Submicron-Sized Nanocomposite Magnetic-Sensitive Carriers: Controllable Organ Distribution and Biological Effects

 ,
,  , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis of Magnetic Nanoparticles
2.2.2. Preparation of Nanocomposite Microcarriers.
2.2.3. In Vitro Biocompatibility
Cell Viability Assay
Interaction of Carriers with the Coagulation Cascade.
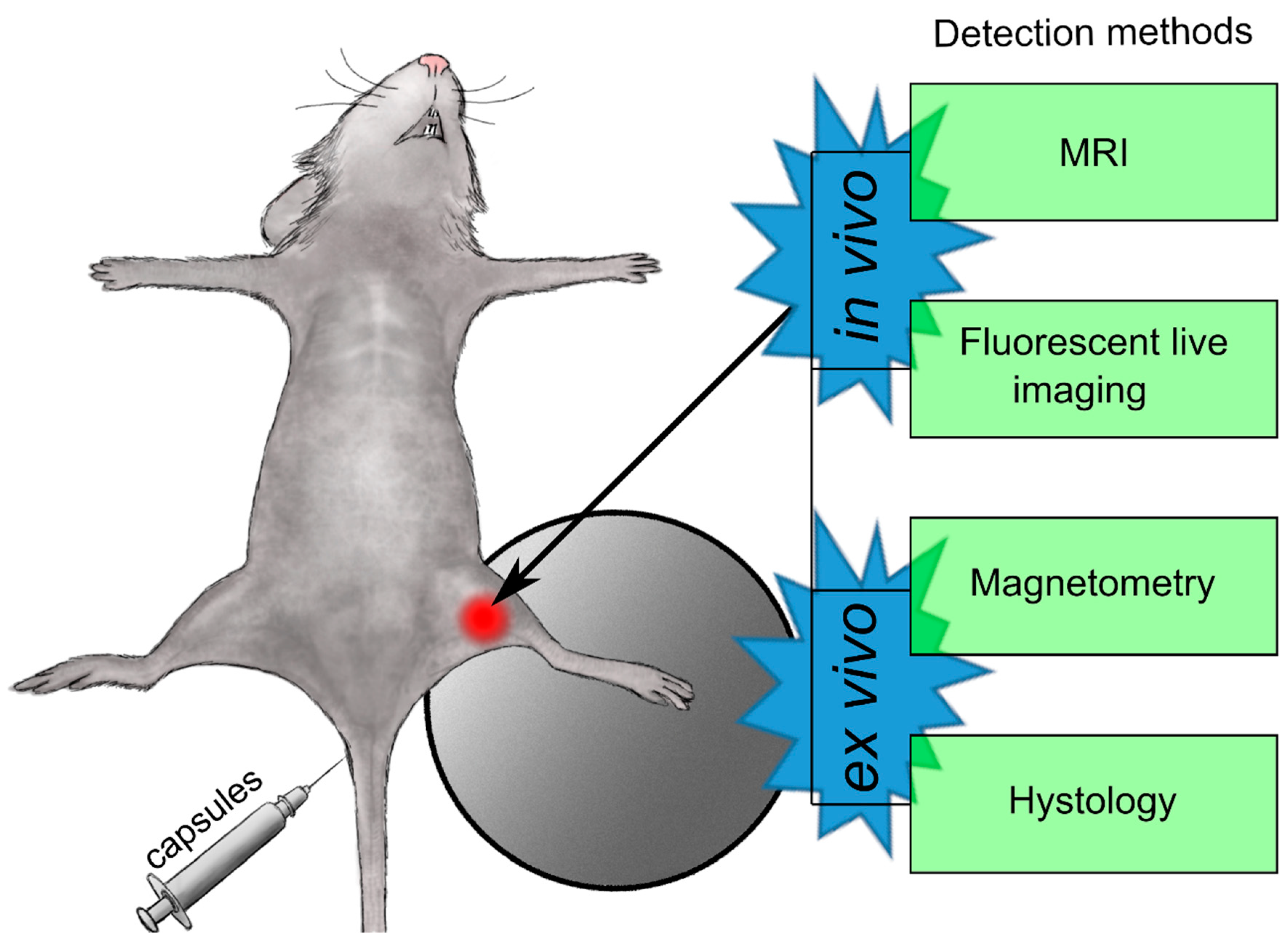
In Vivo Study
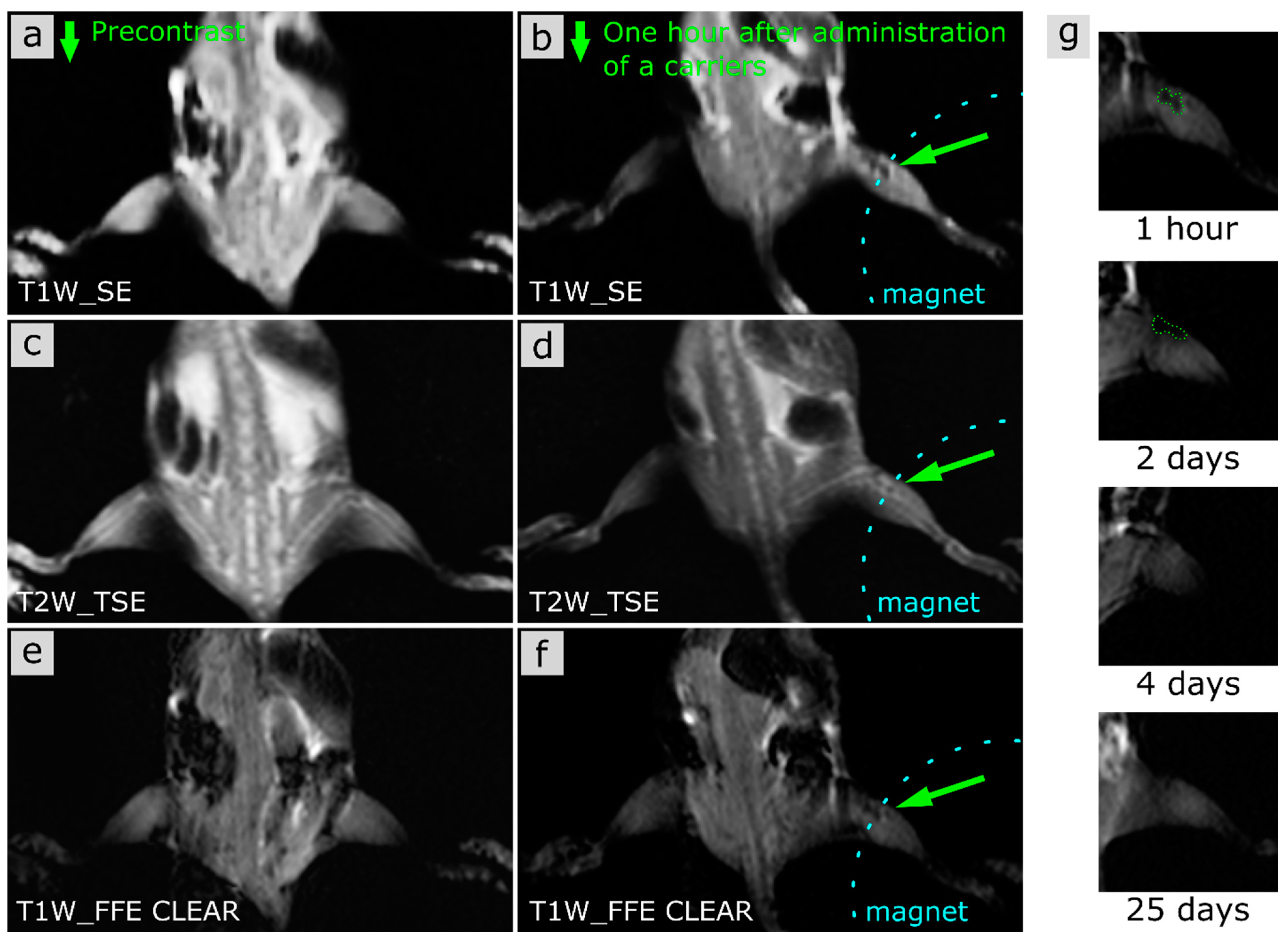
MRI
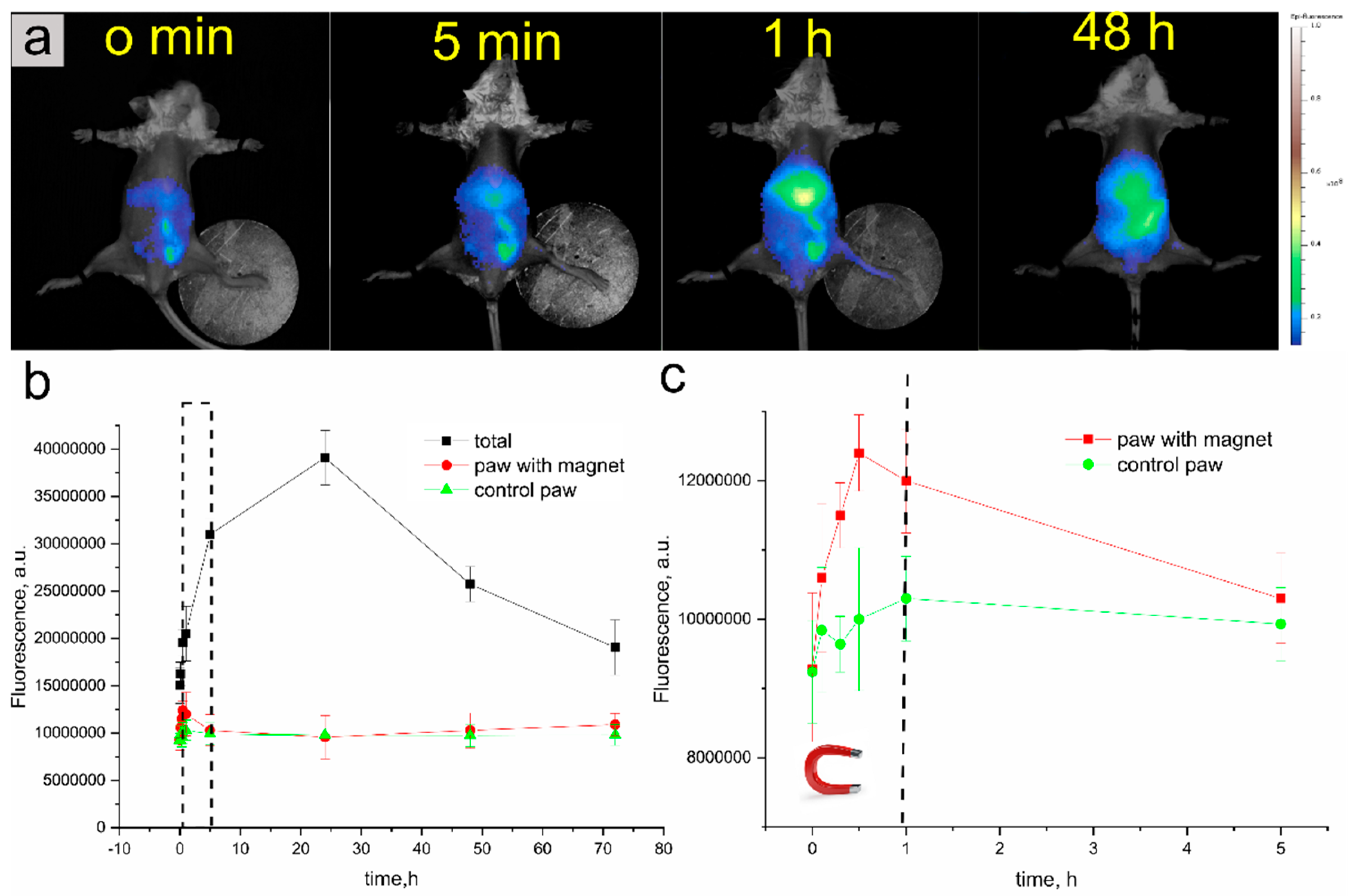
In Vivo Fluorescent Imaging
In Vivo Magnetite Biodistribution Study
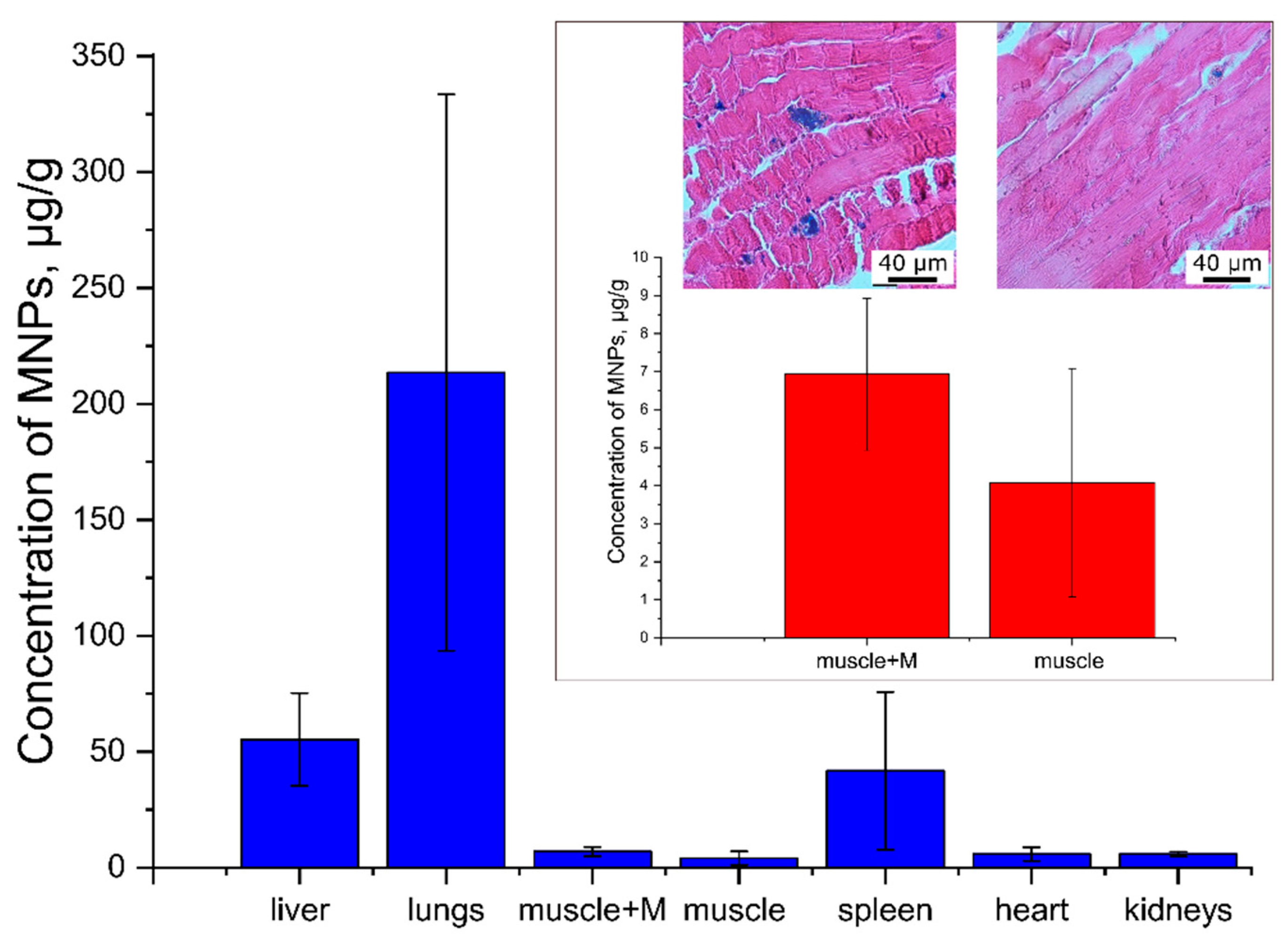
Histological Studies.
