Recyclable Self-Healing Polyurethane Cross-Linked by Alkyl Diselenide with Enhanced Mechanical Properties


 , and
, and
Abstract
:
1. Introduction
2. Experimental
2.1. Material and Reagents
2.2. Synthesis of γ-Butyrothiolactone
2.3. Synthesis of Diselenide- and Disulfide-Functionalized Diamine Cross-Linker
2.4. Synthesis of Di-Isocyanate-Terminated Urethanes 4
2.5. Synthesis of Tri-Isocyanate-Terminated Urethanes 5
2.6. Synthesis of Alkyl Diselenide-Based Polyurethanes
2.7. Characterization
3. Results and Discussion
3.1. Synthesis of Polyurethane
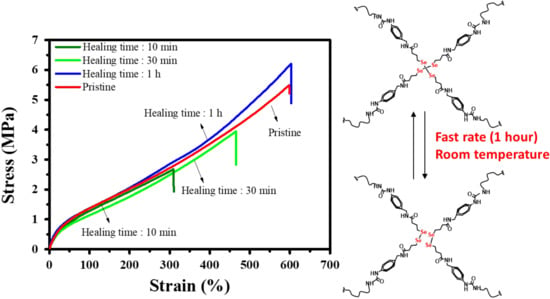
3.2. Mechanical Properties and Healing Properties
3.3. Reprocessing of Diselenide Polyurethanes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hay, J.N.; O’Gara, P. Recent developments in thermoset curing methods. Proc. Inst. Mech. Eng. Part G J. Aerosp. Eng. 2006, 220, 187–195. [Google Scholar] [CrossRef]
- Yourdkhani, M.; Koohbor, B.; Lamuta, C.; Dean, L.M.; Centellas, P.; Ivanoff, D.G.; Robertson, I.D.; White, S.R.; Sottos, N.R. Thermo-Mechanical Properties of Thermoset Polymers and Composites Fabricated by Frontal Polymerization; Springer: Cham, Switzerland, 2019; Volume 5, pp. 89–91. [Google Scholar]
- Kang, H.Y.; Schoenung, J.M. Electronic waste recycling: A review of U.S. infrastructure and technology options. Resour. Conserv. Recycl. 2005, 45, 368–400. [Google Scholar] [CrossRef]
- Garcia, J.M.; Jones, G.O.; Virwani, K.; McCloskey, B.D.; Boday, D.J.; ter Huurne, G.M.; Horn, H.W.; Coady, D.J.; Bintaleb, A.M.; Alabdulrahman, A.M.; et al. Recyclable, strong thermosets and organogels via paraformaldehyde condensation with diamines. Science 2014, 344, 732–735. [Google Scholar] [CrossRef]
- La Scala, J.; Wool, R.P. Property analysis of triglyceride-based thermosets. Polymer 2005, 46, 61–69. [Google Scholar] [CrossRef]
- Martín, C.; Ronda, J.C.; Cádiz, V. Novel flame-retardant thermosets: Diglycidyl ether of bisphenol A as a curing agent of boron-containing phenolic resins. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 1701–1710. [Google Scholar] [CrossRef]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kowsari, K.; Serjouei, A.; Dunn, M.L.; Ge, Q. Reprocessable thermosets for sustainable three-dimensional printing. Nat. Commun. 2018, 9, 1831. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Obadia, M.M.; Mudraboyina, B.P.; Serghei, A.; Montarnal, D.; Drockenmuller, E. Reprocessing and recycling of highly cross-linked ion-conducting networks through transalkylation exchanges of C-N bonds. J. Am. Chem. Soc. 2015, 137, 6078–6083. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, N.; Du Prez, F.E. Fast healing of polyurethane thermosets using reversible triazolinedione chemistry and shape-memory. Macromolecules 2018, 51, 3405–3414. [Google Scholar] [CrossRef]
- Billiet, S.; Hillewaere, X.K.; Teixeira, R.F.; Du Prez, F.E. Chemistry of crosslinking processes for self-healing polymers. Macromol. Rapid Commun. 2013, 34, 290–309. [Google Scholar] [CrossRef] [PubMed]
- Burattini, S.; Greenland, B.W.; Chappell, D.; Colquhoun, H.M.; Hayes, W. Healable polymeric materials: A tutorial review. Chem. Soc. Rev. 2010, 39, 1973–1985. [Google Scholar] [CrossRef]
- Liu, Y.L.; Chuo, T.W. Self-healing polymers based on thermally reversible Diels–Alder chemistry. Polym. Chem. 2013, 4, 2194. [Google Scholar] [CrossRef]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Guan, Z. Olefin metathesis for effective polymer healing via dynamic exchange of strong carbon-carbon double bonds. J. Am. Chem. Soc. 2012, 134, 14226–14231. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Tournilhac, F.; Leibler, L.; Guan, Z. Making insoluble polymer networks malleable via olefin metathesis. J. Am. Chem. Soc. 2012, 134, 8424–8427. [Google Scholar] [CrossRef]
- Imato, K.; Nishihara, M.; Kanehara, T.; Amamoto, Y.; Takahara, A.; Otsuka, H. Self-healing of chemical gels cross-linked by diarylbibenzofuranone-based trigger-free dynamic covalent bonds at room temperature. Angew. Chem. Int. Ed. Engl. 2012, 51, 1138–1142. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, B.; Zhang, X.; Xu, L.; Tao, L.; Li, S.; Wei, Y. A magnetic self-healing hydrogel. Chem. Commun. 2012, 48, 9305–9307. [Google Scholar]
- Liu, F.; Li, F.; Deng, G.; Chen, Y.; Zhang, B.; Zhang, J.; Liu, C.-Y. Rheological images of dynamic covalent polymer networks and mechanisms behind mechanical and self-healing properties. Macromolecules 2012, 45, 1636–1645. [Google Scholar] [CrossRef]
