The Mechanism of the Propagation in the Anionic Polymerization of Polystyryllithium in Non-Polar Solvents Elucidated by Density Functional Theory Calculations. A Study of the Negligible Part Played by Dimeric Ion-Pairs under Usual Polymerization Conditions


Abstract
:
1. Introduction
- (1)
- Polystyryllithium was mainly associated into dimeric species and a small amount of non-associated polystyryllithim species reacted with styrene, as expected. The most stable transition state was the one in which Li was located near the phenyl rings of both styrene and polystyryllithium chain end, and the other transition state in which Li was located near the side chains of both styrene and polystyryllithium chain end, which was previously supposed to be the only transition state, was less stable than the former.
- (2)
- The penultimate unit effect found by one of the authors [22,23], i.e., slower addition of styrene to polystyryllithium with two or more styrene units compared to polystyryllithium with one styrene unit, was confirmed, and the effect was shown to be caused by coordination of the penultimate styrene units of dimeric polystyryllithium to the Li atoms.
- (3)
2. Methods
- (a)
- For (HStLi)2,where H[(HStLi)2] denotes the enthalpy of a particular (HStLi)2, and H[(HStLi)2]0 is the enthalpy of the most stable (HStLi)2 that has the lowest free energy among the studied dimers. For the most stable dimer, ∆Hr = 0, and for other dimers, ∆Hr indicates the extent of instability (in a thermodynamic sense) of the particular dimer, (HStLI)2, with respect to the most stable dimer.∆Hr = H[(HStLi)2] − H[(HStLi)2]0
- (b)
- For St/(HStLi)2 (precursor complexes, transition states, products, etc.).where H[St/(HStLi)2] denotes the enthalpy of a particular St/(HStLi)2 system and H(St) is the enthalpy of styrene. In this case, ∆Hr actually indicates the extent of instability of the particular St/(HStLi)2 system with respect to the starting material (styrene and dimeric (HStLi)2). ∆Hr for the transition state corresponds to the apparent activation energy of the reaction.∆Hr = H[St/(HStLi)2] − [H(St) + H[(HStLi)2]0]
3. Results and Discussion
3.1. Addition of Styrene to (HStLi)2 in the Gas Phase
- (1)
- Structures 1-a to 1-d exhibited sandwich-type features, while 1-e and 1-f were six-membered structures like the four-membered structure of alkyllithium dimers. The former four (HStLi)2 with sandwich-type structures have much lower ∆Hr and ∆Gr values than the latter two six-membered structures.
- (2)
- In 1-a and 1-b, the side chains of the HSt- groups (chain end units) were located near each end of Li−Li one after another, while in 1-c and 1-d, they were located near one end of Li−Li. 1-a and 1-b exhibited lower ∆Hr and ∆Gr when compared to 1-c and 1-d.
- (3)
- 1-a and 1-b were different with respect to the direction of the side chain of the HSt- group, i.e., in the opposite direction for (1-a) and in the same direction for (1-b) with respect to the Li−Li line. The ∆Hr as well as ∆Gr values of 1-a and 1-b were nearly the same. In the case of 1-c vs. 1-d and 1-e vs. 1-f, the situation was essentially similar.