Magnetometry
In Vivo Biocompatibility
- (1)
- MNPs (BSA-TA) carriers diluted in saline solution (n = 10),
- (2)
- (BSA-TA) carriers diluted in saline solution (n = 10)
- (3)
- Saline (0.9% NaCl) as the control (n = 10).
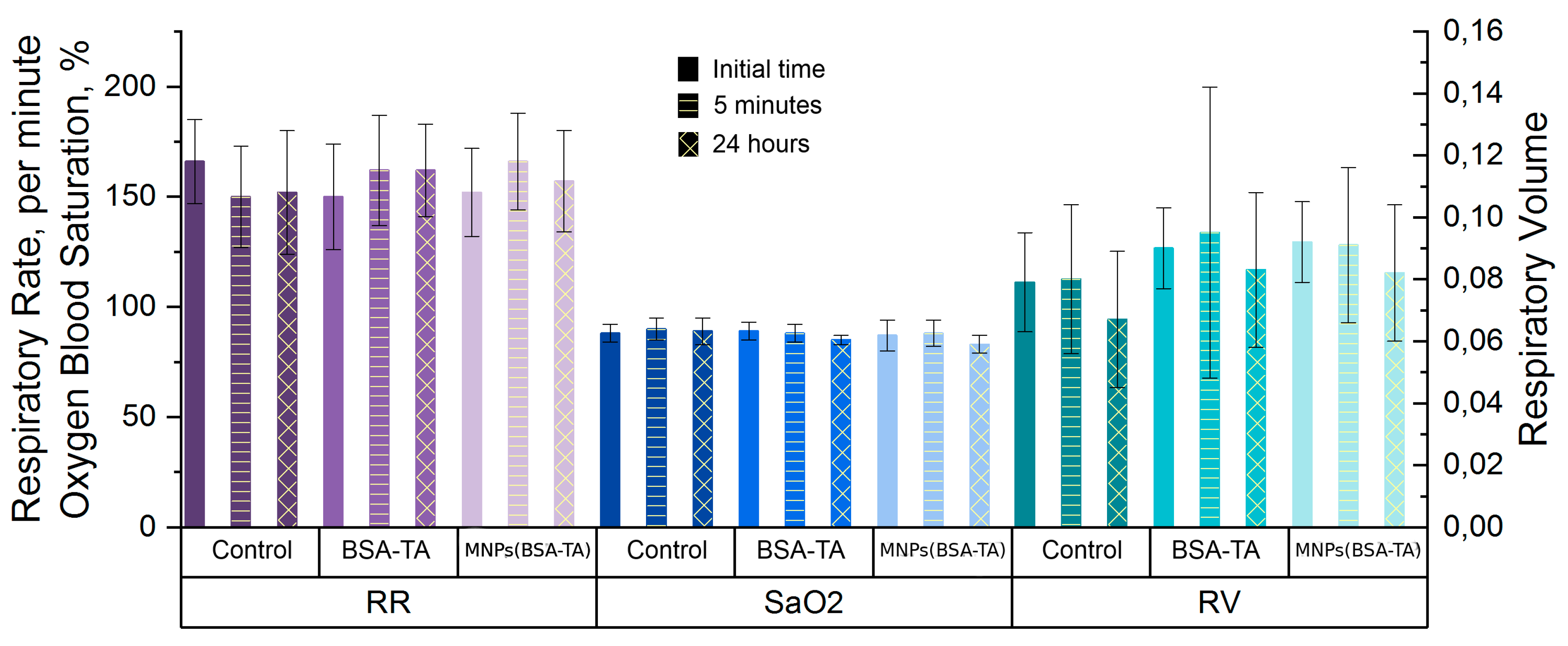
Heart and Lung Function Study.
