Buildup of Multi-Ionic Supramolecular Network Facilitated by In-Situ Intercalated Organic Montmorillonite in 1,2-Polybutadiene


Abstract
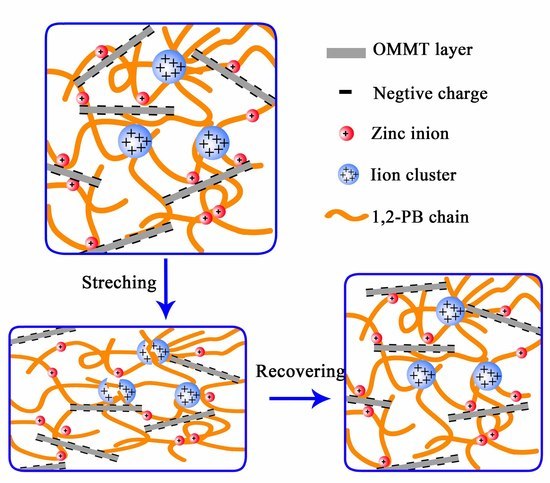
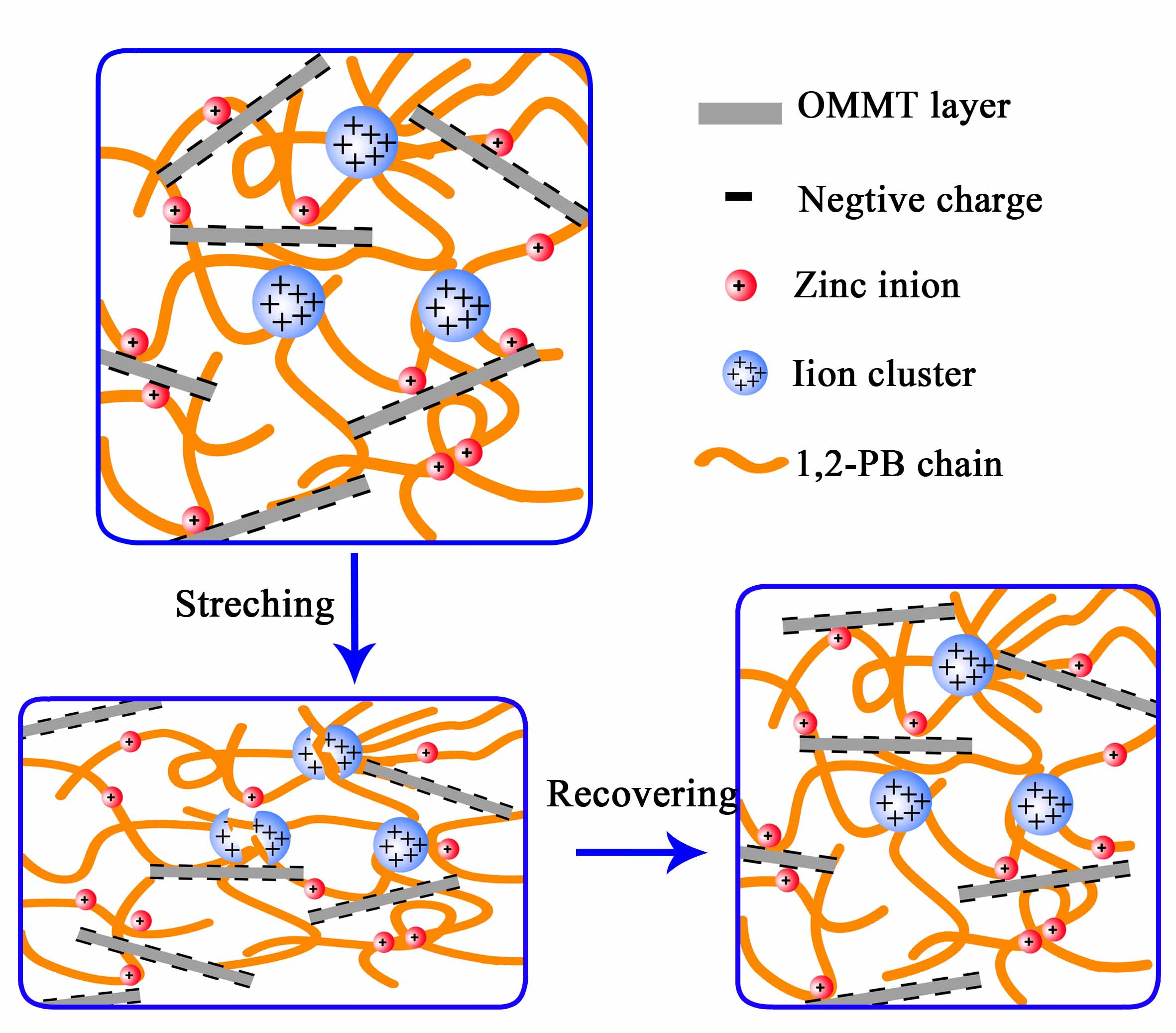
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of OMMT
2.3. Preparation of In-Situ-NC
2.4. Preparation of In-Situ-NC/ZDMA
2.5. Characterization
2.5.1. X-ray Diffraction (XRD)
2.5.2. Gel Permeation Chromatography (GPC)
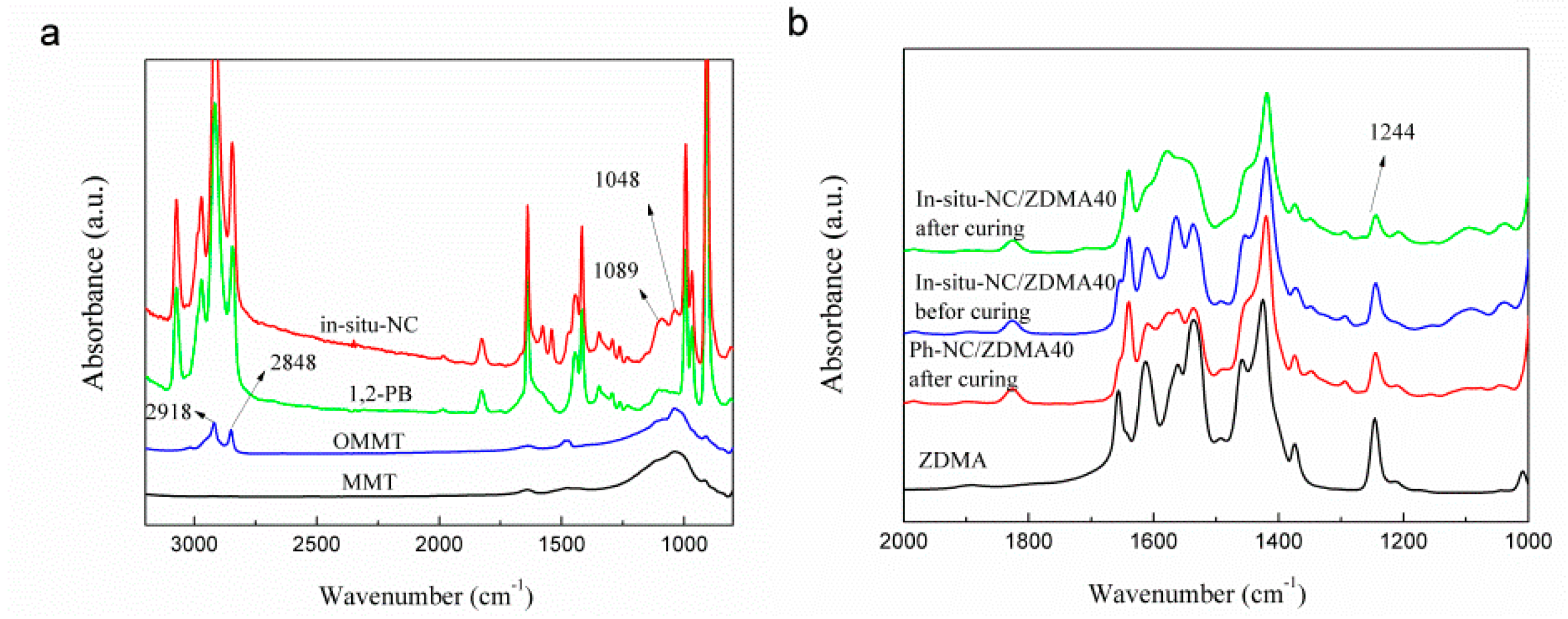
2.5.3. Fourier Transform Infrared Spectroscopy (FTIR)
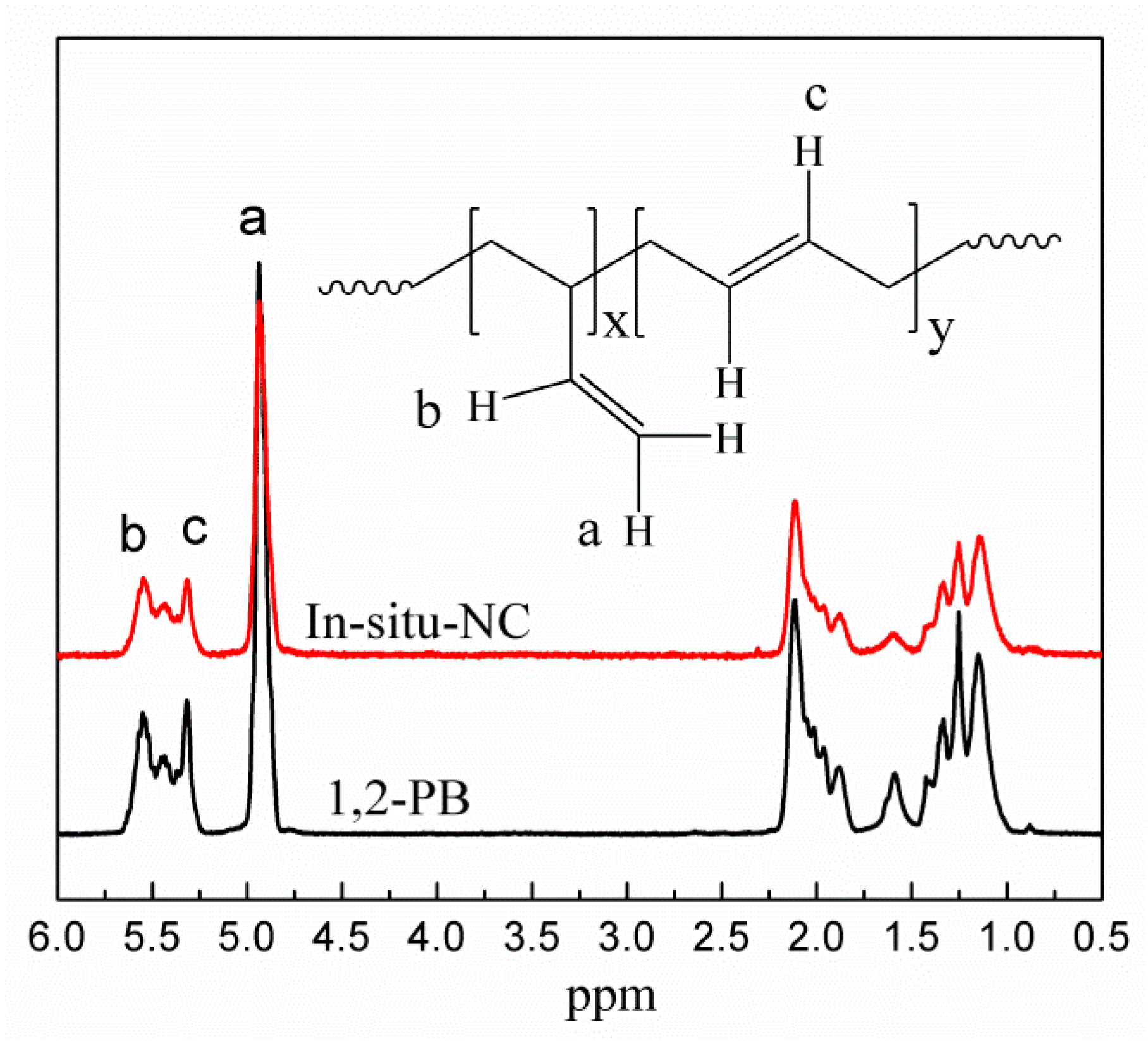
2.5.4. 1H Spectra of Nuclear Magnetic Resonance (1H NMR)
2.5.5. Scanning Electron Microscopy (SEM)
2.5.6. Energy Dispersive Spectroscopy (EDS)
2.5.7. Transmission Electron Microscopy (TEM)
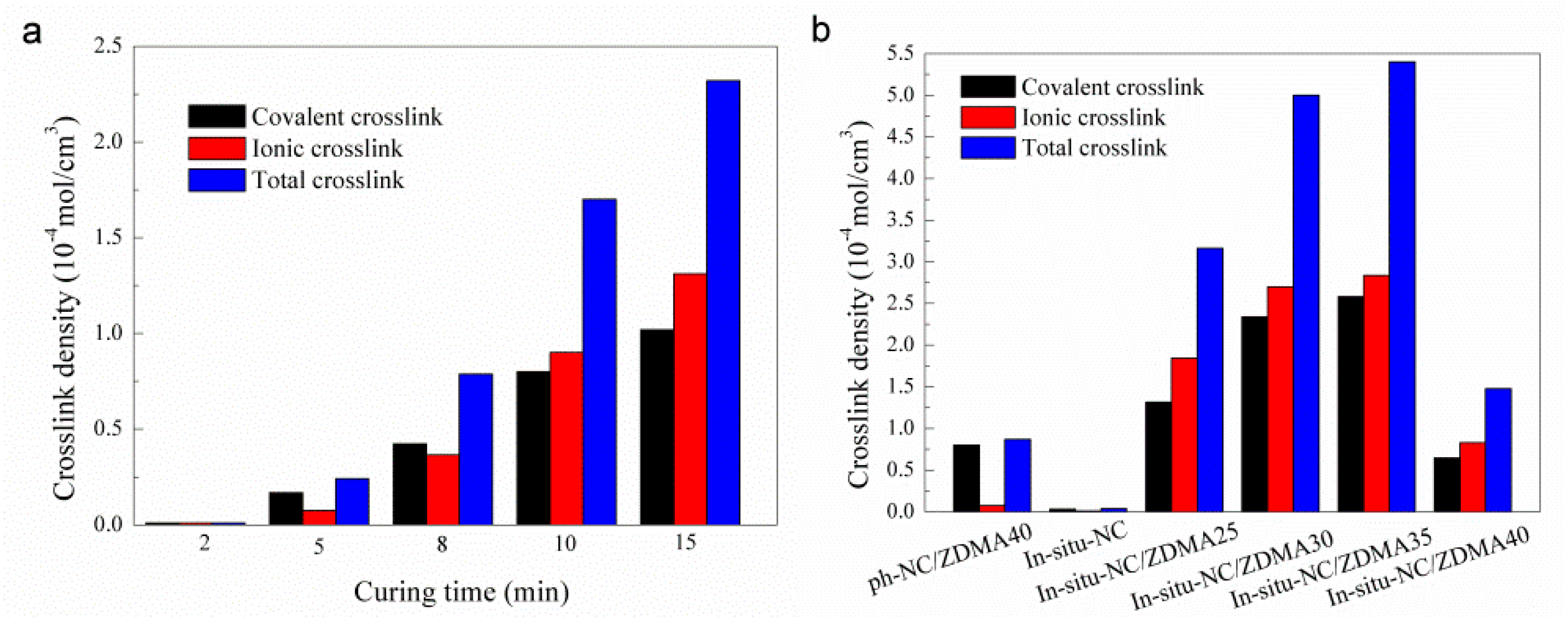
2.5.8. Crosslinking Density
2.5.9. Differential Scanning Calorimetry (DSC)
2.5.10. Dynamic Mechanical Properties (DMA)
2.5.11. Stress-Relaxation Experiment
2.5.12. Hysteresis Loss
2.5.13. Dielectric Performance
2.5.14. Physical and Mechanical Properties
3. Results and Discussion
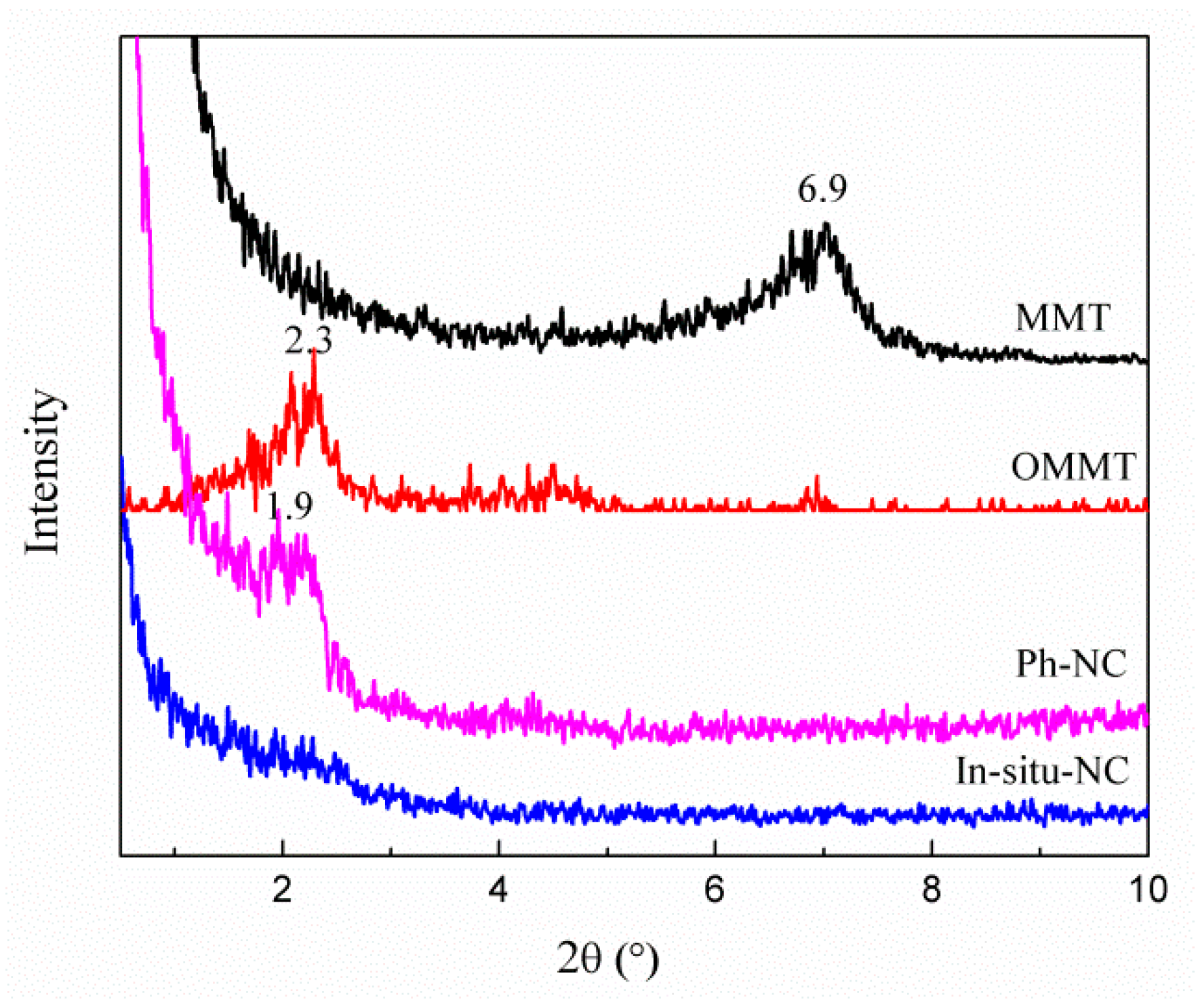
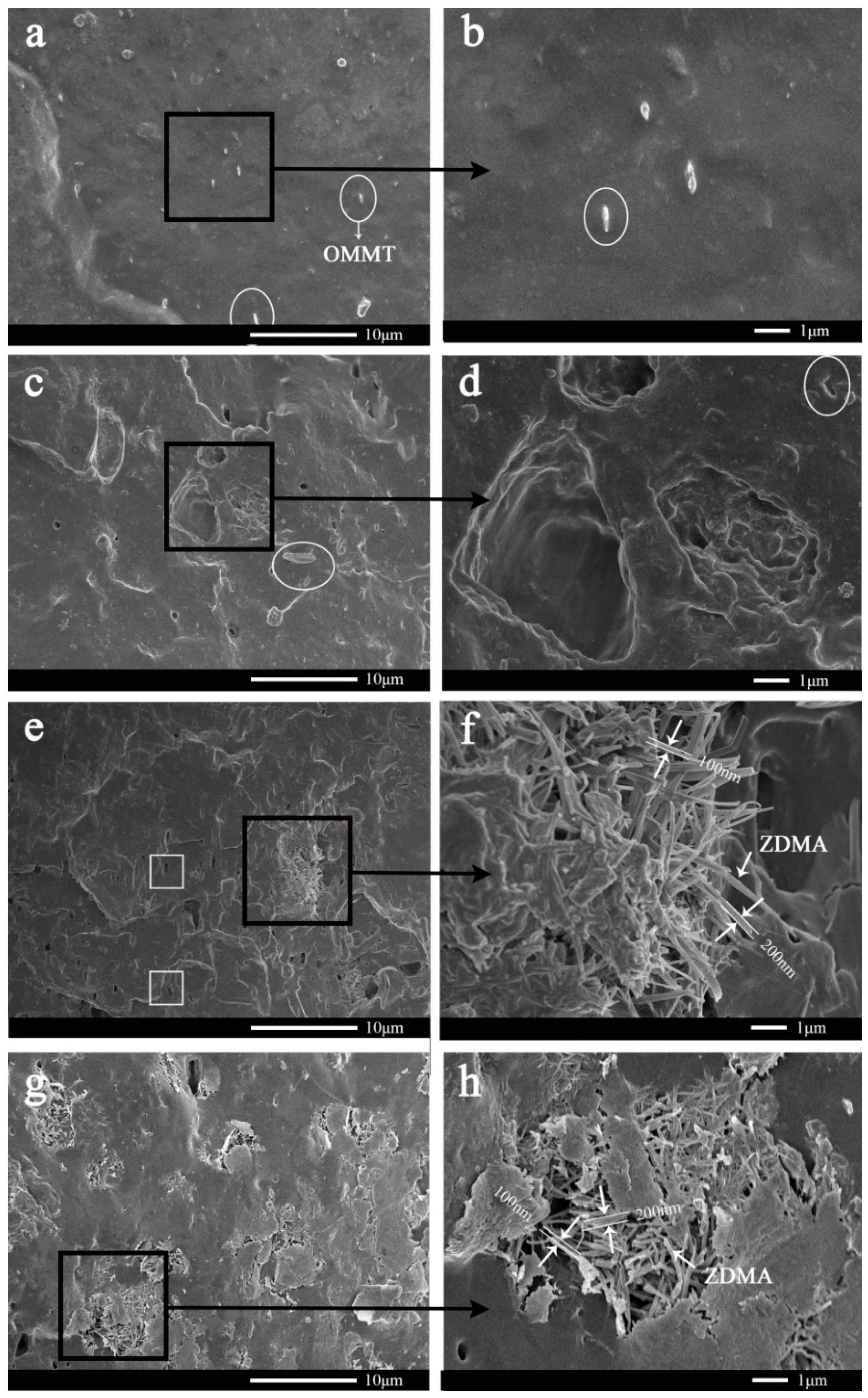
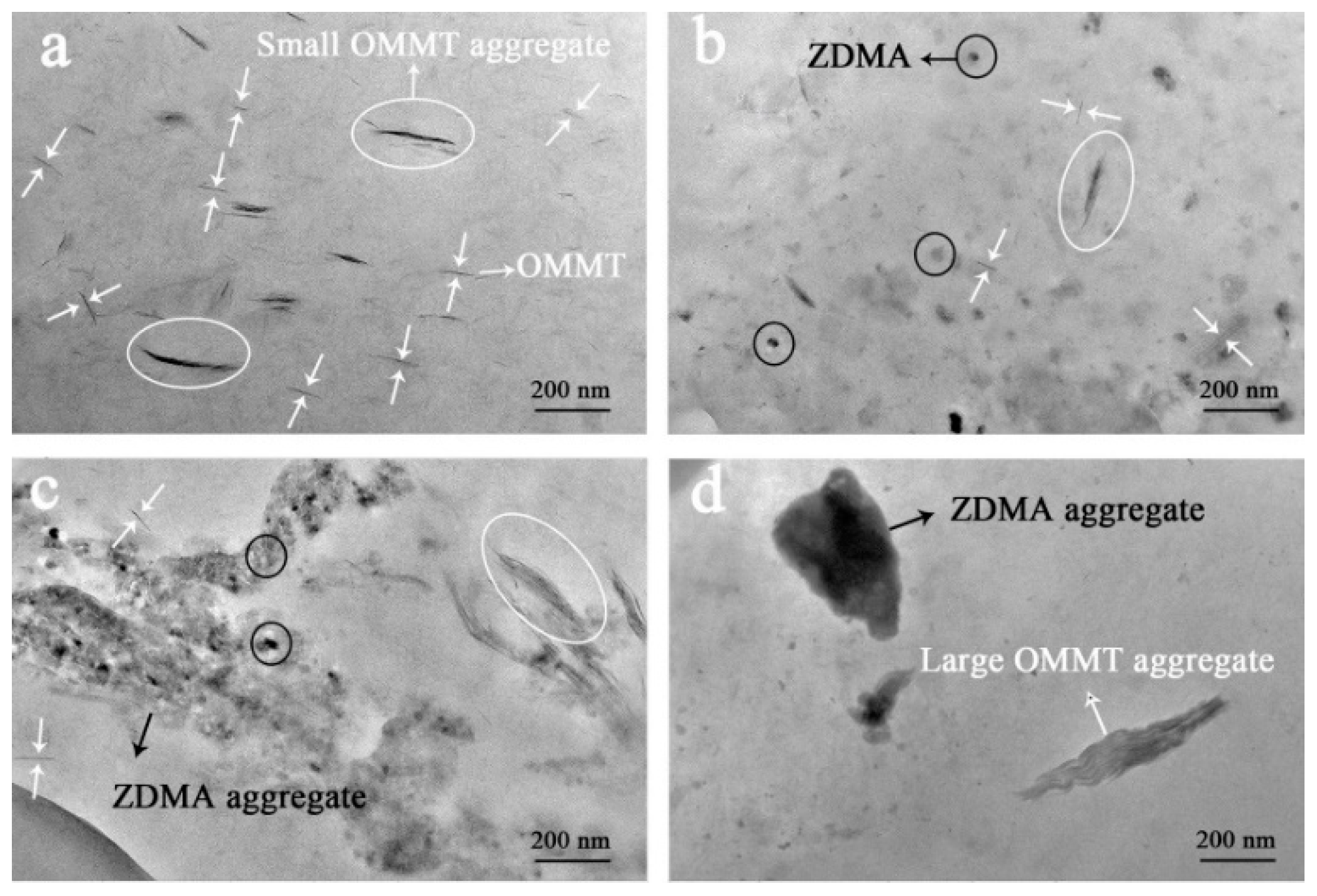
3.1. In-Situ Intercalation of OMMT
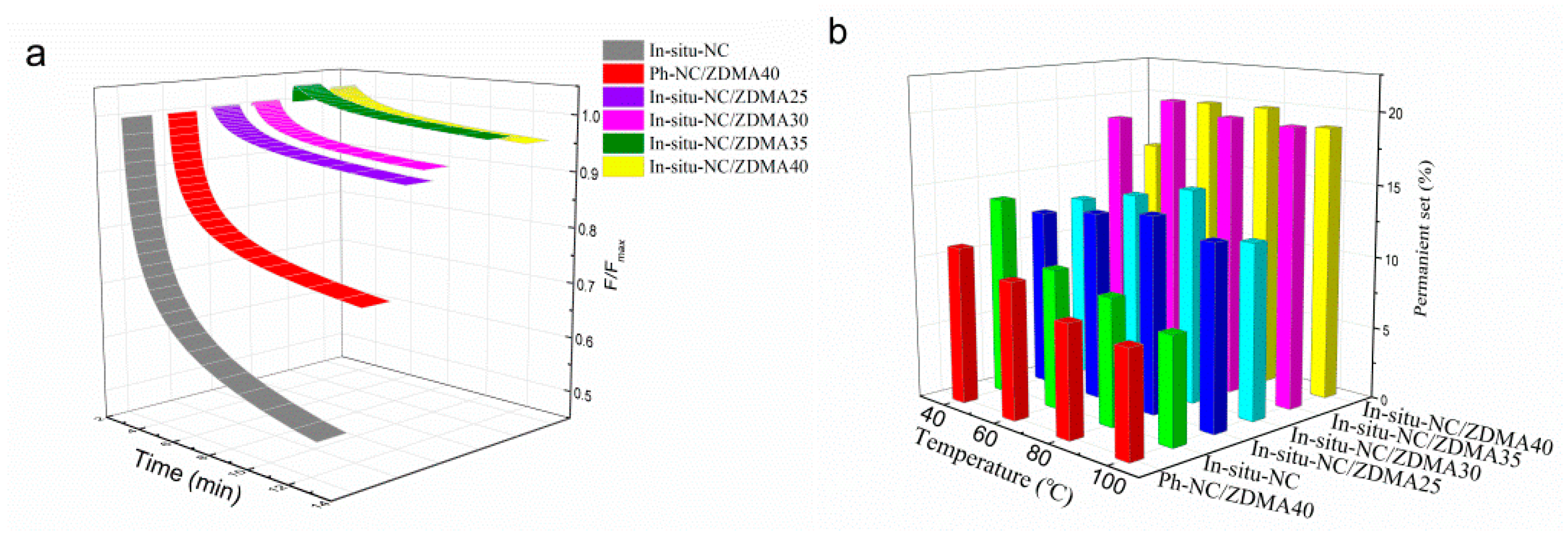
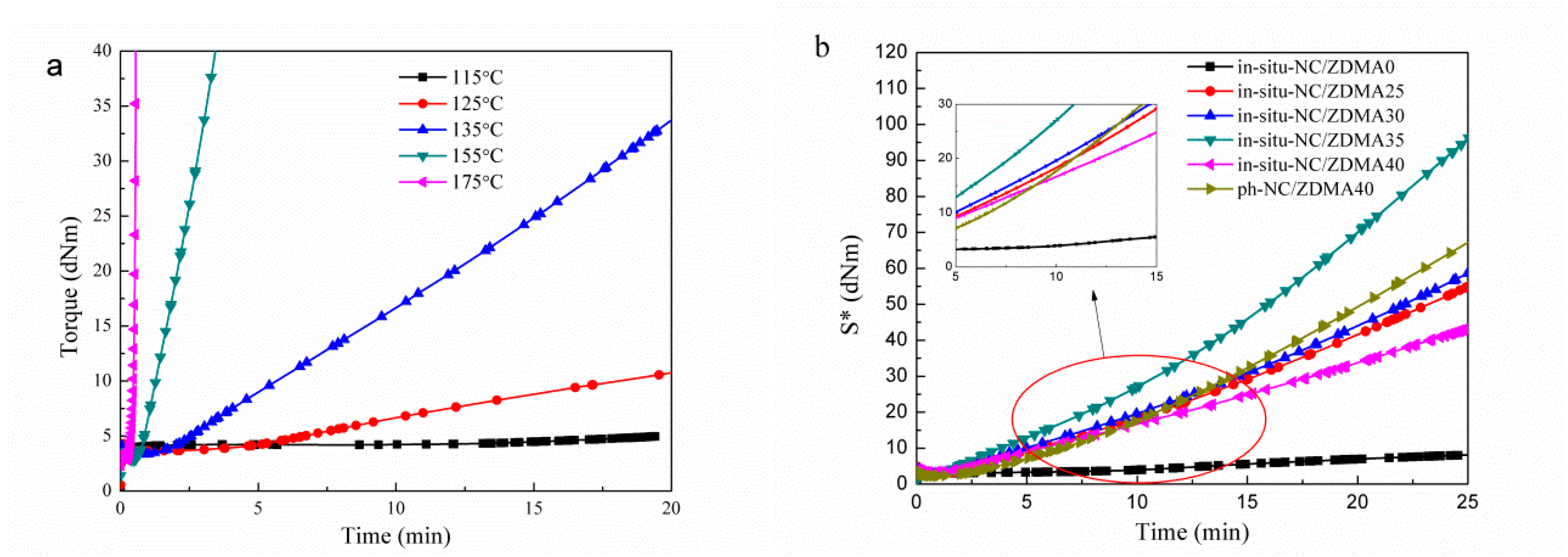
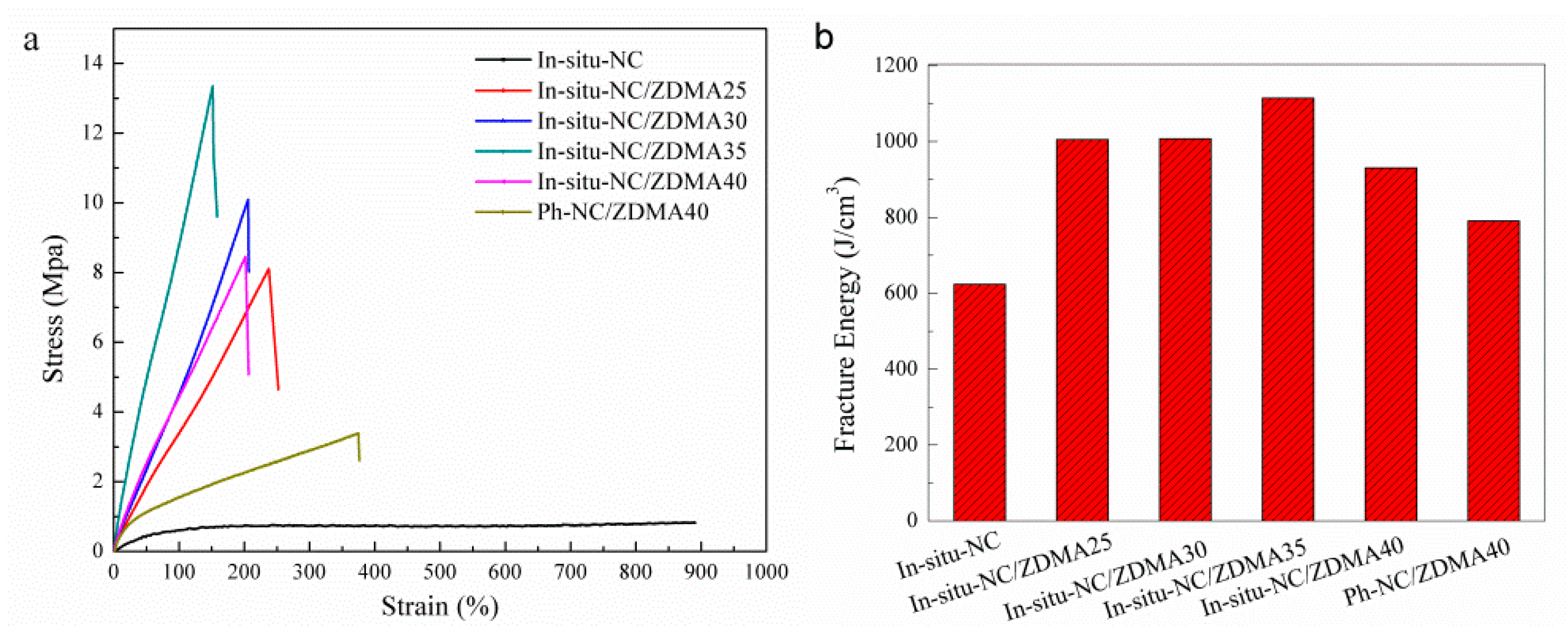
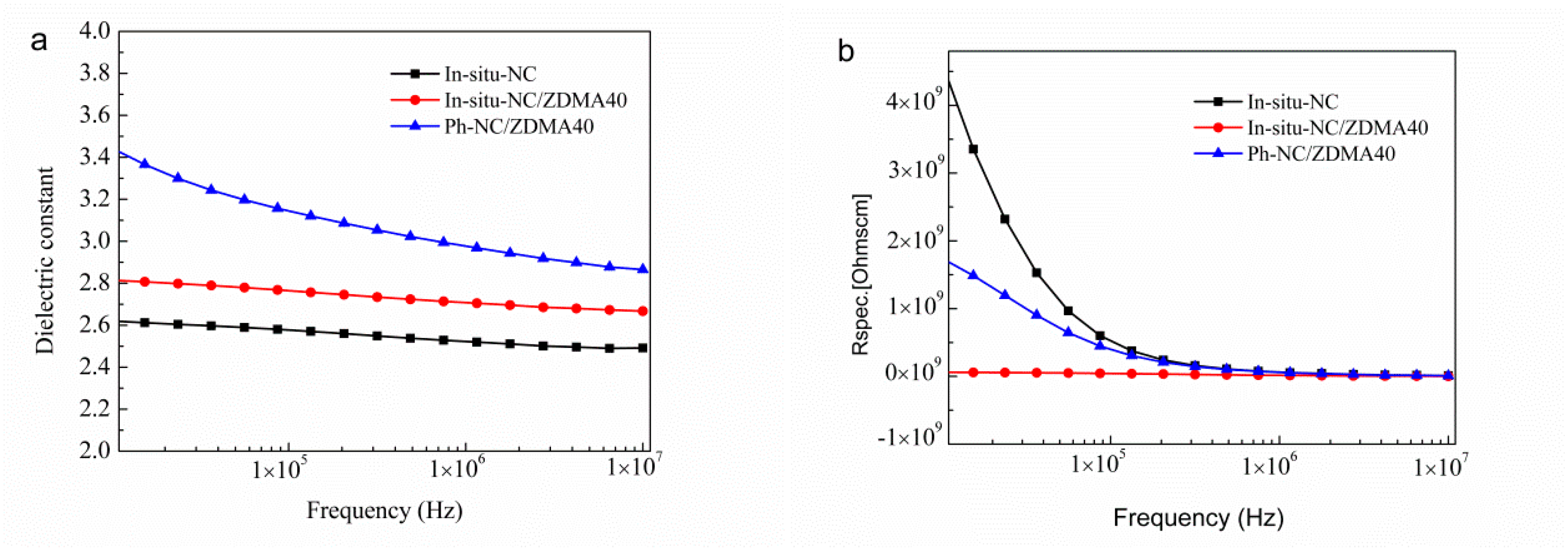
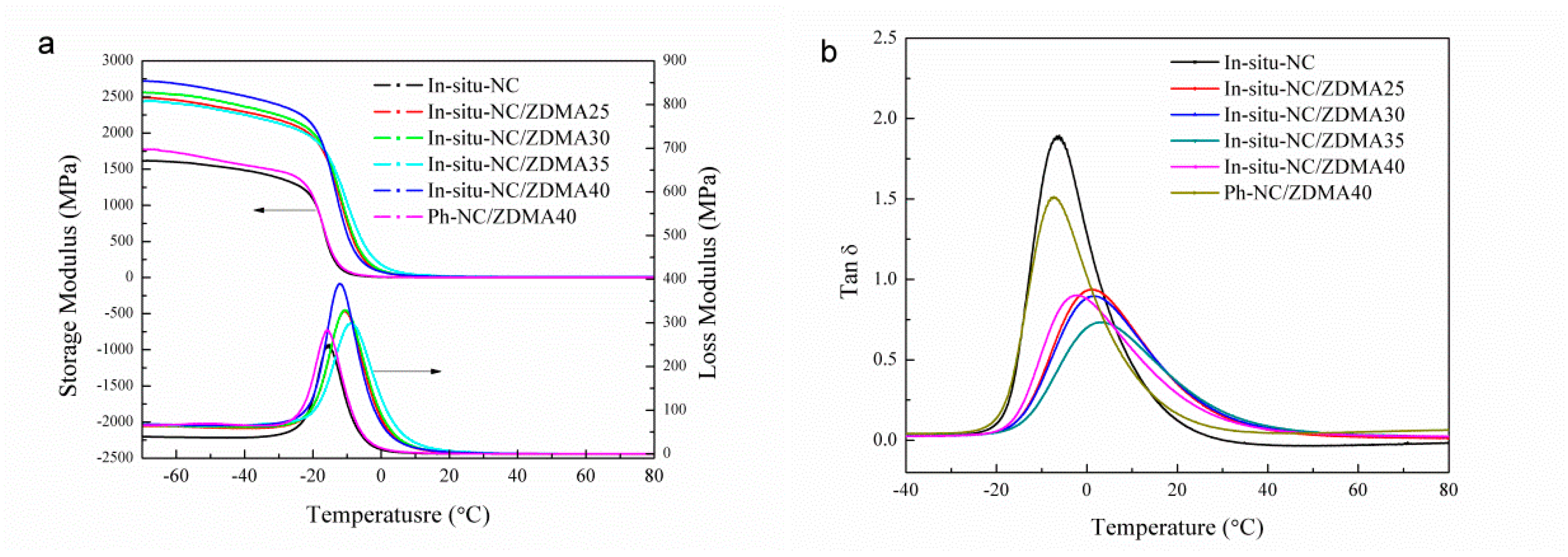
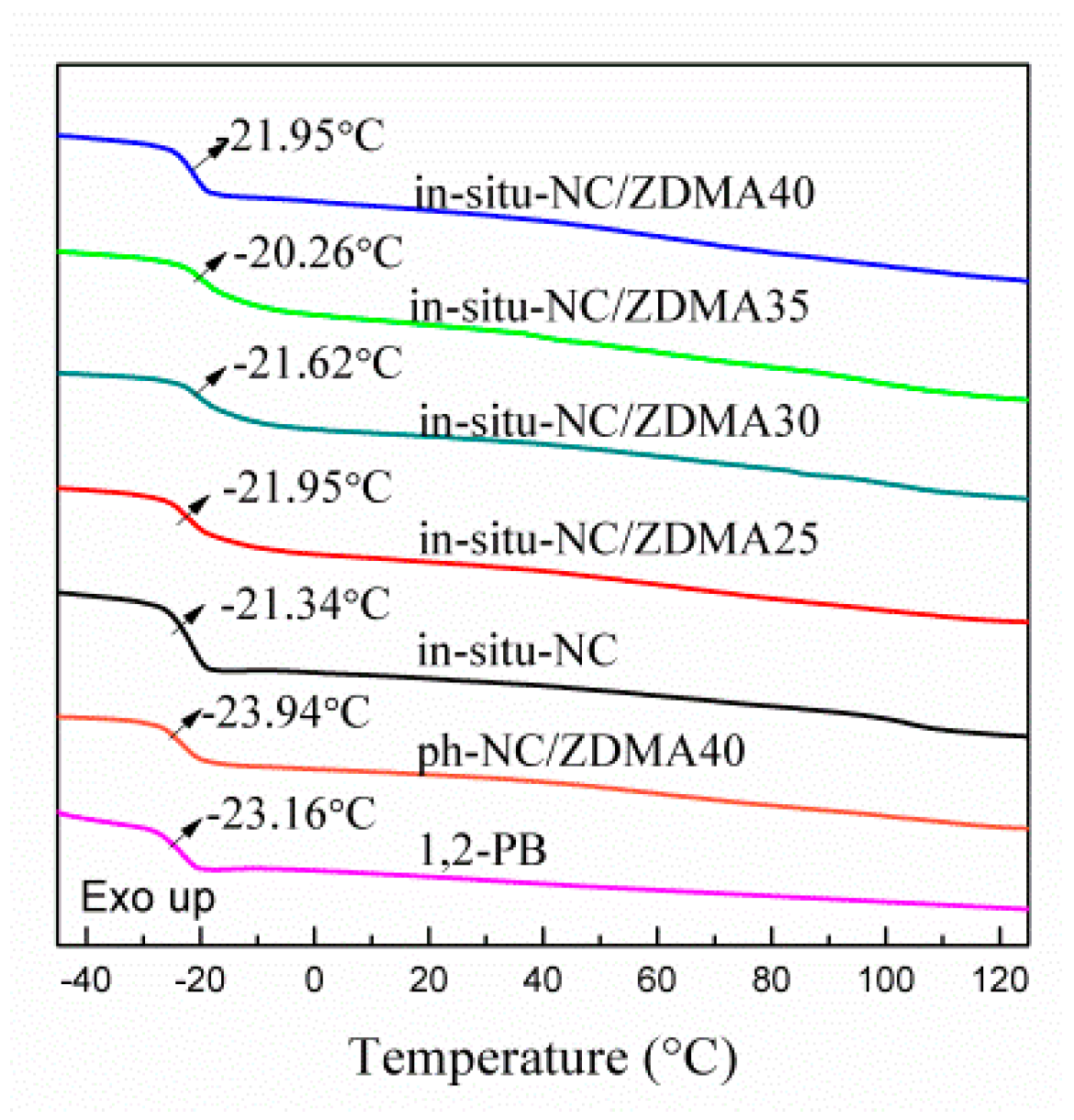
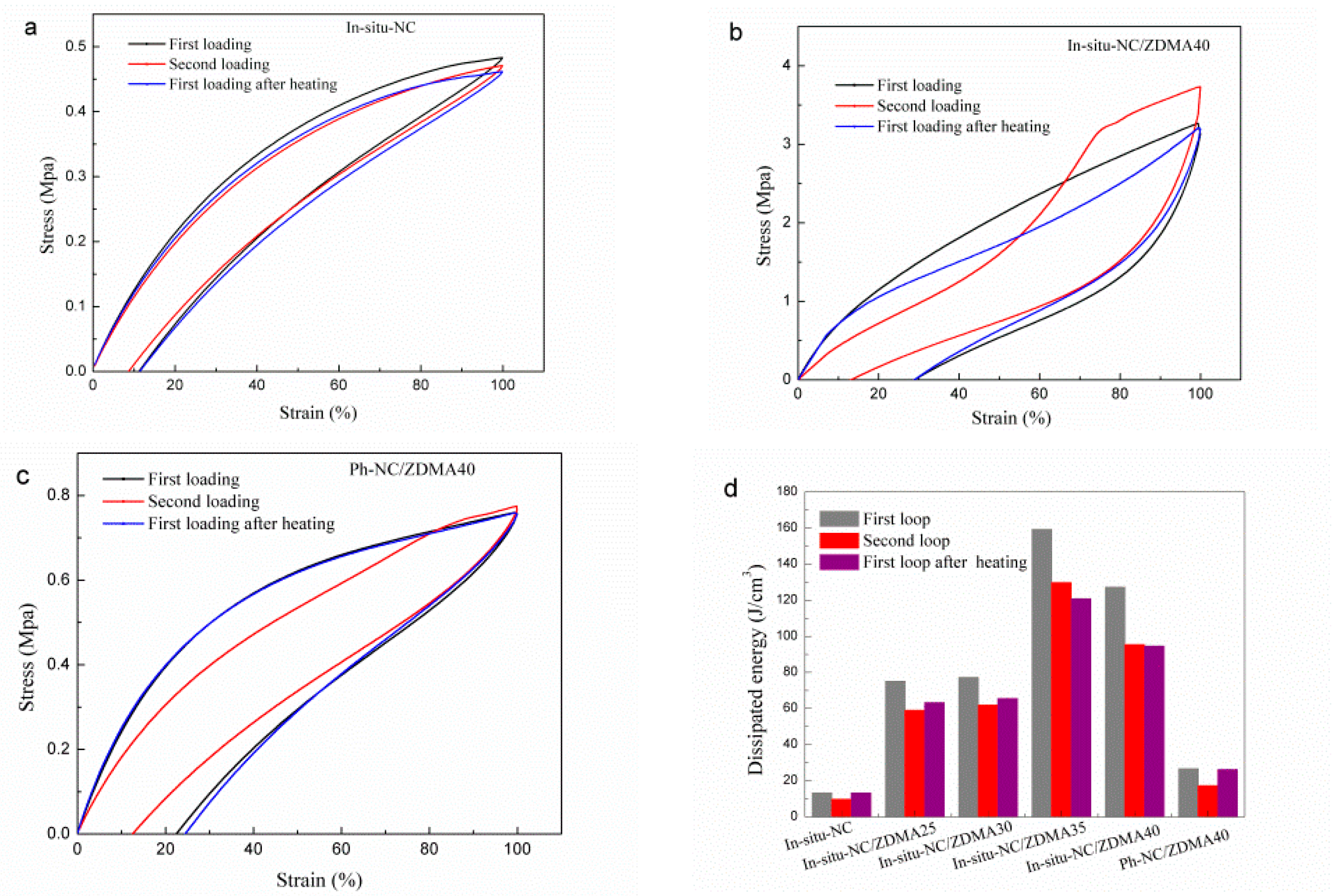
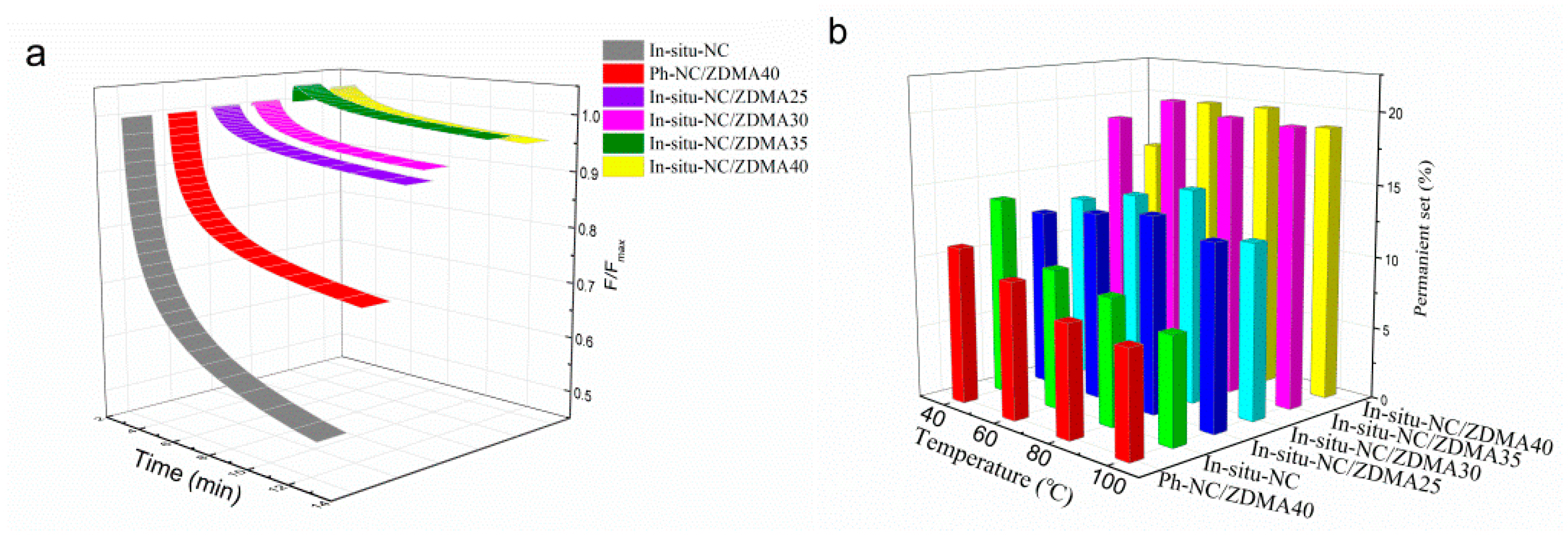
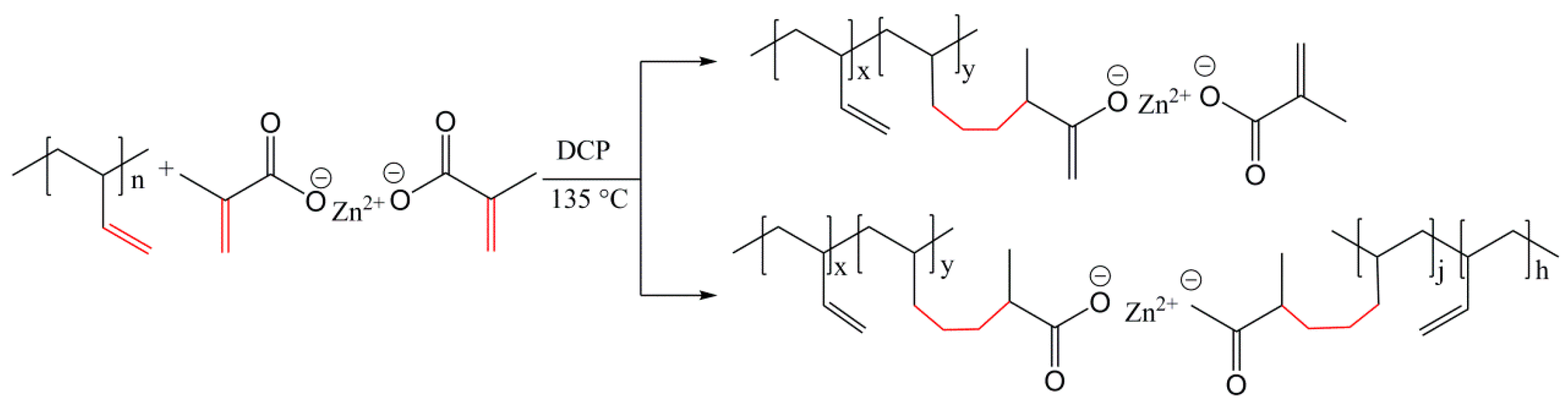
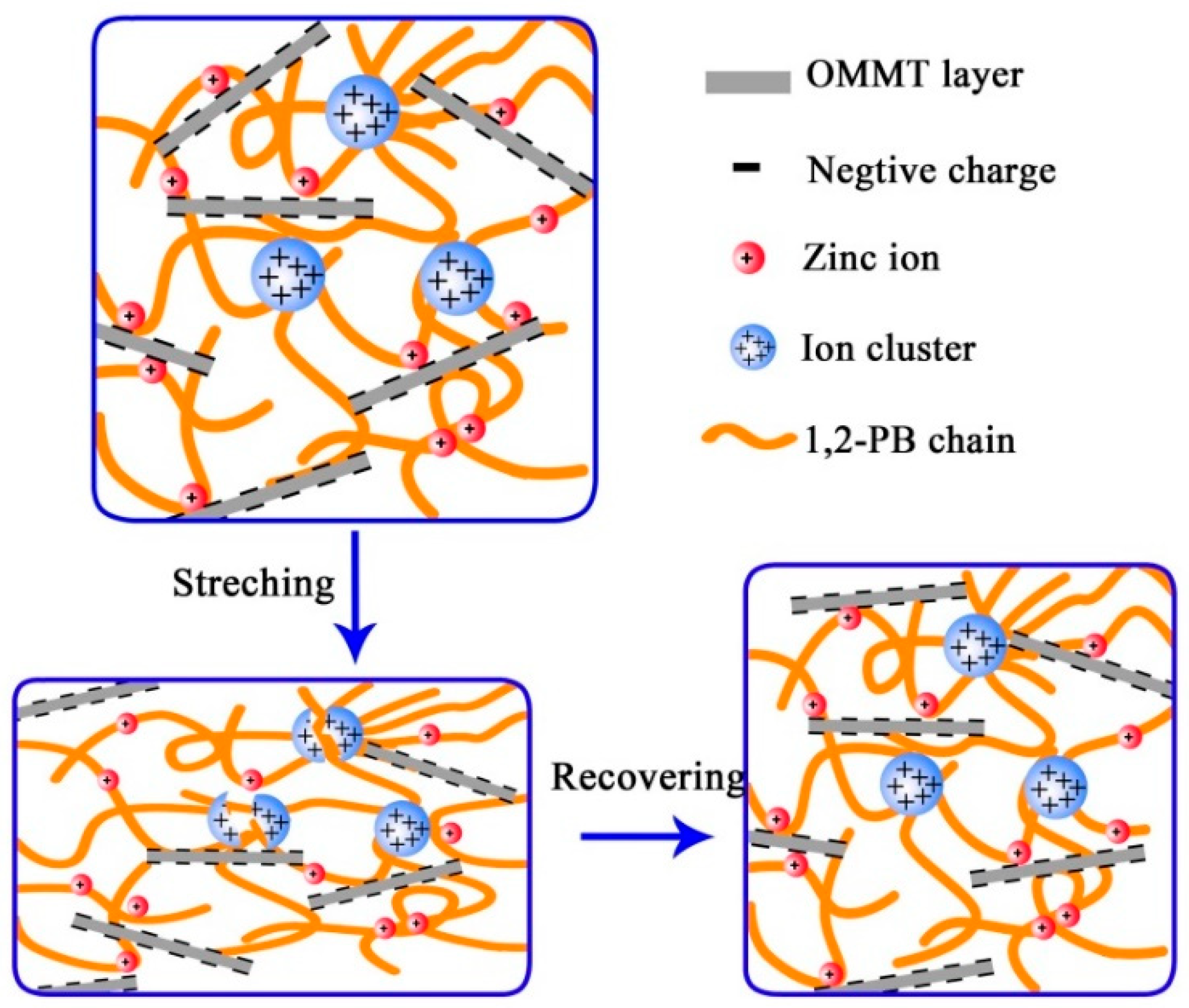