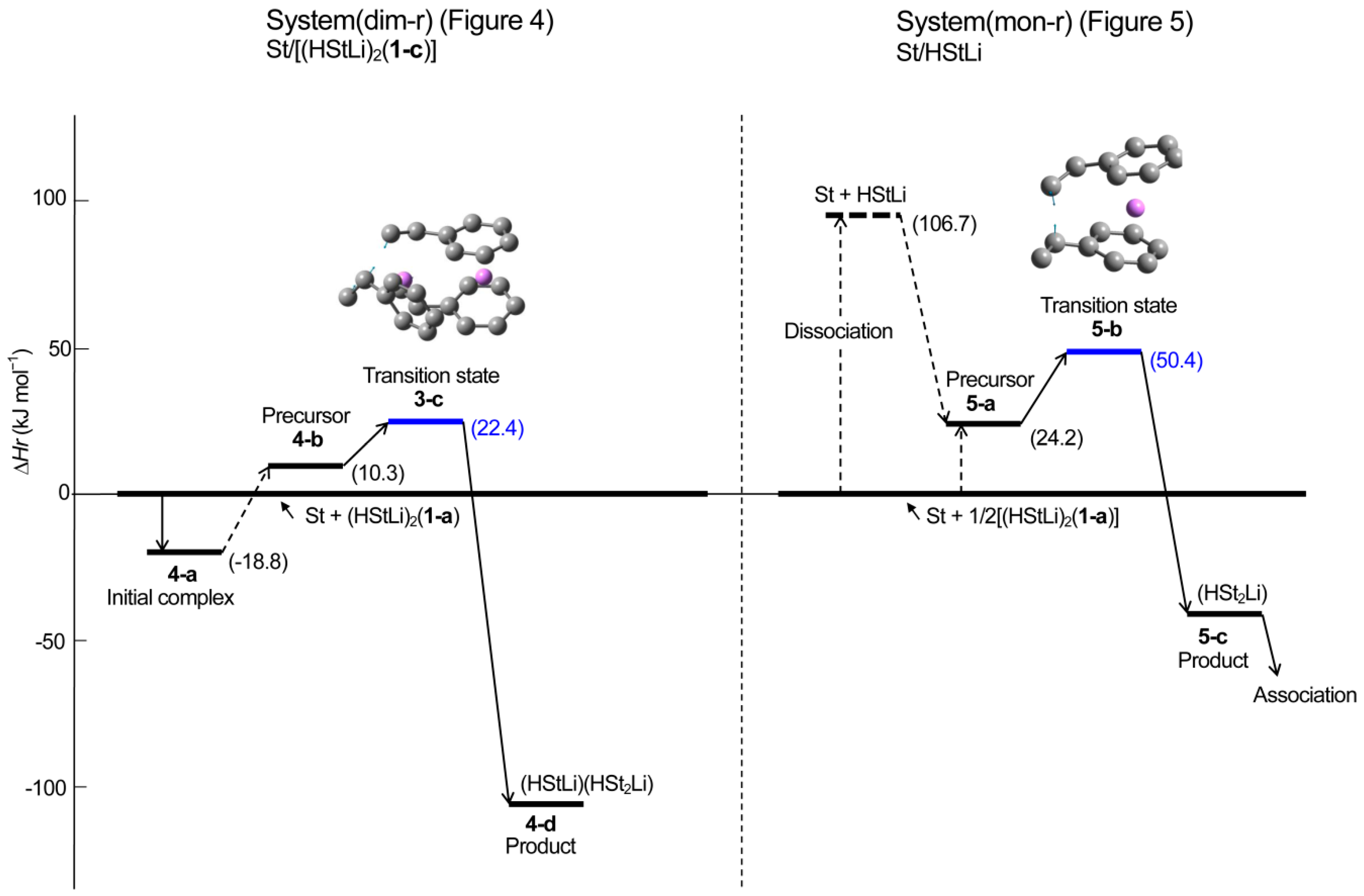
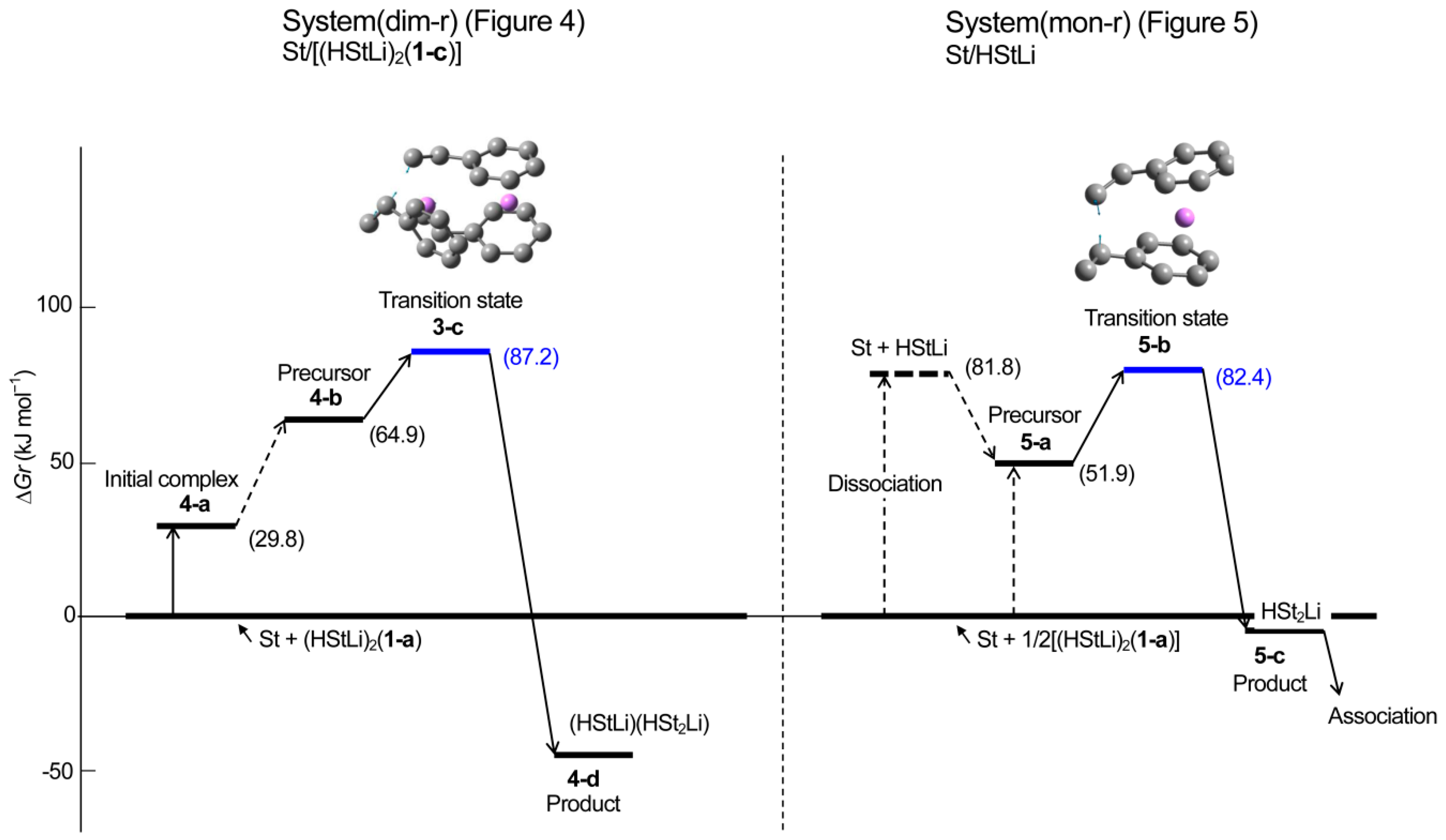
3.2. Addition of Styrene to (HStLi)2 in Cyclohexane
- (1)
- The rate constant of system(mon-r) is larger than that of system(dim-r).km/kd = e−85,700/RT/e−93,300/RT = 21
- (2)
- Usually, high molecular weight polymers are produced using small amounts of initiator, as can be seen in Table 4, that shows conditions of the experiments preformed to obtain the apparent activation energies for the anionic polymerization of styrene as discussed in our previous study. At an initiator concentration of 10−3 mol·L−1 which is approximately the average concentration for these experiments, the effect of initiator concentration is [Init]1/2/[Init] = 33, and Rm/Rd becomes much larger.Rm/Rd = (km/kd)([Init-m]1/2/[Init-d]) = (21)(33) = 690
- (1)
- Although the polymerization of styrene with non-associated polystyryllitnium requires the dissociation of dimeric polystyryllithium for the reaction, its true activation energy for the polymerization reaction is small. Therefore, the polymerization of non-associated polystyryllithium is very rapid. Especially at low catalyst concentrations where high molecular weight polymers are usually obtained, propagation reaction is very powerful.