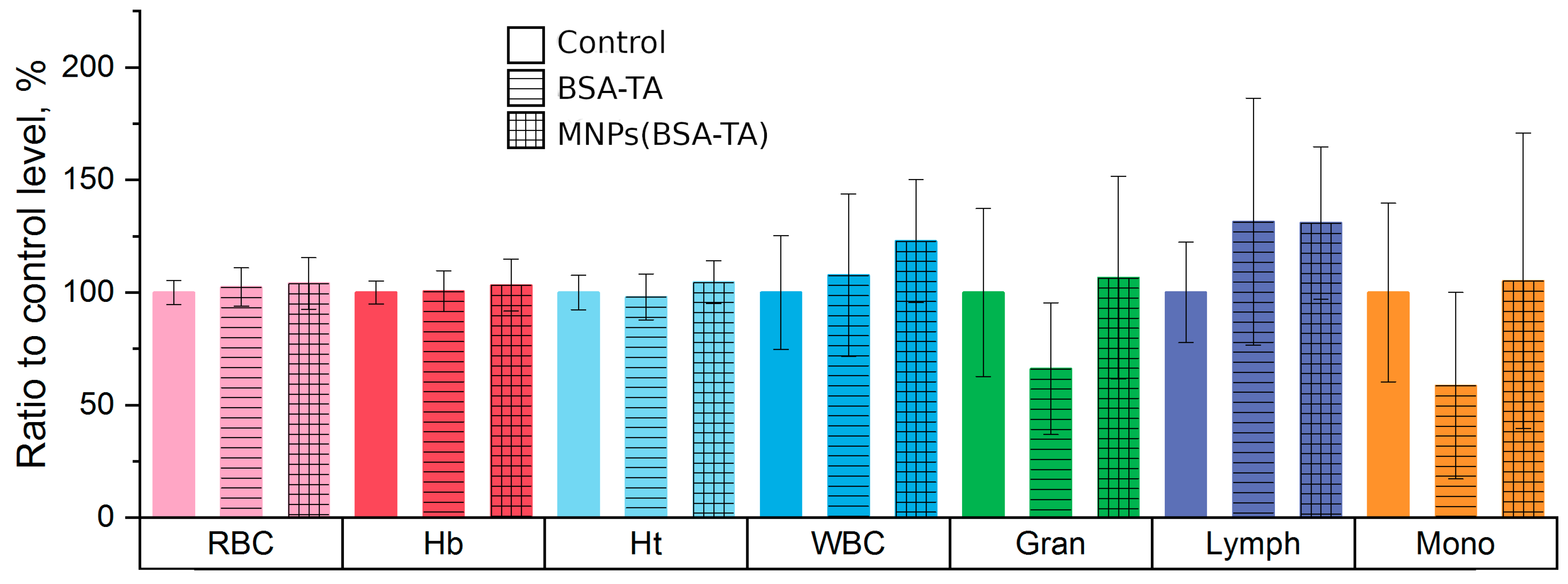
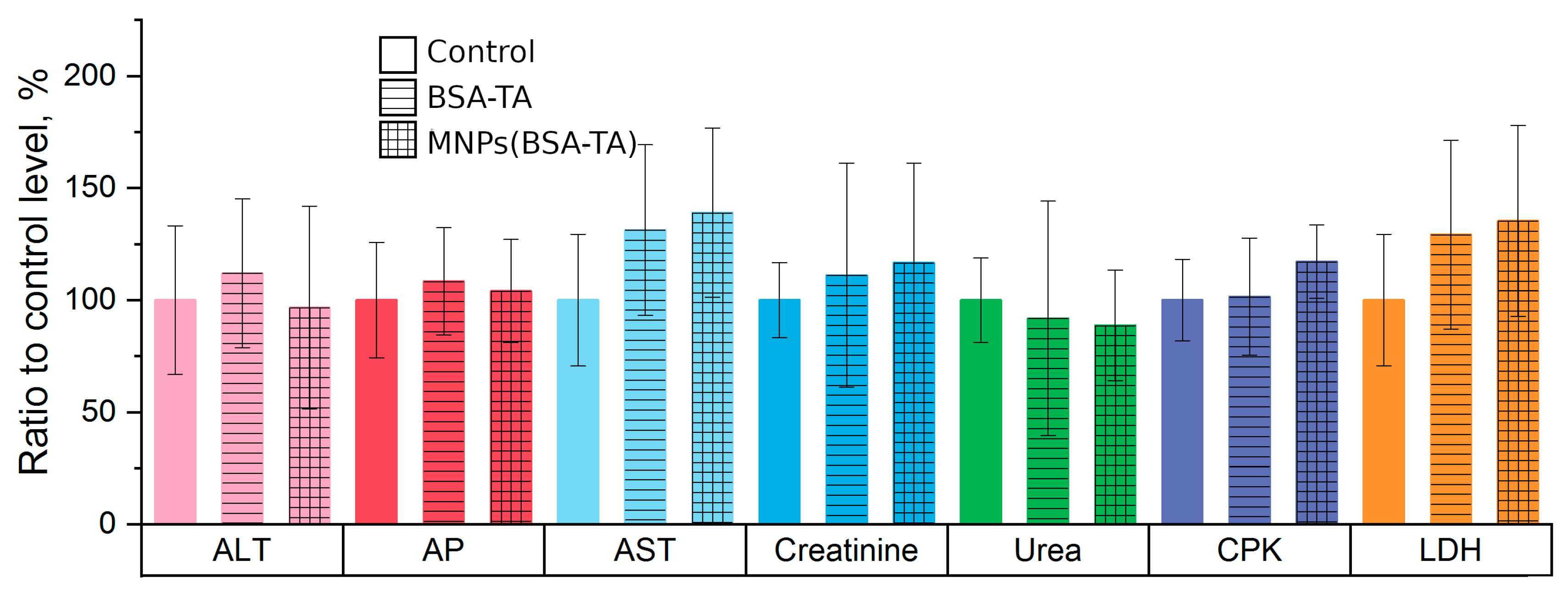
Hematological and Biochemical Parameters.
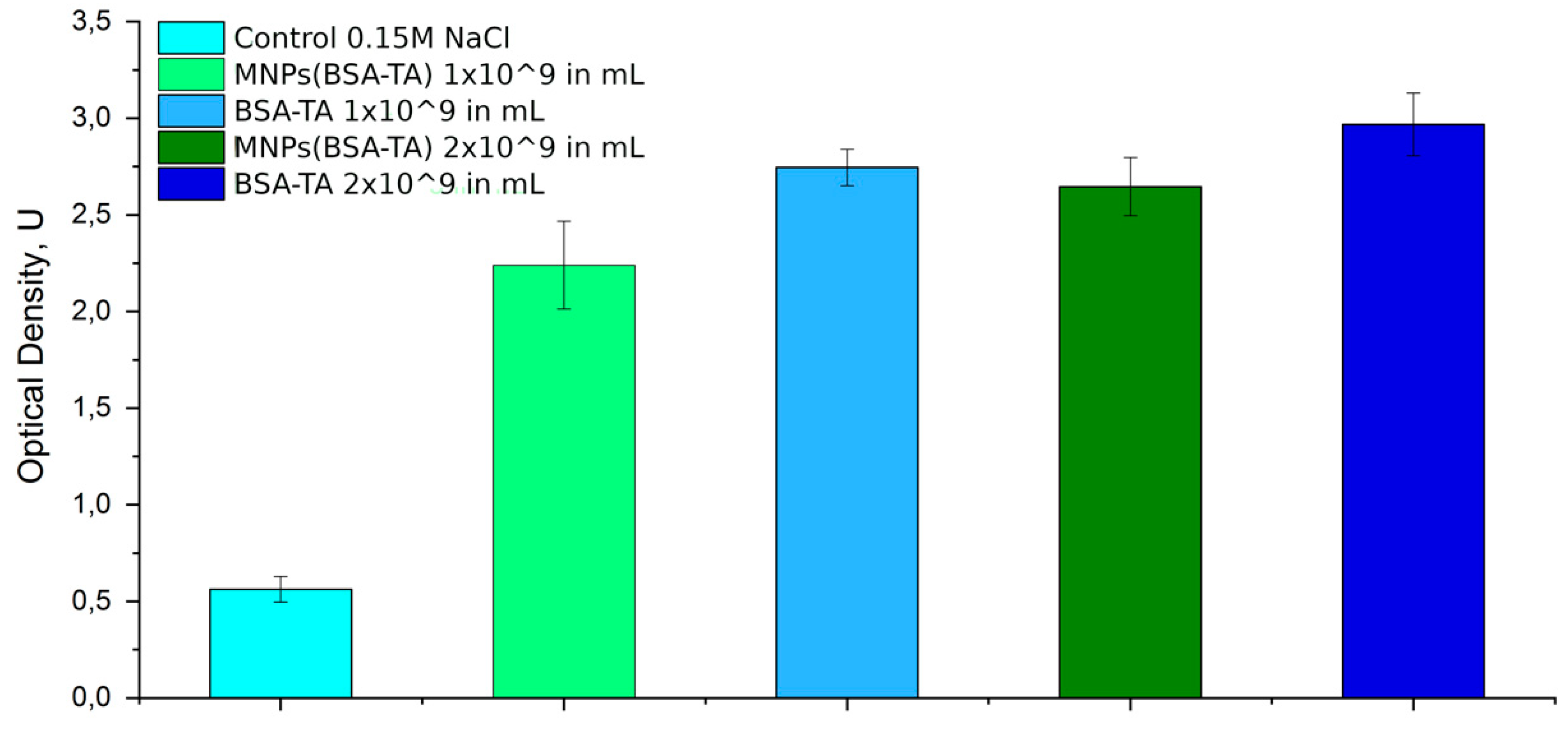
Complement Study
3. Results and Discussion
3.1. Preparation and Characterization of Nanocomposite Carriers
3.2. Organ Distribution
3.2.1. MRI
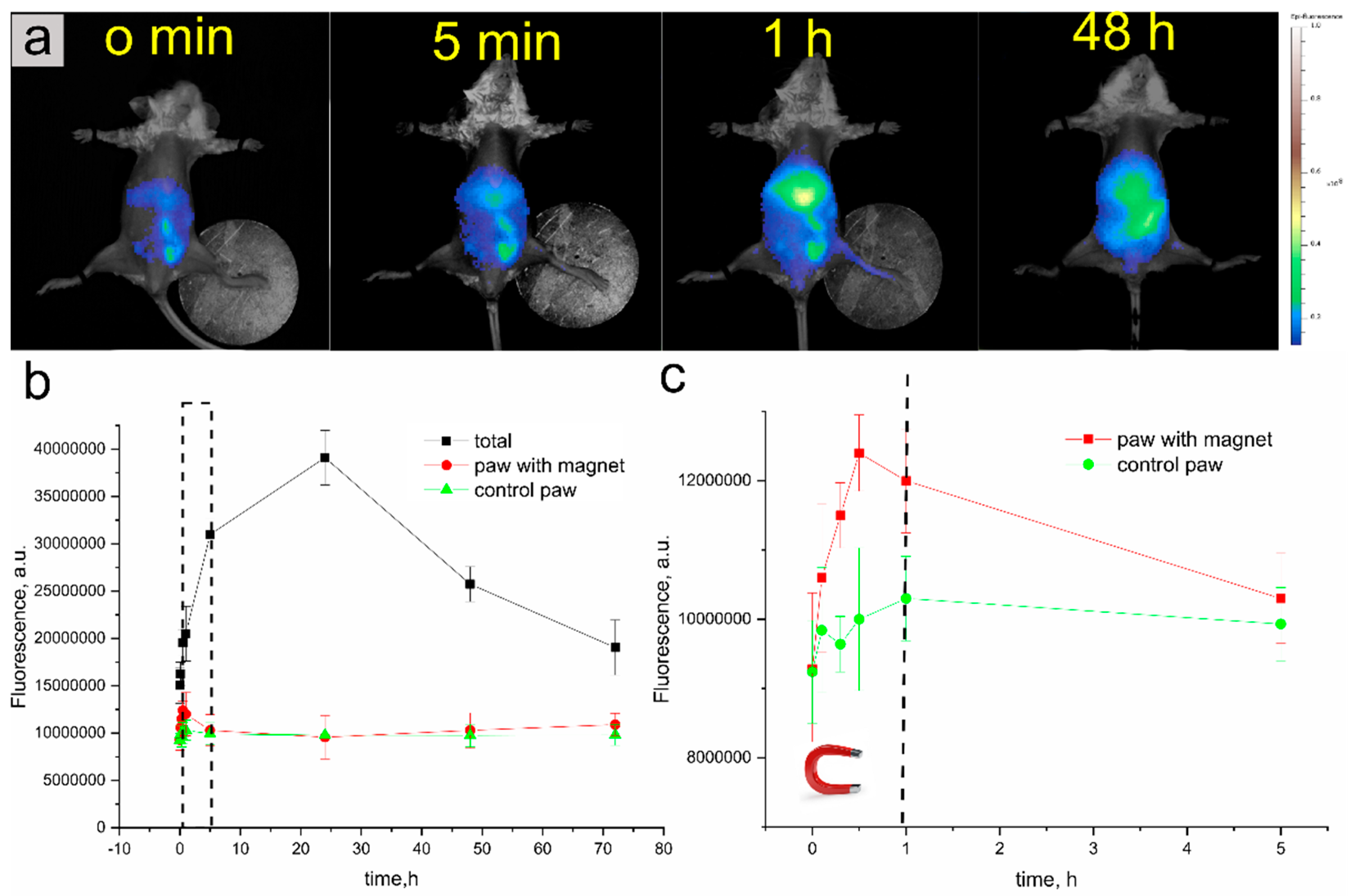
3.2.2. In Vivo Fluorescence Lifetime Imaging
3.2.3. Histological Studies and Magnetometry
3.3. Effect of Carrier Administration on Functional and Biochemical Parameters
3.3.1. Hemocompatibility
3.3.2. Hemostasis
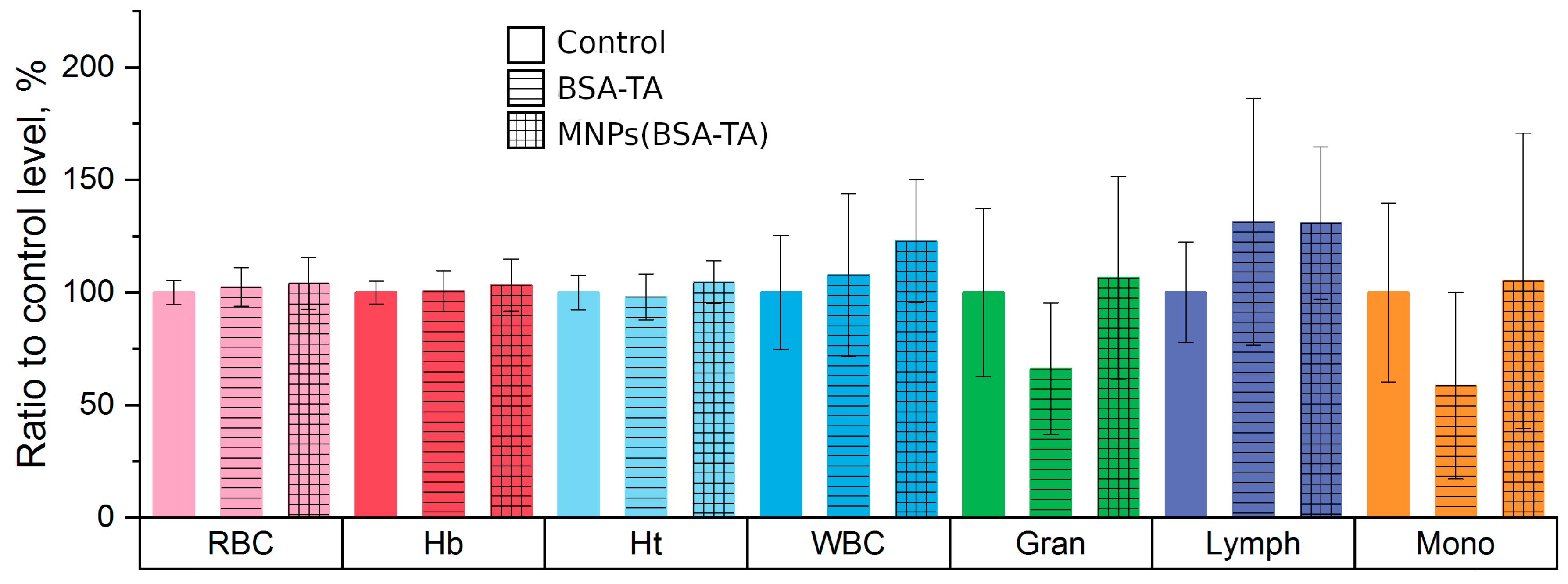
3.3.3. Hematologic Study
3.3.4. Liver and Kidney Function Tests
3.3.5. Lung Function Tests
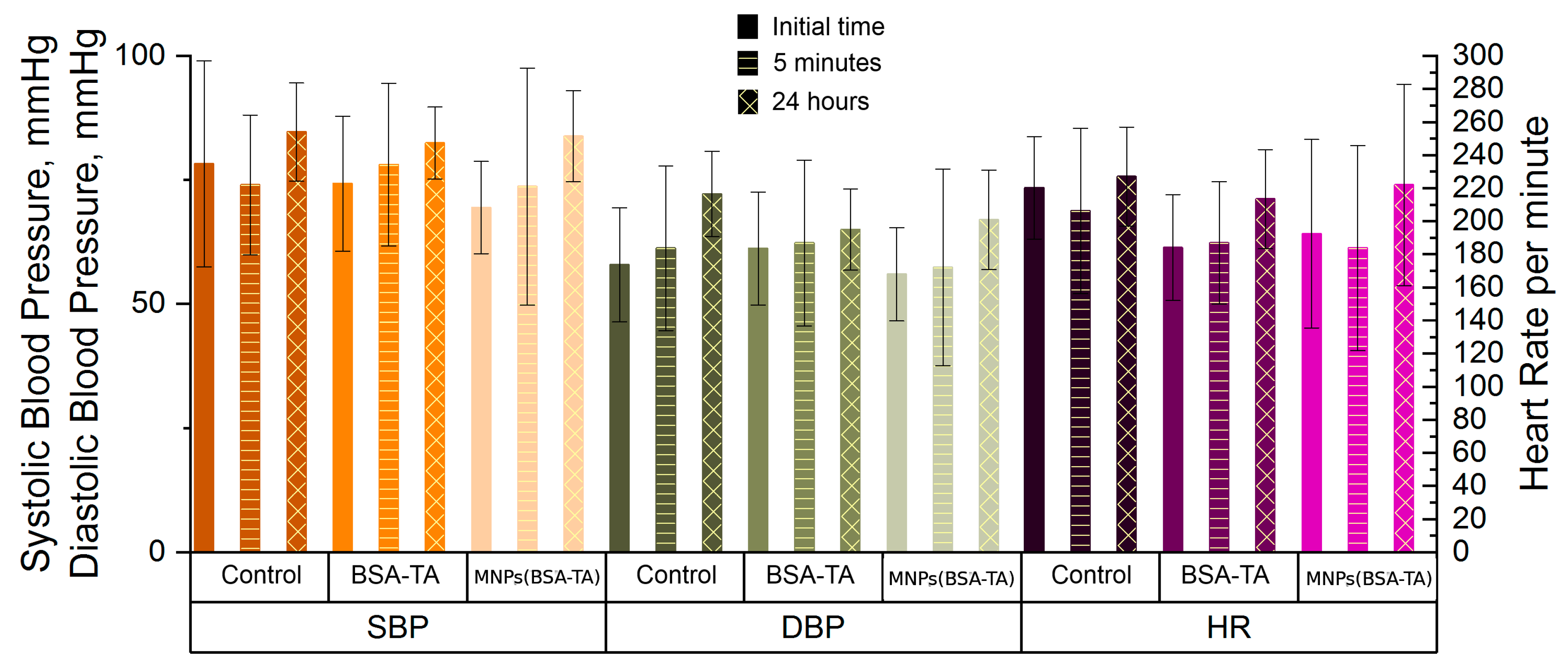
3.3.6. Heart Function Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ivanov, S.; Zhuravsky, S.; Yukina, G.; Tomson, V.; Korolev, D.; Galagudza, M. In Vivo Toxicity of Intravenously Administered Silica and Silicon Nanoparticles. Materials 2012, 5, 1873–1889. [Google Scholar] [CrossRef] [Green Version]
- Gleb, B. Sukhorukov Edwin Donath Sean Davis Heinz Lichtenfeld Frank Caruso Victor I. Popov Helmuth Möhwald Stepwise polyelectrolyte assembly on particle surfaces: A novel approach to colloid design. Polym. Adv. Technol. 1998, 9, 759–767. [Google Scholar]
- Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davis, S.A.; Möhwald, H. Novel Hollow Polymer Shells by Colloid-Templated Assembly of Polyelectrolytes. Angew. Chem. Int. Ed. 1998, 37, 2201–2205. [Google Scholar] [CrossRef]
- Gusliakova, O.; Atochina-Vasserman, E.N.; Sindeeva, O.; Sindeev, S.; Pinyaev, S.; Pyataev, N.; Revin, V.; Sukhorukov, G.B.; Gorin, D.; Gow, A.J. Use of Submicron Vaterite Particles Serves as an Effective Delivery Vehicle to the Respiratory Portion of the Lung. Front. Pharmacol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terentyuk, G.S.; Maslyakova, G.N.; Suleymanova, L.V.; Khlebtsov, B.N.; Kogan, B.Y.; Akchurin, G.G.; Shantrocha, A.V.; Maksimova, I.L.; Khlebtsov, N.G.; Tuchin, V.V. Circulation and distribution of gold nanoparticles and induced alterations of tissue morphology at intravenous particle delivery. J. Biophotonics 2009, 2, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, R.; Moghadam, S.H.; Mohammadyari-Fard, L.; Atyabi, F. Preparation of biodegradable microspheres and matrix devices containing naltrexone. AAPS PharmSciTech 2003, 4, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cock, L.J.; De Koker, S.; De Geest, B.G.; Grooten, J.; Vervaet, C.; Remon, J.P.; Sukhorukov, G.B.; Antipina, M.N. Polymeric Multilayer Capsules in Drug Delivery. Angew. Chem. Int. Ed. 2010, 49, 6954–6973. [Google Scholar] [CrossRef]
- Redhead, H.; Davis, S.; Illum, L. Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: In vitro characterisation and in vivo evaluation. J. Control. Release 2001, 70, 353–363. [Google Scholar] [CrossRef]
