Morphology on Reaction Mechanism Dependency for Twin Polymerization


Abstract
:
1. Introduction
2. Numerical Model, Materials, and Methods
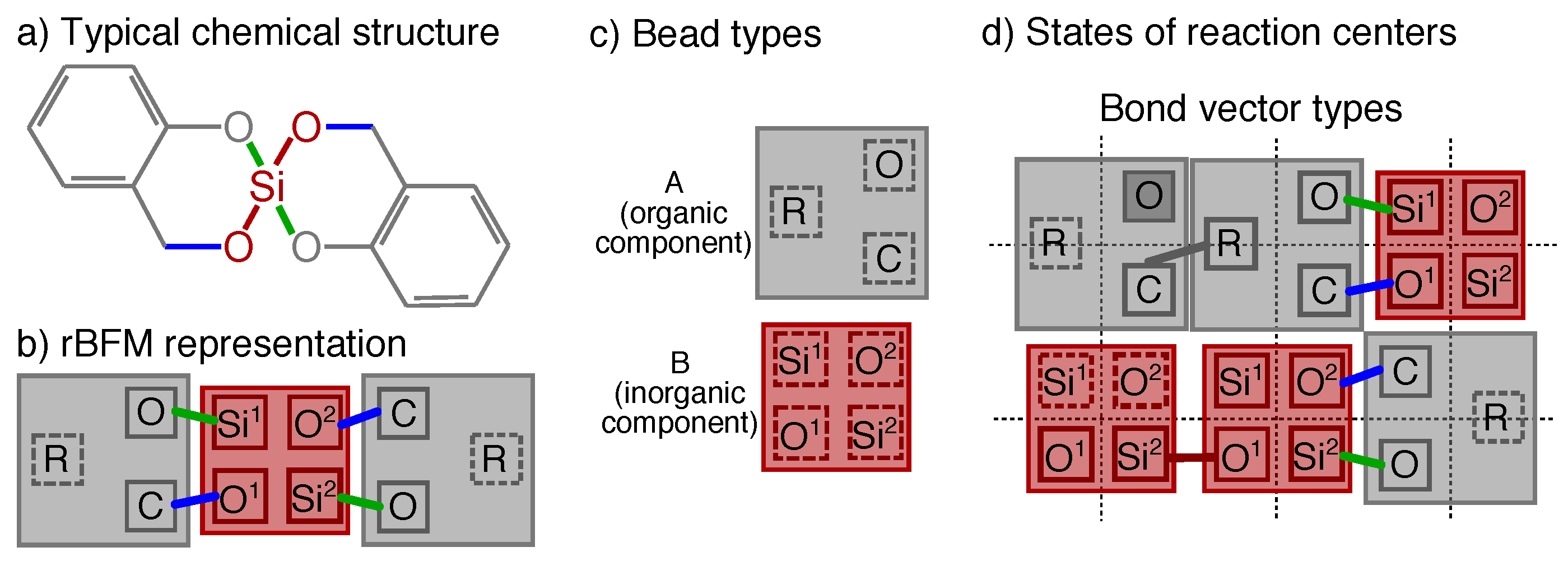
2.1. Reactive Bond Fluctuation Model
- the chosen lattice site is empty,
- all final bond lengths are allowed, and
- the final bonds do not cross each other.
- the reaction is allowed and
- no crossing of bonds occur after the rMC step,
2.2. Twin Polymerization
- cleavage of OSix,
- formation of CR,
- cleavage and formation of COx, and
- formation of SixOx.
2.3. Simulation Details
2.4. Process to Structure Analysis
- the bulk porosity and the specific surface area with A,B,A∪B},
- the radial distribution function with AA,BB} and the local porosity distribution with A,B,A∪B}, as well as
- the percolation probability and the percolation fraction with A,B,A∪B} and d as listed in Table 2.