- (2)
- Dimeric polystyryllithium can polymerize styrene. However, it is not as reactive as non-associated polystyryllithium, although its relative enthalpy is lower because there occurrs no preliminary dissociation in the dimeric species.
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix A.1. Calculated Energies of the Structures Studied in the Gas Phase and Cyclohexane

| Structure Designation | System a | State b | Sym. c | Electronic Energy | ZP−Corrected El. Energy | Enthalpy at 25 °C | Free Energy at 25 °C |
|---|---|---|---|---|---|---|---|
| Hartree | Hartree | Hartree | Hartree | ||||
| Styrene | Cs | −309.509791 | −309.375108 | −309.367380 | −309.406880 | ||
| 1-a | (HStLi)2 | Ci | −635.361832 | −635.066685 | −635.047732 | −635.112274 | |
| 1-b | (HStLi)2 | C2 | −635.362034 | −635.066958 | −635.048066 | −635.111985 | |
| 1-c | (HStLi)2 | C2 | −635.357316 | −635.062952 | −635.043667 | −635.108167 | |
| 1-d | (HStLi)2 | C1 | −635.357150 | −635.062696 | −635.043457 | −635.108156 | |
| 1-e | (HStLi)2 | C1 | −635.340616 | −635.045969 | −635.026702 | −635.093660 | |
| 1-f | (HStLi)2 | C1 | −635.340376 | −635.045695 | −635.026400 | −635.094205 | |
| 2-a | St/[(HstLi)2(1-a)] | trans. st. | C1 | −944.857043 | −944.426327 | −944.400230 | −944.478933 |
| 2-b | St/[(HstLi)2(1-a)] | trans. st. | C1 | −944.857138 | −944.425445 | −944.399662 | −944.477970 |
| 2-c | St/[(HstLi)2(1-a)] | trans. st. | C1 | −944.845308 | −944.413114 | −944.387531 | −944.464941 |
| 2-d | St/[(HstLi)2(1-a)] | trans. st. | C1 | −944.850776 | −944.419815 | −944.393709 | −944.474242 |
| 3-b | St/[(HstLi)2(1-b)] | trans. st. | C1 | −944.858148 | −944.427685 | −944.401521 | −944.480363 |
| 4-a | St/[(HstLi)2(1-c)] | init. com. | C1 | −944.880282 | −944.450194 | −944.422290 | −944.507820 |
| A1-b | ditto | inter. trans. st. | C1 | −844.879390 | −944.449536 | −944.422347 | −944.505178 |
| A1-c | ditto | inter. com. | C1 | −944.880979 | −944.450161 | −944.422734 | −944.505562 |
| A1-d | ditto | inter. trans. st. | C1 | −944.875944 | −944.445103 | −944.418730 | −944.498535 |
| A1-e | ditto | inter. com.. | C1 | −944.876336 | −944.445187 | −944.418088 | −944.499441 |
| A1-f | ditto | inter. trans. st. | C1 | −944.867317 | −944.436903 | −944.410238 | −944.491300 |
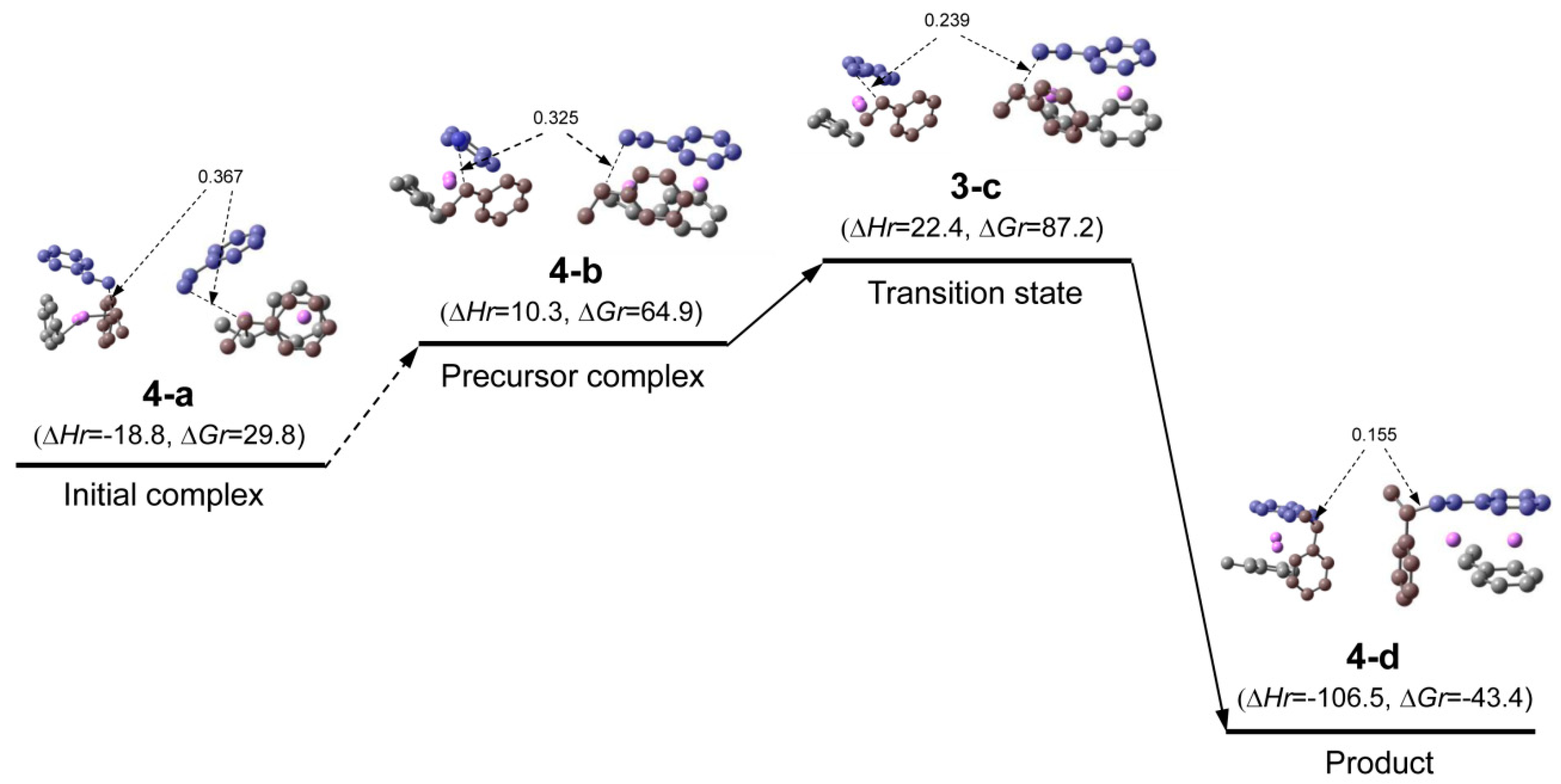
| 4-b | ditto | prec. com. | C1 | −944.868984 | −944.438662 | −944.411171 | −944.494425 |
| 3-c | ditto | trans. st. | C1 | −944.863882 | −944.432619 | −944.406563 | −944.485960 |
| 4-d | ditto | product | C1 | −944.916082 | −944.481597 | −944.455684 | −944.535693 |