3.2. Construction of Multi-Ionic Supramolecular Network
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fratzl, P.; Weinkamer, R. Nature’s hierarchical materials. Prog. Mater. Sci. 2007, 52, 1263–1334. [Google Scholar] [CrossRef] [Green Version]
- Meyers, M.A.; Chen, P.Y.; Lin, Y.M.; Seki, Y. Biological materials: Structure and mechanical properties. Prog. Mater. Sci. 2008, 53, 1–206. [Google Scholar] [CrossRef] [Green Version]
- Satyanarayana, M.S.; Bhowmick, A.K.; Kumar, K.D. Preferentially fixing nanoclays in the phases of incompatible carboxylated nitrile rubber (XNBR)-natural rubber (NR) blend using thermodynamic approach and its effect on physico mechanical properties. Polymer 2016, 99, 21–43. [Google Scholar] [CrossRef]
- Mohana, T.P.; Kuriakoseb, J.; Kanny, K. Effect of nanoclay reinforcement on structure, thermal and mechanical properties of natural rubber-styrene butadine rubber (NR–SBR). J. Ind. Eng. Chem. 2011, 17, 264–270. [Google Scholar] [CrossRef]
- Carretero-Gonzalez, J.; Retsos, H.; Verdejo, R.; Toki, S.; Hsiao, B.S.; Giannelis, E.P.; Lopez-Manchado, M.A. Effect of Nanoclay on Natural Rubber Microstructure. Macromolecules 2008, 41, 6763–6772. [Google Scholar] [CrossRef]
- Espinosa, H.D.; Rim, J.E.; Barthelat, F.; Buehler, M.J. Merger of structure and material in nacre and bone-Perspectives on de novo biomimetic materials. Prog. Mater. Sci. 2009, 54, 1059–1100. [Google Scholar] [CrossRef]
- Andreas, W.; Ingela, B.; Jani-Markus, M.; Janne, R.; Lars, B.; Olli, I. Supramolecular control of stiffness and strength in lightweight high-performance nacre-mimetic paper with fire-shielding properties. Angew. Chem. Int. Ed. 2010, 49, 6448–6453. [Google Scholar]