3. Results and Discussion
3.1. Analysis of the Model Parameters on the Reaction Process
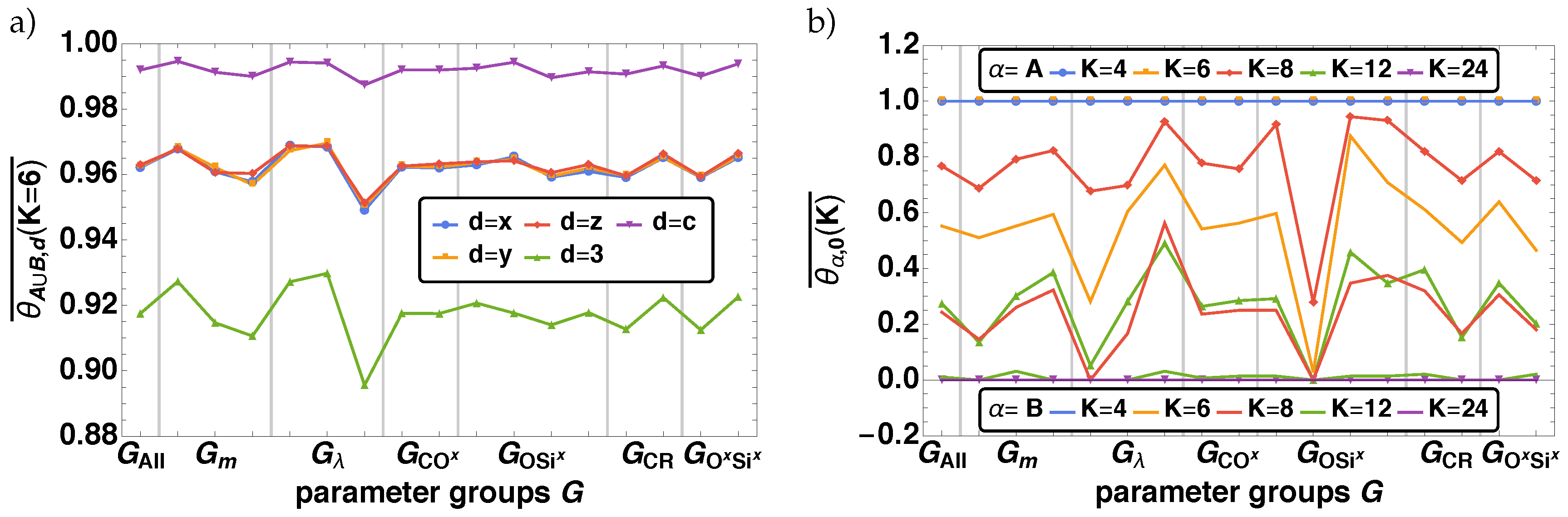
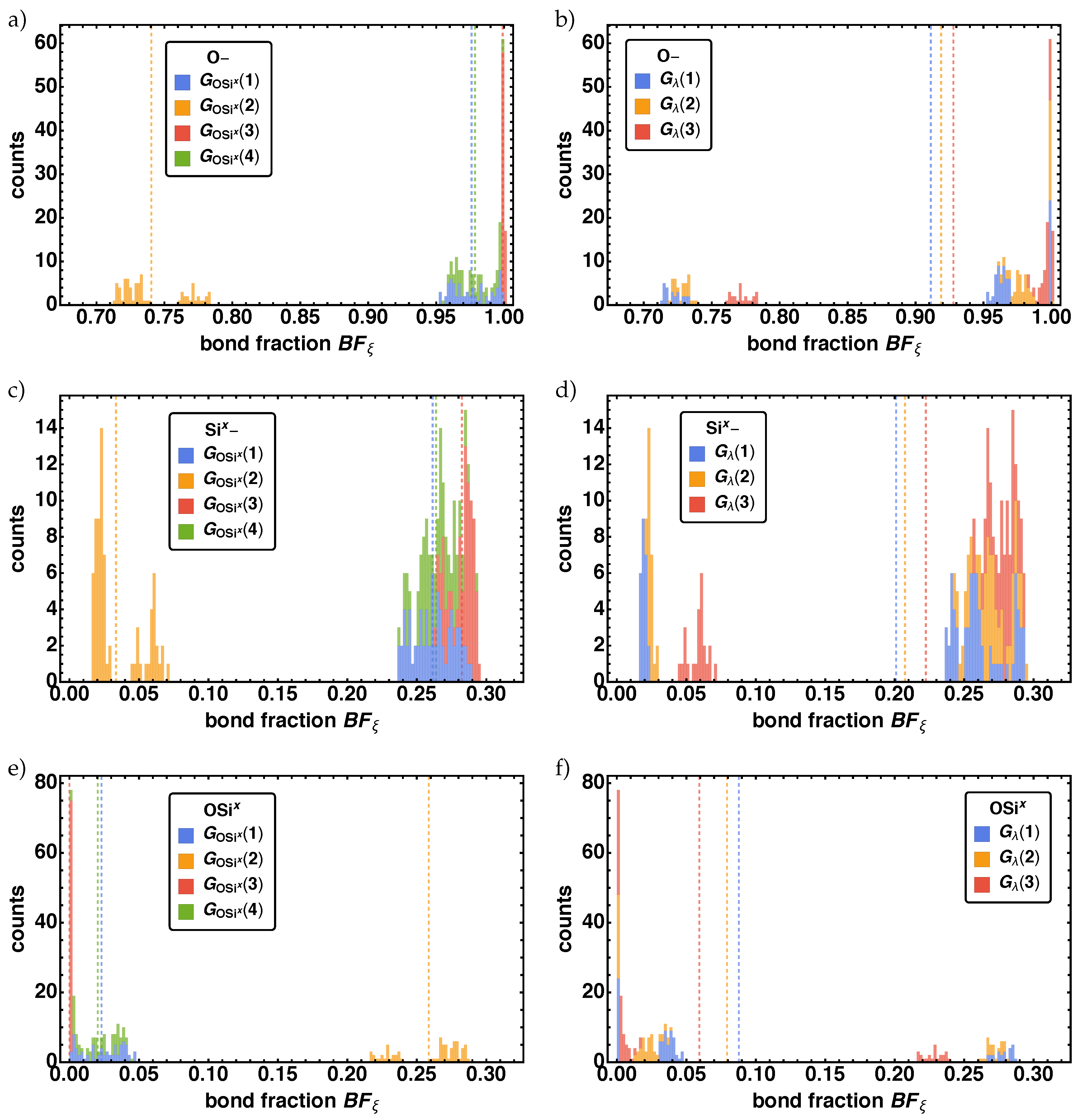
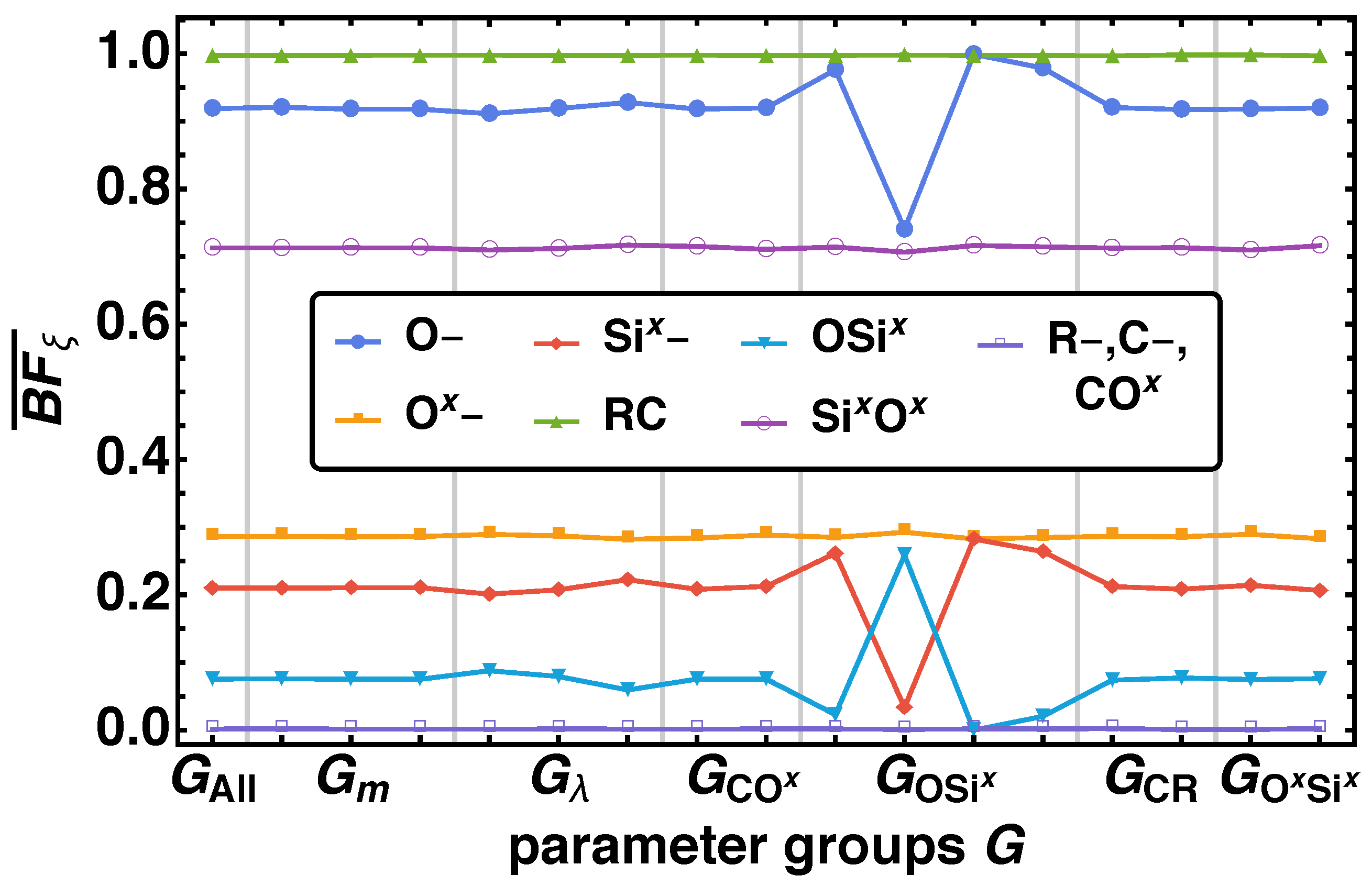
3.1.1. Bond Fraction
- With decreasing m all reactions are shifted to later times.
- With decreasing values of a minor time shift to later times occur for reactions, where the non-bonded reaction centers O–, Ox–, Six–, and the bond vector types OSix, SixOx are involved. Additionally, we find different final values of depending on for O–, Si,– SixOx}.
- With increasing the reactions, where C, R, CR, and COx are related, are shifted to significantly shorter times, whereas the other processes are only influenced in a minor way.
- With decreasing the reaction, where C–, R–, and CR participate, are shifted to significantly later times. The other process steps are not affected.
- The parameter group influences reactions in various ways, which are connected with O–, Ox–, Six–, OSix, and SixOx. In principle these parameter combinations change the duration of the reaction process, so that one can order it by increasing reaction duration. Ordering the results leads to with increasing duration. Similar to , here the influence on the reaction times shows up in the final values of for O–, Six–, SixOx}.
- With decreasing reactions are shifted to later times, where O–, Ox–, Six–, OSix, and SixOx are involved.
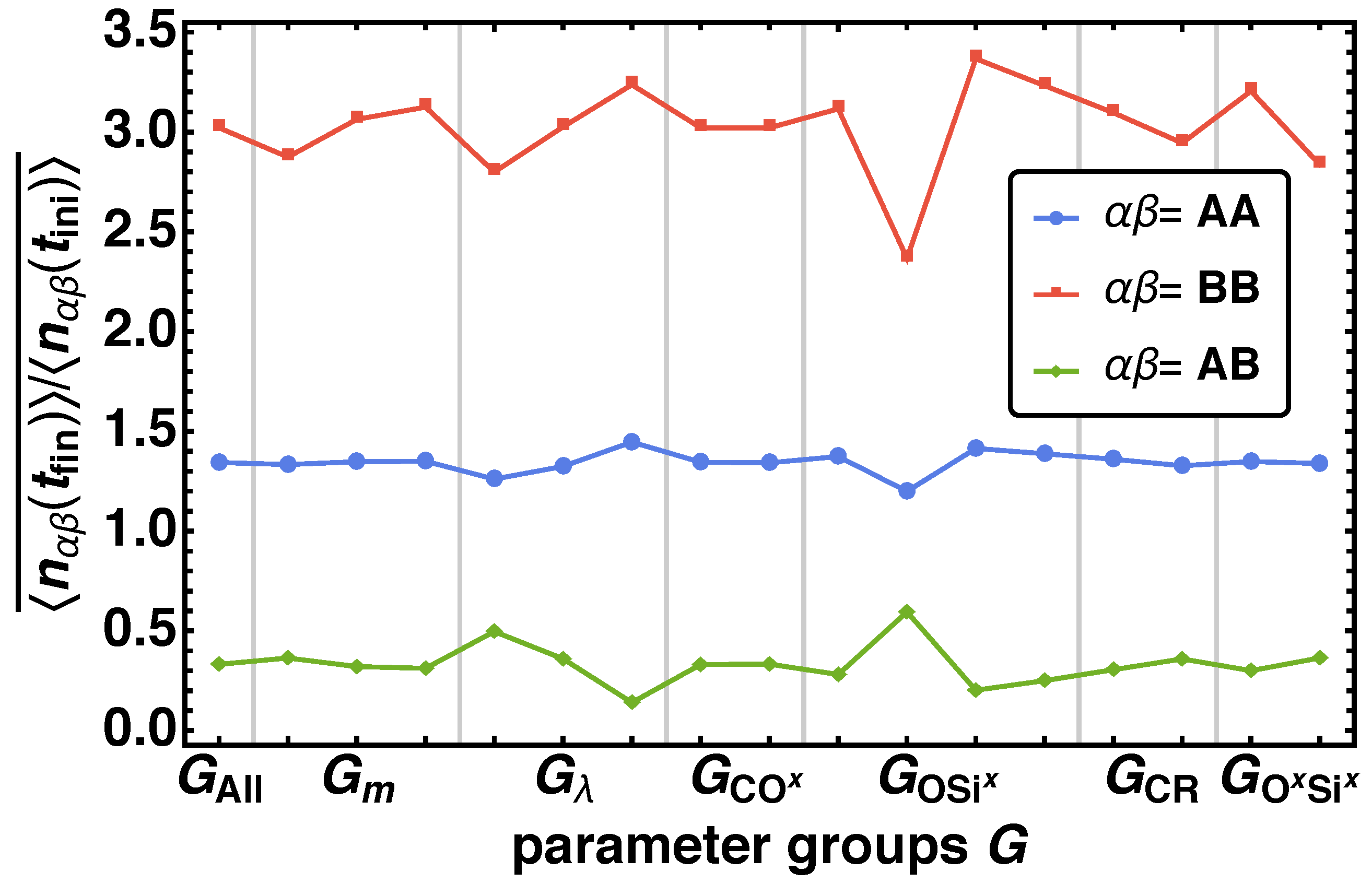
3.1.2. Phase Separation
3.2. Analysis of the Model Parameters on the Structure
3.2.1. Bulk Porosity and Specific Surface Area
3.2.2. Radial Distribution Function