| 3-d | St/[(HstLi)2(1-d)] | trans. st. | C1 | −944.862233 | −944.430290 | −944.404577 | −944.482309 |
| 3-e | St/[(HstLi)2(1-e)] | trans. st. | C1 | −944.857134 | −944.424533 | −944.399110 | −944.476746 |
| 3-f | St/[(HstLi)2(1-f)] | trans. st. | C1 | −944.858296 | −944.426057 | −944.400544 | −944.478525 |
| 5-a | St/HStLi | prec. com. | C1 | −627.182144 | −626.899522 | −626.882012 | −626.943264 |
| 5-b | ditto | trans. st. | C1 | −627.171213 | −626.888516 | −626.872055 | −626.931624 |
| 5-c | ditto | produt | C1 | −627.209000 | −626.922552 | −626.906625 | −626.963788 |
| Structure Designation a | System | State b | Sym. c | Electronic Energy | ZP−Corrected El. Energy | Enthalpy at 25 °C | Free Energy at 25 °C |
|---|---|---|---|---|---|---|---|
| Hartree | Hartree | Hartree | Hartree | ||||
| 8-a | St/[(HStLi)2(1-c)] | trans. st. | C1 | −944.868035 | −944.437159 | −944.410980 | −944.490875 |
| [5-b] | St/HStLi | trans. st. | C1 | −627.174743 | −626.892141 | −626.875730 | −626.934819 |
Appendix A.2. Complete Reaction Pathway for St/[(HStLi)2(1-c)], System(dim-r), in the Gas Phase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Szwarc, M. Carbanions, Living Polymers, and Electron Transfer Processes; John Wiley & Sons Inc.: New York, NY, USA, 1968. [Google Scholar]
- Szwarc, M.; Van Beylen, M. Ionic Polymerization and Living Polymers; Chapman and Hall: New York, NY, USA, 1993. [Google Scholar]
- Bywater, S. Structure and mechanism in anionic polymerization. Prog. Polym. Sci. 1994, 19, 287–316. [Google Scholar] [CrossRef]
- Quirk, R.P.; Zhuo, Q.; Jang, S.H.; Lee, Y.; Lizarraga, G. Principles of Anionic Polymerization: An introduction. ACS Symp. Ser. Am. Chem. Soc. 1998, 696, 2–27. [Google Scholar]
- Hsieh, H.L.; Quirk, R.P. Anionic Polymerization: Principles and Practical Applications; Marcel Dekker Inc.: New York, NY, USA, 1996. [Google Scholar]
- Worsfold, D.J.; Bywater, S. Anionic polymerization of styrene. Can. J. Chem. 1960, 38, 1891–1900. [Google Scholar] [CrossRef]
- Worsfold, D.J.; Bywater, S. Degree of association of polystyryl-, polyisoprenyl- and polybutadienyllithium in hydrocarbon solvents. Macromolecules 1972, 5, 393–397. [Google Scholar] [CrossRef]
- Young, R.N.; Fetters, L.J.; Huang, J.S.; Krishnamoorti, R. Some light on the concept of unreactivity arising from active center association in anionic polymerizations. Polym. Int. 1994, 33, 217–231. [Google Scholar] [CrossRef]