- Jackson, A.P.; Vincent, J.F.V.; Turner, R.M. The Mechanical Design of Nacre. Proc. R. Soc. Lond. Ser. 1988, 234, 415–440. [Google Scholar]
- Li, H.; Oberhauser, A.F.; Fowler, S.B.; Clarke, J.; Fernandez, J.M. Atomic force microscopy reveals the mechanical design of a modular protein. Proc. Natl. Acad. Sci. USA 2000, 97, 6527–6531. [Google Scholar] [CrossRef] [Green Version]
- Etienne, D.; Yulan, C.; Markus, B.; Sijbesma, R.P.; Costantino, C. Toughening elastomers with sacrificial bonds and watching them break. Science 2014, 344, 186–189. [Google Scholar]
- Damien, M.; Francois, T.; Manuel, H.; Jean-Luc, C.; Ludwik, L. Versatile one-pot synthesis of supramolecular plastics and self-healing rubbers. J. Am. Chem. Soc. 2009, 131, 7966–7967. [Google Scholar]
- Philippe, C.; Francois, T.; Corinne, S.Z.; Ludwik, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977. [Google Scholar]
- Chen, Y.; Kushner, A.M.; Williams, G.A.; Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 2012, 4, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Son, D.; Wang, G.I.N.; Liu, Y.; Lopez, J.; Kim, Y.; Jin, Y.O.; Katsumata, T.; Mun, J.; Lee, Y. Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin. Adv. Sci. 2018, 30, 1706846. [Google Scholar] [CrossRef] [PubMed]
- Mark, B.; Liming, T.; Kumpfer, J.R.; Duncan, A.J.; Beyer, F.L.; Fiore, G.L.; Rowan, S.J.; Christoph, W. Optically healable supramolecular polymers. Nature 2011, 472, 334. [Google Scholar]