3.2.3. Local Porosity Distribution
3.2.4. Percolation
4. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BFM | bond fluctuation model |
| rBFM | reactive bond fluctuation model |
| RDF | radial distribution function |
| nMC, rMC | non-reactive Monte Carlo, reactive Monte Carlo |
References
- Higgins, B.A.; Brittain, W.J. Polycarbonate carbon nanofiber composites. Eur. Polym. J. 2005, 41, 889–893. [Google Scholar] [CrossRef]
- Chemtob, A.; Belon, C.; Croutxé-Barghorn, C.; Rigolet, S.; Vidal, L.; Brendlé, J.; Mandle, J.; Blanchard, N. Tandem Cationic and Sol–Gel Photopolymerizations of a Vinyl Ether Alkoxysilane. Polym. Eng. Sci. 2011, 51, 1466–1475. [Google Scholar] [CrossRef]
- Ni, L.; Moreau, N.; Chemtob, A.; Croutxé-Barghorn, C. Organic–inorganic tandem route to polymer nanocomposites: kinetic products versus thermodynamic products. J. Sol-Gel Sci. Technol. 2012, 64, 500–509. [Google Scholar] [CrossRef]
- Rodrígez-Tobías, H.; Morales, G.; Rodríguez-Fernández, O.; Acuña, P. Mechanical and UV-Shielding Properties of In Situ Synthesized Poly(acrylonitrile-butadiene-styrene)/Zinc Oxide Nanocomposites. J. Appl. Polym. Sci. 2013, 127, 4708–4718. [Google Scholar] [CrossRef]
- Wang, Y.; Jabarin, S.A. Novel preparation method for enhancing nanoparticle dispersion and barrier properties of poly(ethylene terephthalate) and poly(m-xylylene adipamide). J. Appl. Polym. Sci. 2013, 129, 1455–1465. [Google Scholar] [CrossRef]
- Ebert, T.; Seifert, A.; Spange, S. Twin polymerization—A new principle for hybrid material synthesis. Macromol. Rapid Commun. 2015, 36, 1623–1639. [Google Scholar] [CrossRef]