- Asmatulu, R.; Zalich, M.A.; Claus, R.O.; Riffle, J.S. Synthesis, characterization and targeting of biodegradable magnetic nanocomposite particles by external magnetic fields. J. Magn. Magn. Mater. 2005, 292, 108–119. [Google Scholar] [CrossRef]
- Lübbe, A.S.; Bergemann, C.; Brock, J.; McClure, D.G. Physiological aspects in magnetic drug-targeting. J. Magn. Magn. Mater. 1999, 194, 149–155. [Google Scholar] [CrossRef]
- Rudge, S.; Kurtz, T.; Vessely, C.; Catterall, L.; Williamson, D. Preparation, characterization, and performance of magnetic iron–carbon composite microparticles for chemotherapy. Biomaterials 2000, 21, 1411–1420. [Google Scholar] [CrossRef]
- Jalil, R.; Nixon, J.R. Biodegradable poly(lactic acid) and poly(lactide-co-glycolide) microcapsules: Problems associated with preparative techniques and release properties. J. Microencapsul. 1990, 7, 297–325. [Google Scholar] [CrossRef] [PubMed]
- Washington, K.E.; Kularatne, R.N.; Karmegam, V.; Biewer, M.C.; Stefan, M.C. Stimuli-responsive poly (ε-caprolactone)s for drug delivery applications. Wood Head Publ. Ser. Biomater. 2018, 1, 501–529. [Google Scholar]
- Fattal, E.; Vauthier, C.; Aynié, I.; Nakada, Y.; Lambert, G.; Malvy, C.; Couvreur, P. Biodegradable polyalkylcyanoacrylate nanoparticles for the delivery of oligonucleotides. J. Control. Release 1998, 53, 137–143. [Google Scholar] [CrossRef]
- Wu, X.S. Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers: Part III. Drug delivery application. Artif. Cells blood Substit. Biotechnol. 2004, 32, 575–591. [Google Scholar] [CrossRef]
- Li, S. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J. Biomed. Mater. Res. 1999, 483, 342–353. [Google Scholar] [CrossRef]
- Nechifor, C.D.; Dorohoi, D.O.; Ciobanu, C. The influence of gamma radiations on physico-chemical properties of some polymer membranes. Rom. Reports Phys. 2009, 54, 349–359. [Google Scholar]
- Hunter, A.C. Molecular hurdles in polyfectin design and mechanistic background to polycation induced cytotoxicity. Adv. Drug Deliv. Rev. 2007, 58, 1523–1531. [Google Scholar] [CrossRef]
- Lomova, M.V.; Brichkina, A.I.; Kiryukhin, M.V.; Vasina, E.N.; Pavlov, A.M.; Gorin, D.A.; Sukhorukov, G.B.; Antipina, M.N. Multilayer capsules of bovine serum albumin and tannic acid for controlled release by enzymatic degradation. ACS Appl. Mater. Interfaces 2015, 7, 11732–11740. [Google Scholar] [CrossRef]
- Kozlovskaya, V.; Zavgorodnya, O.; Chen, Y.; Ellis, K.; Tse, H.M.; Cui, W.; Thompson, J.A.; Kharlampieva, E. Ultrathin polymeric coatings based on hydrogen-bonded polyphenol for protection of pancreatic islet cells. Adv. Funct. Mater. 2012, 22, 3389–3398. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Novoselova, M.V.; Lim, S.H.; Pyataev, N.A.; Pinyaev, S.I.; Kulikov, O.A.; Sindeeva, O.A.; Mayorova, O.A.; Murney, R.; Antipina, M.N.; et al. Formulation for Oral Delivery of Lactoferrin Based on Bovine Serum Albumin and Tannic Acid Multilayer Microcapsules. Sci. Rep. 2017, 7, 44159. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Vandelli, M.A.; Rivasi, F.; Guerra, P.; Forni, F.; Arletti, R. Gelatin microspheres crosslinked with d,l-glyceraldehyde as a potential drug delivery system: Preparation, characterisation, in vitro and in vivo studies. Int. J. Pharm. 2001, 215, 175–184. [Google Scholar] [CrossRef]