- Fetters, L.J.; Balsara, N.P.; Huang, J.S.; Jeon, H.S.; Almdal, K.; Lin, M.Y. Aggregation in Living Polymer Solutions by Light and Neutron Scattering: A Study of Model Ionomers. Macromolecules 1995, 28, 4996–5005. [Google Scholar] [CrossRef]
- Fetters, L.J.; Huang, J.S.; Young, R.N. An Evaluation of the Concept of Unreactivity Arising from Head-Group Aggregation in Anionic Polymerizations. J. Polym. Sci. Part A 1996, 34, 1517–1527. [Google Scholar] [CrossRef]
- Fetters, L.J.; Huang, J.S.; Stellbrink, J.; Willner, L.; Richter, D. Association behavior of living anionic lipophobic head-groups in hydrocarbon mileau. Macromol. Symp. 1997, 121, 1–26. [Google Scholar] [CrossRef]
- Stellbrink, J.; Willner, L.; Jucknischke, O.; Richter, D.; Lindner, P.; Fetters, L.J.; Huang, J.S. Self-Assembling Behavior of Living Polymers. Macromolecules 1998, 31, 4189–4197. [Google Scholar] [CrossRef]
- Balsara, N.P.; Fetters, L.J. Reevaluation of Light Scattering from Living Polymer Solutions. Macromolecules 1999, 32, 5147–5148. [Google Scholar] [CrossRef]
- Mishima, E.; Matsumiya, Y.; Yamago, S.; Watanabe, M. Kinetics of Living Anionic Polymerization of Polystyrenyl Lithium in Cyclohexane. Polym. J. 2008, 40, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Bywater, S. Active center association in anionic polymerization. Its relation to the mechanism of polymerization of dienes in non-polar solvents. Polym. Int. 1995, 38, 325–333. [Google Scholar] [CrossRef]
- Arest-Yakubovich, A.A. On the kinetics of Lithium-Initiated Anionic Polymerization in Nonpolar Solvents. J. Polym. Sci. Part A 1997, 35, 3613–3615. [Google Scholar] [CrossRef]
- Bywater, S. Comments on the Determination of Absolute Rate Constants in Anionic Polymerization. J. Polym. Sci. Part A 1998, 36, 1065–1068. [Google Scholar] [CrossRef]
- Bywater, S. Active Center Aggregation in Lithium-Based Anionic Polymerization. Are Very Large Aggregates Present? Macromolecules 1998, 31, 6010–6013. [Google Scholar] [CrossRef]
- Szwarc, M. Treatment of Experimental Data. J. Polym. Sci. Part A 1999, 37, 873–875. [Google Scholar] [CrossRef]
- Morita, H.; Van Beylen, M. New Vistas on the Anionic Polymerization of Styrene in Non-Polar Solvents by Means of Density Functional Theory. Polymers 2016, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlaer, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition states: Two new functionals and systematic testing of four M-06 class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Walckiers, E.; Van Beylen, M. Penultimate unit effect in anionic polymerization of styrene. In Proceedings of the Macromolecular Preprints XXIII Iupac Congress, Boston, MA, USA, 26–30 July 1971; pp. 1199–1206. [Google Scholar]