- Grund, S.; Kempe, P.; Baumann, G.; Seifert, A.; Spange, S. Zwillingspolymerisation: Ein Weg zur Synthese von Nanokompositen. Angew. Chem. 2007, 119, 636–640. [Google Scholar] [CrossRef]
- Spange, S.; Kempe, P.; Seifert, A.; Auer, A.A.; Ecorchard, P.; Lang, H.; Falke, M.; Hietschold, M.; Pohlers, A.; Hoyer, W.; et al. Nanocomposites with Sturcture Domains of 0.5 to 3 nm by Polymerization of Silicon Spiro Compounds. Angew. Chem. Int. Ed. 2009, 48, 8254–8258. [Google Scholar] [CrossRef]
- Huster, C.; Nagel, K.; Spange, S.; Prehl, J. A reactive bond fluctuation model (rBFM) for twin polymerization: Comparison of simulated morphologies with experimental data. Chem. Phys. Lett. 2018, 713, 145–148. [Google Scholar] [CrossRef]
- Spange, S.; Grund, S. Nanostructured Organic-Inorganic Composite Materials by Twin Polymerization of Hybrid Monomers. Adv. Mater. 2009, 21, 2111–2116. [Google Scholar] [CrossRef]
- Brückner, J.; Thieme, S.; Böttger-Hiller, F.; Bauer, I.; Grossmann, H.T.; Strubel, P.; Althues, H.; Spange, S.; Kaskel, S. Carbon-Based Anodes for Lithium Sulfur Full Cells with High Cycle Stability. Adv. Funct. Mater. 2014, 24, 1284–1289. [Google Scholar] [CrossRef]
- Kitschke, P.; Walter, M.; Rüffer, T.; Seifert, A.; Speck, F.; Seyller, T.; Spange, S.; Lang, H.; Auer, A.A.; Kovalenko, M.V.; Mehring, M. Porous Ge@C materials via twin polymerization of germanium(II) salicyl alcoholates for Li-ion batteries. J. Mater. Chem. A 2016, 4, 2705–2719. [Google Scholar] [CrossRef]
- Spange, S.; Mehring, M. (Eds.) Twin Polymerization—New Strategy for Hybrid Materials; DeGruyter: Berlin/Boston, Germany, 2018. [Google Scholar]
