalose inhibits A53T Mutant α-Synuclein overexpression and neurotoxicity in transduced PC12 Cells. Molecules 2017, 22, 1293. [Google Scholar] [CrossRef] [Green Version]
- Lan, D.M.; Liu, F.T.; Zhao, J.; Chen, Y.; Wu, J.J.; Ding, Z.T.; Yue, Z.Y.; Ren, H.M.; Jiang, Y.P.; Wang, J. Effect of trehalose on PC12 cells overexpressing wild-type or A53T mutant α-synuclein. Neurochem. Res. 2012, 37, 2025–2032. [Google Scholar] [CrossRef]
- Perucho, J.; Gómez, A.; Muñoz, M.P.; de Yébenes, J.G.; Mena, M.Á.; Casarejos, M.J. Trehalose rescues glial cell dysfunction in striatal cultures from HD R6/1 mice at early postnatal development. Mol. Cell. Neurosci. 2016, 74, 128–145. [Google Scholar] [CrossRef]
- Rodríguez-Navarro, J.A.; Rodríguez, L.; Casarejos, M.J.; Solano, R.M.; Gómez, A.; Perucho, J.; Cuervo, A.M.; García de Yébenes, J.; Mena, M.A. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 2010, 39, 423–438. [Google Scholar] [CrossRef]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; Van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [Green Version]
- Belzile, J.-P.; Sabalza, M.; Craig, M.; Clark, E.; Morello, C.S.; Spector, D.H. Trehalose, an mTOR-Independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J. Virol. 2016, 90, 1259–1277. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.L.; Higgins, C.B.; Heitmeier, M.R.; Kraft, T.E.; Qian, X.; Crowley, J.R.; Hyrc, K.L.; Beatty, W.L.; Yarasheski, K.E.; Hruz, P.W.; et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef] [PubMed]
- Kurita, K.; Hirakawa, N.; Iwakura, Y. Synthetic polymers containing sugar residues, 3. Direct addition polymerization of D-cellobiose and diisocyanates to form novel polyurethanes. Die Makromol. Chem. 1979, 180, 855–858. [Google Scholar] [CrossRef]
- Bat, E.; Lau, U.Y.; Maynard, H.D.; Hedrick, J.L.; Lee, J.; Lin, E.-W. Trehalose Glycopolymers as Excipients for Protein Stabilization. Biomacromolecules 2013, 14, 2561–2569. [Google Scholar]
- Lau, U.Y.; Pelegri-O’Day, E.M.; Maynard, H.D. Synthesis and biological evaluation of a degradable trehalose glycopolymer prepared by RAFT polymerization. Macromol. Rapid Commun. 2018, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pelegri-O’Day, E.M.; Paluck, S.J.; Maynard, H.D. Substituted polyesters by thiol-ene modification: Rapid diversification for therapeutic protein stabilization. J. Am. Chem. Soc. 2017, 139, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, K.M.; Maynard, H.D. Site-specific insulin-trehalose glycopolymer conjugate by grafting from strategy improves bioactivity. ACS Macro Lett. 2018, 7, 324–329. [Google Scholar] [CrossRef]
- Xue, L.; Ma, J.; Reineke, T.M.; Kelkar, S.S.; Wang, X.; Madsen, L.A. A theranostic polycation containing trehalose and lanthanide chelate domains for siRNA delivery and monitoring. RSC Adv. 2015, 5, 74102–74106. [Google Scholar] [CrossRef]
- Boyle, W.S.; Senger, K.; Tolar, J.; Reineke, T.M. Heparin enhances transfection in concert with a trehalose-based polycation with challenging cell types. Biomacromolecules 2017, 18, 56–67. [Google Scholar] [CrossRef]
- Tolstyka, Z.P.; Phillips, H.; Cortez, M.; Wu, Y.; Ingle, N.; Bell, J.B.; Hackett, P.B.; Reineke, T.M. Trehalose-based block copolycations promote polyplex stabilization for lyophilization and in vivo pDNA Delivery. ACS Biomater. Sci. Eng. 2016, 2, 43–55. [Google Scholar] [CrossRef]