- Kersey, F.R.; Loveless, D.M.; Craig, S.L. A hybrid polymer gel with controlled rates of cross-link rupture and self-repair. J. R. Soc. Interface 2007, 4, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Li, C.H.; Wang, C.; Keplinger, C.; Zuo, J.L.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y.; Lissel, F.; Linder, C. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef]
- Liu, J.; Wang, S.; Tang, Z.; Huang, J.; Guo, B.; Huang, G. Bioinspired engineering of two different types of sacrificial bonds into chemically cross-linked cis-1,4-polyisoprene toward a high-performance elastomer. Macromolecules 2016, 49, 8593–8604. [Google Scholar] [CrossRef]
- Kalista, S.J.; Ward, T.C.; Oyetunji, Z. Self-Healing of Poly(Ethylene-co-Methacrylic Acid) Copolymers Following Projectile Puncture. Mech. Adv. Mater. Struc. 2007, 14, 391–397. [Google Scholar] [CrossRef]
- Ye, X.; Huang, H.; Peng, X. Synthesis of self-healing waterborne polyurethanes containing sulphonate groups. RSC Adv. 2017, 7, 20093–20100. [Google Scholar] [Green Version]
- Bin Ihsan, A.; Sun, T.L.; Kurokawa, T.; Karobi, S.N.; Nakajima, T.; Nonoyama, T.; Roy, C.K.; Luo, F.; Gong, J.P. Self-healing behaviors of tough polyampholyte hydrogels. Macromolecules 2016, 49, 4245–4252. [Google Scholar] [CrossRef]
- Lin, S.T.; Takayuki, K.; Shinya, K.; Abu Bin, I.; Taigo, A.; Koshiro, S.; Md Anamul, H.; Tasuku, N.; Ping, G.J. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 2013, 12, 932. [Google Scholar]
- Zhang, H.J.; Sun, T.L.; Zhang, A.K.; Ikura, Y.; Nakajima, T.; Nonoyama, T.; Kurokawa, T.; Ito, O.; Ishitobi, H.; Gong, J.P. Tough Physical Double-Network Hydrogels Based on Amphiphilic Triblock Copolymers. Adv. Mater. 