- Auer, A.A.; Richter, A.; Berezkin, A.V.; Guseva, D.V.; Spange, S. Theoretical Study of Twin Polymerization—From Chemical Reactivity to Structure Formation. Macromol. Theory Simul. 2012, 21, 615–628. [Google Scholar] [CrossRef]
- Tchernook, I.; Prehl, J.; Friedrich, J. Quantum chemical investigation of the counter anion in the acid catalyzed initiation of 2,2’-spirobi[4H-1,3,2-benzodioxasiline] polymerization. Polymer 2015, 60, 241–251. [Google Scholar] [CrossRef]
- Schönfelder, T.; Friedrich, J.; Prehl, J.; Seeger, S.; Spange, S.; Hoffmann, K.H. Reactive force field for electrophilic substitution at an aromatic system in twin polymerization. Chem. Phys. 2014, 440, 119–126. [Google Scholar] [CrossRef]
- Prehl, J.; Schönfelder, T.; Friedrich, J.; Hoffmann, K.H. Site Dependent Atom Type ReaxFF for the Proton-Catalyzed Twin Polymerization. J. Phys. Chem. C 2017, 121, 15984–15992. [Google Scholar] [CrossRef]
- Hoffmann, K.H.; Prehl, J. Modeling the structure formation process of twin polymerization. Reac. Kinet. Mech. Cat. 2018, 123, 367–383. [Google Scholar] [CrossRef]
- Fiedler, B.; Friedrich, J.; Prehl, J. Modeling and simulation of nanostructure formation of TP. In Twin Polymerization—New Strategy for Hybrid Materials Synthesis, 1st ed.; Stefan, S., Mehring, M., Eds.; Walter de Gruyter: Berlin, Germany, 2018; Chapter 3.3; pp. 135–166. [Google Scholar]
- Carmesin, I.; Kremer, K. The Bond Fluctuation Method: A new effective algorithm for the dynamics of polymers in all spatial dimensions. Macromolecules 1988, 21, 2819–2823. [Google Scholar] [CrossRef]
- Shaffer, J.S. Effects of chain topology on polymer dynamics: Bulk melts. J. Comput. Phys. 1994, 101, 4205–4213. [Google Scholar] [CrossRef]