- Burek, M.; Waśkiewicz, S.; Lalik, A.; Student, S.; Bieg, T.; Wandzik, I. Thermoresponsive microgels containing trehalose as soft matrices for 3D cell culture. Biomater. Sci. 2017, 5, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, B.; Jayawardana, K.W.; Hao, N.; Jayawardena, H.S.N.; Langer, R.; Jaklenec, A.; Yan, M. Magnetic multivalent trehalose glycopolymer nanoparticles for the detection of mycobacteria. Adv. Healthc. Mater. 2016, 5, 2007–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burek, M.; Kowalczyk, M.; Czuba, Z.P.; Krol, W.; Pilawka, R.; Waskiewicz, S. Poly(N-isopropylacrylamide) hydrogels cross-linked by α,α-trehalose diacetals as thermo-responsive and acid-degradable carriers for drug delivery. Polym. Degrad. Stab. 2016, 129, 296–305. [Google Scholar] [CrossRef]
- Burek, M.; Czuba, Z.P.; Waskiewicz, S. Novel acid-degradable and thermo-sensitive poly(N-isopropylacrylamide) hydrogels cross-linked by α,α-trehalose diacetals. Polymer 2014, 55, 6460–6470. [Google Scholar] [CrossRef]
- Burek, M.; Waśkiewicz, S.; Lalik, A.; Wandzik, I. Hydrogels with novel hydrolytically labile trehalose-based crosslinks: Small changes-big differences in degradation behavior. Polym. Chem. 2018, 9, 3721–3726. [Google Scholar] [CrossRef]
- Burek, M.; Kubic, K.; Nabiałczyk, I.; Waśkiewicz, S.; Wandzik, I. Study on protein release from hydrolytically degradable hydrogels governed by substituent effects in trehalose-based crosslinker and network properties. Eur. Polym. J. 2019, 111, 123–133. [Google Scholar] [CrossRef]
- Burek, M.; Wandzik, I. Synthetic hydrogels with covalently incorporated saccharides studied for biomedical applications-15 year overview. Polym. Rev. 2018, 58, 537–586. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogel | DMAM or AM/MTx in The Feed (w/w) A | Yield (%) | Calcd. Content of Trehalose in the Monomers Feed (wt%) | Detd. Content of Trehalose in the Hydrogel (wt%) | Detd. Content of Trehalose Originating from crosslinker B (wt%) | ESR | Degradation Time at 37 °C |
|---|---|---|---|---|---|---|---|
| Acid-labile hydrogels | |||||||
| DMAM-MT1-0 | 100/0 | 88 | 2.4 | 1.7 | 1.7 | 20.6 | 54 h * |
| DMAM-MT1-15 | 85/15 | 83 | 7.6 | 6.1 | 2.1 | 19.7 | 60 h * |
| DMAM-MT1-30 | 70/30 | 81 | 12.9 | 11.4 | 2.5 | 19.1 | 66 h * |
| DMAM-MT2-0 | 100/0 | 88 | 2.6 | 2.0 | 2.0 | 19.6 | 78 h * |
| DMAM-MT2-15 | 85/15 | 85 | 11.5 | 8.5 | 2.3 | 17.7 | 90 h * |
| DMAM-MT2-30 | 70/30 | 80 | 20.3 | 16.4 | 3.0 | 17.0 | 114 h * |
| Alkali-labile hydrogels | |||||||
| AM-MT3-0 | 100/0 | 96 | 5.7 | 4.6 | ND | 16.9 | 7 days ** |
| AM-MT3-25 | 75/25 | 96 | 25.6 | 22.7 | ND | 17.0 | 9 days ** |
| AM-MT3-50 | 50/50 | 91 | 45.6 | 39.9 | ND | 17.2 | 11 days ** |
| AM-MT3-75 | 25/75 | 87 | 65.6 | 51.7 | ND | 22.8 | 12 days ** |
| AM-MT3-50-1.5x | 50/50 | 84 | 44.8 | 39.6 | ND | 13.8 | 12 days ** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burek, M.; Wandzik, I. Trehalose-Rich, Degradable Hydrogels Designed for Trehalose Release under Physiologically Relevant Conditions. Polymers 2019, 11, 2027. https://doi.org/10.3390/polym11122027
Burek M, Wandzik I. Trehalose-Rich, Degradable Hydrogels Designed for Trehalose Release under Physiologically Relevant Conditions. Polymers. 2019; 11(12):2027. https://doi.org/10.3390/polym11122027
Chicago/Turabian StyleBurek, Małgorzata, and Ilona Wandzik. 2019. "Trehalose-Rich, Degradable Hydrogels Designed for Trehalose Release under Physiologically Relevant Conditions" Polymers 11, no. 12: 2027. https://doi.org/10.3390/polym11122027
APA StyleBurek, M., & Wandzik, I. (2019). Trehalose-Rich, Degradable Hydrogels Designed for Trehalose Release under Physiologically Relevant Conditions. Polymers, 11(12), 2027. https://doi.org/10.3390/polym11122027