2016, 28, 4884–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burattini, S.; Colquhoun, H.M.; Fox, J.D.; Friedmann, D.; Greenland, B.W.; Harris, P.J.; Hayes, W.M.M.E.; Rowan, S.J. A self-repairing, supramolecular polymer system: Healability as a consequence of donor-acceptor π-π- stacking interactions. Chem. Commun. 2009, 44, 6717–6719. [Google Scholar] [CrossRef]
- Fox, J.; Wie, J.J.; Greenland, B.W.; Burattini, S.; Hayes, W.; Colquhoun, H.M.; Mackay, M.E.; Rowan, S.J. High-strength, healable, supramolecular polymer nanocomposites. J. Am. Chem. Soc. 2012, 134, 5362–5368. [Google Scholar] [CrossRef] [PubMed]
- Paramita, D.; Andreas, W. Ionic supramolecular bonds preserve mechanical properties and enable synergetic performance at high humidity in water-borne, self-assembled nacre-mimetics. Nanoscale 2013, 5, 9348–9356. [Google Scholar]
- Kai, L.; Lv, Q.; Jing, H. Study on damping properties of HVBR/EVM blends prepared by in situ polymerization. Polym. Test. 2017, 60, 321–325. [Google Scholar]
- Jing, H.; Kai, L.; Wang, Z.; Geng, J.; Xin, W. Effect of vinyl and phenyl group content on the physical and dynamic mechanical properties of HVBR and SSBR. J. Appl. Polym. Sci. 2018, 135, 45975. [Google Scholar]
- Zhang, W.; Huang, B.; Ai, D.U.; Yao, W.; Yang, S.; Wang, M. Properties of TPI/HVBR/SBR blends. China Rubber Ind. 2002, 49, 69–72. [Google Scholar]
- Jinhui, L.; Jieting, G.; Kai, L.; Lingli, Z.; Zhaobo, W.; Jing, H. Effect of electronegativity and steric hindrance of the cocatalyst on the activity and selectivity of butadiene polymerization catalyzed by molybdenum. J. Appl. Polym. Sci. 2019, 136, 46906. [Google Scholar]
- Cong, Y.; Ying, L.; Jing, H. Vulcanization formula study on high vinyl polybutadiene rubber synthesized by molybdenum catalyst system. Qilu Petrochem. Technol. 2014, 42, 9–12. [Google Scholar]