- Sahin, S.; Selek, H.; Ponchel, G.; Ercan, M.T.; Sargon, M.; Hinçal, A.; Kaş, H. Preparation, characterization and in vivo distribution of terbutaline sulfate loaded albumin microspheres. J. Control. Release 2002, 82, 345–358. [Google Scholar] [CrossRef]
- Zhao, D.; Zhao, X.; Zu, Y.; Li, J.; Zhang, Y.; Jiang, R.; Zhang, Z. Preparation, characterization, and in vitro targeted delivery of folate-decorated paclitaxel-loaded bovine serum albumin nanoparticles. Int. J. Nanomed. 2010, 5, 669–677. [Google Scholar] [Green Version]
- Yu, Z.; Yu, M.; Zhang, Z.; Hong, G.; Xiong, Q. Bovine serum albumin nanoparticles as controlled release carrier for local drug delivery to the inner ear. Nanoscale Res. Lett. 2014, 9, 343. [Google Scholar] [CrossRef]
- Singh, P.; Singh, H.; Castro-Aceituno, V.; Ahn, S.; Kim, Y.J.; Yang, D.C. Bovine serum albumin as a nanocarrier for the efficient delivery of ginsenoside compound K: Preparation, physicochemical characterizations and in vitro biological studies. RSC Adv. 2017, 7, 15397–15407. [Google Scholar] [CrossRef]
- Ilinskaya, A.N.; A Dobrovolskaia, M. Nanoparticles and the blood coagulation system. Part II: Safety concerns. Nanomedicine 2013, 8, 969–981. [Google Scholar] [CrossRef]
- Soares, S.; Mateus, N.; De Freitas, V. Interaction of Different Polyphenols with Bovine Serum Albumin (BSA) and Human Salivary α-Amylase (HSA) by Fluorescence Quenching. J. Agric. Food Chem. 2007, 55, 6726–6735. [Google Scholar] [CrossRef]
- German, S.V.; Novoselova, M.V.; Bratashov, D.N.; Demina, P.A.; Atkin, V.S.; Voronin, D.V.; Khlebtsov, B.N.; Parakhonskiy, B.V.; Sukhorukov, G.B.; Gorin, D.A. High-efficiency freezing-induced loading of inorganic nanoparticles and proteins into micron- and submicron-sized porous particles. Sci. Rep. 2018, 8, 17763. [Google Scholar] [CrossRef] [PubMed]
- Trushina, D.B.; Bukreeva, T.V.; Antipina, M.N. Size-Controlled Synthesis of Vaterite Calcium Carbonate by the Mixing Method: Aiming for Nanosized Particles. Cryst. Growth Des. 2016, 16, 1311–1319. [Google Scholar] [CrossRef]
- Yu, C.-J.; Wu, S.-M.; Tseng, W.-L. Magnetite Nanoparticle-Induced Fluorescence Quenching of Adenosine Triphosphate–BODIPY Conjugates: Application to Adenosine Triphosphate and Pyrophosphate Sensing. Anal. Chem. 2013, 85, 8559–8565. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Belting, M.; Sandgren, S.; Wittrup, A. Nuclear delivery of macromolecules: Barriers and carriers. Adv. Drug Deliv. Rev. 2005, 57, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Non-viral gene delivery systems. Curr. Opin. Biotechnol. 2002, 13, 128–131. [Google Scholar] [CrossRef]