- Zheng, P.; McCarthy, T.J. A surprise from 1954: Siloxane equilibration is a simple, robust, and obvious polymer self-healing mechanism. J. Am. Chem. Soc. 2012, 134, 2024–2027. [Google Scholar] [CrossRef]
- Martin, R.; Rekondo, A.; Echeberria, J.; Cabanero, G.; Grande, H.J.; Odriozola, I. Room temperature self-healing power of silicone elastomers having silver nanoparticles as crosslinkers. Chem. Commun. 2012, 48, 8255–8257. [Google Scholar] [CrossRef] [PubMed]
- Lafont, U.; van Zeijl, H.; van der Zwaag, S. Influence of cross-linkers on the cohesive and adhesive self-healing ability of polysulfide-based thermosets. ACS Appl. Mater. Interfaces 2012, 4, 6280–6288. [Google Scholar] [CrossRef]
- Kildaht, N.K. Bond energy data summarized. J. Chem. Educ. 1995, 72, 423. [Google Scholar] [CrossRef]
- Lei, Z.Q.; Xiang, H.P.; Yuan, Y.J.; Rong, M.Z.; Zhang, M.Q. Room-temperature self-healable and remoldable cross-linked polymer based on the dynamic exchange of disulfide bonds. Chem. Mater. 2014, 26, 2038–2046. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.-P. Self-healing and shape memory linear polyurethane based on disulfide linkages with excellent mechanical property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-healing materials based on disulfide links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; de Luzuriaga, A.R.; Cabanero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horizons. 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior toughness and fast self-healing at room temperature engineered by transparent elastomers. Adv. Mater. 2018, 30, 1705145. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Visible-light-induced self-healing diselenide-containing polyurethane elastomer. Adv. Mater. 2015, 27, 7740–7745. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Dynamic diselenide bonds: Exchange reaction induced by visible light without catalysis. Angew. Chem. Int. Ed. Engl. 2014, 53, 6781–6785. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Aguirresarobe, R.H.; Irusta, L.; Ruipérez, F.; Matxain, J.M.; Pan, X.; Aramburu, N.; Mecerreyes, D.; Sardon, H.; Zhu, J. Aromatic diselenide crosslinkers to enhance the reprocessability and self-healing of polyurethane thermosets. Polym. Chem. 2017, 8, 3641–3646. [Google Scholar] [CrossRef]
- Sashida, H.; Nakayama, A.; Kaname, M. A new one-pot synthetic method for selenium-containing medium-sized α,β-unsaturated cyclic ketones. Synthesis 2008, 2008, 3229–3236. [Google Scholar] [CrossRef]
- Pan, X.; Driessen, F.; Zhu, X.; Du Prez, F.E. Selenolactone as a building block toward dynamic diselenide-containing polymer architectures with controllable topology. ACS Macro Lett. 2017, 6, 89–92. [Google Scholar] [CrossRef]
- Chen, Y.; Kushner, A.M.; Williams, G.A.; Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 2012, 4, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhang, Y.; Gao, L.; Bai, T.; Wang, W.; Cui, Y.; Liu, W. A Mechanically strong, highly stable, thermoplastic, and self-healable supramolecular polymer hydrogel. Adv. Mater. 2015, 27, 3566–3571. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Takahashi, A.; Ohishi, T.; Goseki, R.; Otsuka, H. Enhancement of the stimuli-responsiveness and photo-stability of dynamic diselenide bonds and diselenide-containing polymers by neighboring. Polymer 2018, 154, 281–290. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [PPG-2NCO]/[PPG-3NCO] | Strain (%) | Stress (MPa) | Toughness (MPa) |
|---|---|---|---|
| 0:1 | 560.00 | 10.52 | 30.07 |
| 0.25:1 | 723.57 | 11.91 | 41.63 |
| 0.50:1 | 801.42 | 9.98 | 40.26 |
| 0.75:1 | 804.48 | 8.64 | 34.09 |
| 1:1 | 598.00 | 5.21 | 16.92 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Y.; An, X.; Huang, X.; Pan, X.; Zhu, J.; Zhu, X. Recyclable Self-Healing Polyurethane Cross-Linked by Alkyl Diselenide with Enhanced Mechanical Properties. Polymers 2019, 11, 773. https://doi.org/10.3390/polym11050773
Qian Y, An X, Huang X, Pan X, Zhu J, Zhu X. Recyclable Self-Healing Polyurethane Cross-Linked by Alkyl Diselenide with Enhanced Mechanical Properties. Polymers. 2019; 11(5):773. https://doi.org/10.3390/polym11050773
Chicago/Turabian StyleQian, Yuqing, Xiaowei An, Xiaofei Huang, Xiangqiang Pan, Jian Zhu, and Xiulin Zhu. 2019. "Recyclable Self-Healing Polyurethane Cross-Linked by Alkyl Diselenide with Enhanced Mechanical Properties" Polymers 11, no. 5: 773. https://doi.org/10.3390/polym11050773
APA StyleQian, Y., An, X., Huang, X., Pan, X., Zhu, J., & Zhu, X. (2019). Recyclable Self-Healing Polyurethane Cross-Linked by Alkyl Diselenide with Enhanced Mechanical Properties. Polymers, 11(5), 773. https://doi.org/10.3390/polym11050773