- Deng, Z.F.; Guo, L.Y.; Ling, X.U.; Hua, J. Dynamic Property of Molybdenum-catalyzed High 1,2-Polybutadiene Rubber. China Rubber Ind. 2011, 4, 227–230. [Google Scholar]
- Wang, L.F.; Wang, W.; Guo, B.S.; Li, M.; Lin, C.Y.; Wang, D.L. Research advance of 1,2-polybutadiene. China Elastom. 2009, 3, 70–75. [Google Scholar]
- Hua, J.; Liu, J.; Wang, X.; Yue, Z.; Yang, H.; Geng, J.; Ding, A. Structure and properties of a cis-1,4-polybutadiene/organic montmorillonite nanocomposite prepared via in-situ polymerization. J. Macromol. Sci. B 2017, 56, 451–461. [Google Scholar] [CrossRef]
- Hua, J.; Chen, F.; Tian, Y.; Wang, X.; Cao, K.; Xu, L. Structures and Properties of 1,2-Polybutadiene/Organic Montmorillonite Nanocomposites Prepared by In-situ Polymerization. J. Macromol. Sci. B 2012, 51, 2034–2048. [Google Scholar] [CrossRef]
- Xu, C.; Cao, L.; Lin, B.; Liang, X.; Chen, Y. Design of self-Healing supramolecular rubbers by introducing ionic cross-links into natural rubber via a controlled vulcanization. ACS Appl. Mater. Interfaces 2016, 8, 17728–17737. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cao, L.; Huang, X.; Chen, Y.; Fu, L. Self-healing natural rubber with tailorable mechanical properties based on ionic supramolecular hybrid network. ACS Appl. Mater. Interfaces 2017, 9, 29363–29373. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Xin, W.; Jing, H. Grafting of Butadiene-Acrylonitrile Copolymer onto High Vinyl Polybutadiene by Copolymerization. Adv. Polym. Technol. 2016. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Mechanics of Swelling of Network Structures. J. Chem. Phys. 1950, 18, 108–111. [Google Scholar] [CrossRef]