- Dils, J.; Van Beylen, M. Penultimate unit effects in anionic polymerization. The anionic polymerization of 1,1-diphenylalkali salts with styrene. In Proceedings of the International Symposium on Macromolecule, Dublin, Ireland, 17–22 July 1977; pp. 69–76. [Google Scholar]
- Ohlinger, R.; Banderman, F. Kinetik der Wachstumstreaktionen der Copolymerisation von Butadien und Styrol mit Lithiumorganylen. Makromol. Chem. 1980, 181, 1935–1947. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schiegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schiegel, H.B. Reaction path following in mass-weighed internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the revision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Auguste, S.; Edwards, H.G.M.; Johnson, A.F.; Meszena, Z.G. Anionic polymerization of styrene and butadiene initiated by n-butyllithium in ethylbenzene: Determination of the propagation rate constants using Raman spectroscopy and gel permeation chromatography. Polymer 1996, 37, 3665–3673. [Google Scholar] [CrossRef]
- Frischknecht, A.L.; Scott, T.M. Model for the aggregation state of living anionic polymers. J. Chem. Phys. 2001, 114, 1032–1050. [Google Scholar] [CrossRef]
- Stellbrink, J.; Willner, L.; Richter, D.; Fetters, L.J.; Huang, J.S. Self-Assembling Behavior of Butadienyllithium Headgroups in Benzene via SANS Measurement. Macromolecules 1999, 32, 5321–5329. [Google Scholar] [CrossRef]





| Item | Detail | 2-a | 2-b | 2-c | 2-d |
|---|---|---|---|---|---|
| Coordination of styrene with one Li (Li(1) a) or two Li atoms | Two Li | Two Li | Two Li | One Li | |
| Arrangement | Direction in which the side chain of styrene approaches the reacting HSt- group | Opposite | Opposite | Same | Same |
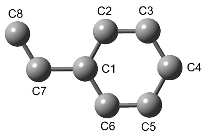
| Portion of the styrene phenyl group b coordinating with Li(2) a | C2−C3 | C5−C6 | C2−C3 | ||
| Distance between the reacting C atoms c (nm) | 0.252 | 0.238 | 0.206 | 0.240 | |
| Results | Distance between the Li atoms (nm) | 0.305 | 0.289 | 0.340 | 0.351 |
| ∆Gr of the transition state (kJ·mol−1) | 105.6 | 108.1 | 142.4 | 117.9 |
| Item | Detail | 2-a | 3-b | 3-c | 3-d | 3-e | 3-f |
|---|---|---|---|---|---|---|---|
| (HStLi)2 | Type of (HStLi)2 | 1-a | 1-b | 1-c | 1-d | 1-e | 1-f |
| ∆Gr for (HStLi)2 (kJ·mol−1) | 0 | 0.8 | 10.8 | 10.8 | 48.9 | 47.4 | |
| Coordination of styrene with one Li (Li(1) a) or two Li atoms | Two Li | Two Li | Two Li | Two Li | Two Li | Two Li | |