- Veiseh, O.; Gunn, J.W.; Zhang, M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv. Drug Deliv. Rev. 2010, 62, 284–304. [Google Scholar] [CrossRef] [Green Version]
- Kozlova, A.A.; Burilova, E.A.; Khlebtsov, B.N.; Amirov, R.R.; German, S.V.; Bratashov, D.N.; Navolokin, N.A.; Lomova, M.V.; Novoselova, M.V.; Zyev, V.V.; et al. In vitro and in vivo MRI visualization of nanocomposite biodegradable microcapsules with tunable contrast. Phys. Chem. Chem. Phys. 2016, 18, 32238–32246. [Google Scholar]
- Davis, S. Biomédical applications of nanotechnology—Implications for drug targeting and gene therapy. Trends Biotechnol. 1997, 15, 217–224. [Google Scholar] [CrossRef]
- Nishioka, Y.; Yoshino, H. Lymphatic targeting with nanoparticulate system. Adv. Drug Deliv. Rev. 2001, 47, 55–64. [Google Scholar] [CrossRef]
- Goodwin, S.; Peterson, C.; Hoh, C.; Bittner, C. Targeting and retention of magnetic targeted carriers (MTCs) enhancing intra-arterial chemotherapy. J. Magn. Magn. Mater. 1999, 194, 132–139. [Google Scholar] [CrossRef]
- Voronin, D.V.; Sindeeva, O.A.; Kurochkin, M.A.; Mayorova, O.; Fedosov, I.V.; Semyachkina-Glushkovskaya, O.; Gorin, D.A.; Tuchin, V.V.; Sukhorukov, G.B. In Vitro and in Vivo Visualization and Trapping of Fluorescent Magnetic Microcapsules in a Bloodstream. ACS Appl. Mater. Interfaces 2017, 9, 6885–6893. [Google Scholar] [CrossRef] [PubMed]
- Leakakos, T.; Ji, C.; Lawson, G.; Peterson, C.; Goodwin, S. Intravesical administration of doxorubicin to swine bladder using magnetically targeted carriers. Cancer Chemother. Pharmacol. 2003, 51, 445–450. [Google Scholar] [PubMed]
- Kohler, N.; Sun, C.; Fichtenholtz, A.; Gunn, J.; Fang, C.; Zhang, M. Methotrexate-Immobilized Poly(ethylene glycol) Magnetic Nanoparticles for MR Imaging and Drug Delivery. Small 2006, 2, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-K.; Lim, S.-J. Recent progress in drug delivery systems for anticancer agents. Arch. Pharmacal Res. 2002, 25, 229–239. [Google Scholar] [CrossRef]
- Sun, C.; Lee, J.S.; Zhang, M. Magnetic Nanoparticles in MR Imaging and Drug Delivery. Adv. Drug Deliv. Rev. 2008, 60, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Grasset, F.; Duguet, E. Magnetic nanoparticle design for medical diagnosis and therapy. J. Mater. Chem. 2004, 14, 2161–2175. [Google Scholar]
- Dobrovolskaia, M.A.; McNeil, S.E. Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines. J. Control. Release 2013, 172, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-H.; Ye, M.-K.; Kim, H.-S.; Kang, H.-S. The effects of nano-silver on the proliferation and cytokine expression by peripheral blood mononuclear cells. Int. Immunopharmacol. 2007, 7, 1813–1818. [Google Scholar] [CrossRef]
- Cho, W.-S.; Cho, M.; Jeong, J.; Choi, M.; Cho, H.-Y.; Han, B.S.; Kim, S.H.; Kim, H.O.; Lim, Y.T.; Chung, B.H.; et al. Acute toxicity and pharmacokinetics of 13 nm-sized PEG-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 2009, 236, 16–24. [Google Scholar] [CrossRef]