- Subramanian, G.; Shanbhag, S. On the relationship between two popular lattice models for polymer melts. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef]
- Paul, W.; Pistoor, N. A mapping of realistic onto abstract polymer models and an application to two bisphenol polycarbonates. Macromolecules 1994, 27, 1249–1255. [Google Scholar] [CrossRef]
- Tries, V.; Paul, W.; Baschnagle, J.; Binder, K. Modeling polyethylene with the bond fluctuation model. J. Chem. Phys. 1997, 106, 738–748. [Google Scholar] [CrossRef]
- Malik, R.; Hall, C.K.; Genzer, J. Phase Separation Dynamics for a Polymer Blend Compatibilized by Protein-like Copolymers: A Monte Carlo Simulation. Macromolecules 2011, 44, 8284–8293. [Google Scholar] [CrossRef]
- Rubio, A.M.; Storey, M.; Lodge, J.F.M.; Freire, J.J. Dynamics of Bond-Fluctuation Model Chains in Good and Theta Solvents. Macromol. Theory Simul. 2002, 11, 171–183. [Google Scholar] [CrossRef]
- Nakagawa, N.; Maeda, S.; Ishii, S.; Ohno, K. A Monte Carlo Simulation of the Formation of Micelles in a Ternary System of Water, Oil and Amphiphilic Polymers. Mat. Trans. 2007, 48, 653–657. [Google Scholar] [CrossRef] [Green Version]
- Müller, M. Miscibility behavior and single chain properties in polymer blends: a bond fluctuation model study. Macromol. Theory Simul. 1999, 8, 343–374. [Google Scholar] [CrossRef]
- Kempe, P.; Löschner, T.; Adner, D.; Spange, S. Selective ring opening of 4H-1,3,2-benzodioxasiline twin monomers. New J. Chem. 2011, 35, 2735–2739. [Google Scholar] [CrossRef]