| Arrangement | Direction in which the side chain of styrene approaches the reacting HSt- group b | Opp. | Opp. | Opp. | Opp. | Opp. | Opp. |
| Portion of the styrene phenyl group c coordinating with Li(2) a | C2−C3 | C2−C3 | C5−C6 | C2−C3 | C5−C6 | C5−C6 | |
| Results | Distance between the reacting C atoms d (nm) | 0.252 | 0.248 | 0.239 | 0.224 | 0.227 | 0.225 |
| Distance between the Li atoms (nm) | 0.305 | 0.306 | 0.352 | 0.318 | 0.246 | 0.247 | |
| ∆Gr of the transition state (kJ·mol−1) | 105.6 | 101.9 | 87.2 | 96.8 | 113.3 | 106.7 |

| Item | In the Gas Phase | In Cyclohexane a | ||||
|---|---|---|---|---|---|---|
| Structure | ∆Hr | ∆Gr | Structure b | ∆Hrch | ∆Grch | |
| kJ·mol−1 | kJ·mol−1 | kJ·mol−1 | kJ·mol−1 | |||
| St/[(HStLi)2(1-c)] (system(dim-r)) | 3-c | 22.4 | 87.2 | 8-a | 27.6 | 93.3 |
| St/HStLi (system(mon-r)) | 5-b | 50.4 | 82.4 | [5-b] | 51.1 | 85.7 |
| Experimental Conditions | Results | |||||
|---|---|---|---|---|---|---|
| Solvent | Styrene Concentration | Initiator Concentration | Temperature | Rate of Reaction a | Activation Energy b | |
| mol·L−1 × 10−3 | mol·L−1 × 10−3 | °C | kJ·mol−1 | |||
| Worsfold et al. [6] | Benzene | 1.9–28 | 0.016–39 | 10–30.3 | k[St][Init]0.5 | 59.8 c |
| Ohlinger et al. [24] | Toluene | 0.12–15 | 0.1–15 | 20–50 | k[St][Init]0.5 | 59.9 |
| Auguste et al [31] | Ethylbenzene | 860–97 | 0.09–70 | 25–70 | k[St][Init]05 | 75 ± 8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morita, H.; Van Beylen, M. The Mechanism of the Propagation in the Anionic Polymerization of Polystyryllithium in Non-Polar Solvents Elucidated by Density Functional Theory Calculations. A Study of the Negligible Part Played by Dimeric Ion-Pairs under Usual Polymerization Conditions. Polymers 2019, 11, 1022. https://doi.org/10.3390/polym11061022
Morita H, Van Beylen M. The Mechanism of the Propagation in the Anionic Polymerization of Polystyryllithium in Non-Polar Solvents Elucidated by Density Functional Theory Calculations. A Study of the Negligible Part Played by Dimeric Ion-Pairs under Usual Polymerization Conditions. Polymers. 2019; 11(6):1022. https://doi.org/10.3390/polym11061022
Chicago/Turabian StyleMorita, Hideo, and Marcel Van Beylen. 2019. "The Mechanism of the Propagation in the Anionic Polymerization of Polystyryllithium in Non-Polar Solvents Elucidated by Density Functional Theory Calculations. A Study of the Negligible Part Played by Dimeric Ion-Pairs under Usual Polymerization Conditions" Polymers 11, no. 6: 1022. https://doi.org/10.3390/polym11061022