- Arroyo, M.; López-Manchado, M.A.; Herrero, B. Organo-montmorillonite as substitute of carbon black in natural rubber compounds. Polymer 2003, 44, 2447–2453. [Google Scholar] [CrossRef]
- Bala, P.; Samantaray, B.K.; Srivastava, S.K.; Nando, G.B. Organomodified montmorillonite as filler in natural and synthetic rubber. J. Appl. Polym. Sci. 2004, 92, 3583–3592. [Google Scholar] [CrossRef]
- Ping, Z.; Fei, Z.; Yuan, Y.; Shi, X.; Zhao, S. Network evolution based on general-purpose diene rubbers/sulfur/TBBS system during vulcanization (I). Polymer 2010, 51, 257–263. [Google Scholar]
- Bai, Y.; Cheng, Z.Y.; Bharti, V.; Xu, H.S.; Zhang, Q.M. High-dielectric-constant ceramic- powder polymer composites. Appl. Phys. Lett. 2000, 76, 3804–3806. [Google Scholar] [CrossRef]
- Hill, L.W. Calculation of crosslink density in short chain networks. Prog. Org. Coat. 1997, 31, 235–243. [Google Scholar] [CrossRef]
- Basu, D.; Das, A.; Stöckelhuber, K.W.; Jehnichen, D.; Formanek, P.; Sarlin, E.; Vuorinen, J.; Heinrich, G. Evidence for an in situ developed polymer phase in ionic elastomers. Macromolecules 2014, 47, 3436–3450. [Google Scholar] [CrossRef]
- Rief, M.; Gautel, M.; Oesterhelt, F.; Fernandez, J.M.; Gaub, H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 1997, 276, 1109–1112. [Google Scholar] [CrossRef]
- Tang, Z.; Jing, H.; Guo, B.; Zhang, L.; Fang, L. Bioinspired engineering of sacrificial metal–ligand bonds into elastomers with supramechanical performance and adaptive recovery. Macromolecules 2016, 49, 1781–1789. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ph-NC/ZDMA40 | In-Situ-NC | In-Situ-NC/ZDMA25 | In-Situ-NC/ZDMA30 | In-Situ-NC/ZDMA35 | In-Situ-NC/ZDMA40 |