- Kitschke, P.; Auer, A.A.; Löschner, T.; Seifert, A.; Spange, S.; Rüffer, T.; Lang, H.; Mehring, M. Microporous Carbon and Mesoporous Silica by Use of Twin Polymerization: An Integrated Experimental and Theoretical Approach to Precursor Reactivity. ChemPlusChem 2014, 79, 1009–1023. [Google Scholar] [CrossRef]
- Biswal, B.; Manwart, C.; Hilfer, R. Three-Dimensional Local Porosity Analysis of Porous Media. Physica A 1998, 255, 221–241. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Parameter (Variable) | Symbol | Analyzed Values |
|---|---|---|
| COx cleavage | ||
| OSix cleavage | ||
| OSix formation | ||
| CR formation | ||
| SixOx formation | ||
| ratio rMCS/nMCS | m | |
| attraction parameter |
| Index d | Meaning | Index d | Meaning |
|---|---|---|---|
| x | x-direction | 3 | -direction |
| y | y-direction | c | -direction |
| z | z-direction | 0 | -direction |
| Model Parameter | Notation | Model Parameter | Notation |
|---|---|---|---|
| average over all | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prehl, J.; Huster, C. Morphology on Reaction Mechanism Dependency for Twin Polymerization. Polymers 2019, 11, 878. https://doi.org/10.3390/polym11050878
Prehl J, Huster C. Morphology on Reaction Mechanism Dependency for Twin Polymerization. Polymers. 2019; 11(5):878. https://doi.org/10.3390/polym11050878
Chicago/Turabian StylePrehl, Janett, and Constantin Huster. 2019. "Morphology on Reaction Mechanism Dependency for Twin Polymerization" Polymers 11, no. 5: 878. https://doi.org/10.3390/polym11050878
APA StylePrehl, J., & Huster, C. (2019). Morphology on Reaction Mechanism Dependency for Twin Polymerization. Polymers, 11(5), 878. https://doi.org/10.3390/polym11050878