|---|---|---|---|---|---|---|
| Tensile strength (Mpa) | 2.82 | 0.99 | 7.98 | 8.10 | 11.40 | 8.45 |
| Tensile stress at 100% elongation (MPa) | 1.14 | 0.60 | 4.51 | 3.37 | 5.82 | 4.24 |
| Elongation at break (%) | 375 | 800 | 205 | 237 | 213 | 202 |
| Tearing strength (kN/m) | 28.05 | 11.87 | 23.28 | 25.74 | 31.23 | 29.48 |
| Shore A hardness (°) | 67 | 45 | 59 | 61 | 66 | 65 |
| Abrasion loss (cm3·1.6 km−1) | 0.38 | 0.28 | 0.33 | 0.34 | 0.35 | 0.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Li, D.; Zhao, X.; Geng, J.; Hua, J.; Wang, X. Buildup of Multi-Ionic Supramolecular Network Facilitated by In-Situ Intercalated Organic Montmorillonite in 1,2-Polybutadiene. Polymers 2019, 11, 492. https://doi.org/10.3390/polym11030492
Liu J, Li D, Zhao X, Geng J, Hua J, Wang X. Buildup of Multi-Ionic Supramolecular Network Facilitated by In-Situ Intercalated Organic Montmorillonite in 1,2-Polybutadiene. Polymers. 2019; 11(3):492. https://doi.org/10.3390/polym11030492
Chicago/Turabian StyleLiu, Jinhui, Di Li, Xiangshuai Zhao, Jieting Geng, Jing Hua, and Xin Wang. 2019. "Buildup of Multi-Ionic Supramolecular Network Facilitated by In-Situ Intercalated Organic Montmorillonite in 1,2-Polybutadiene" Polymers 11, no. 3: 492. https://doi.org/10.3390/polym11030492
APA StyleLiu, J., Li, D., Zhao, X., Geng, J., Hua, J., & Wang, X. (2019). Buildup of Multi-Ionic Supramolecular Network Facilitated by In-Situ Intercalated Organic Montmorillonite in 1,2-Polybutadiene. Polymers, 11(3), 492. https://doi.org/10.3390/polym11030492