- Igarashi, E. Factors affecting toxicity and efficacy of polymeric nanomedicines. Toxicol. Appl. Pharmacol. 2008, 229, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, G.; Griffin, J.I.; Brenneman, B.; Banda, N.K.; Holers, V.M.; Backos, D.S.; Wu, L.; Moghimi, S.M.; Simberg, D. Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat. Nanotechnol. 2017, 12, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Blunk, T.; Hochstrasser, D.F.; Sanchez, J.-C.; Müller, B.W.; Müller, R.H. Colloidal carriers for intravenous drug targeting: Plasma protein adsorption patterns on surface-modified latex particles evaluated by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis 1993, 14, 1382–1387. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [Green Version]
- Ekdahl, K.N.; Soveri, I.; Hilborn, J.; Fellström, B.; Nilsson, B. Cardiovascular disease in haemodialysis: Role of the intravascular innate immune system. Nat. Rev. Nephrol. 2017, 13, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Campagne, M.V.L.; Wiesmann, C.; Brown, E.J. Macrophage complement receptors and pathogen clearance. Cell. Microbiol. 2007, 9, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Weiszhár, Z.; Czúcz, J.; Révész, C.; Rosivall, L.; Szebeni, J.; Rozsnyay, Z. Complement activation by polyethoxylated pharmaceutical surfactants: Cremophor-EL, Tween-80 and Tween-20. Eur. J. Pharm. Sci. 2012, 45, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Storm, G. Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem. Biophys. Res. Commun. 2015, 468, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.; Szebeni, J.; Liebes, L.; Rafique, N.M.; Alving, C.R.; Muggia, F.M.; Savay, S. Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil(R)): Possible role in hypersensitivity reactions. Ann. Oncol. 2003, 14, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Bedocs, P.; Rozsnyay, Z.; Weiszhár, Z.; Urbanics, R.; Rosivall, L.; Cohen, R.; Garbuzenko, O.; Bathori, G.; Tóth, M.; et al. Liposome-induced complement activation and related cardiopulmonary distress in pigs: Factors promoting reactogenicity of Doxil and AmBisome. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hempel, J.C.; Poppelaars, F.; Gaya da Costa, M.; Franssen, C.F.; de Vlaam, T.P.; Daha, M.R.; Berger, S.P.; Seelen, M.A.; Gaillard, G.C. Distinct in vitro Complement Activation by Various Intravenous Iron Preparations. Am. J. Nephrol. 2017, 45, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C.; Vernon, K. Complement Activation-Related Pseudo-Allergy: A Fresh Look at Hypersensitivity Reactions to Intravenous Iron. Am. J. Nephrol. 2017, 45, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, E.F. Activation of the blood coagulation system during gram-negative infections and endotoxemias. Fortschr. Med. 1975, 93, 1072–1076. [Google Scholar] [PubMed]
- Herrmann, I.K.; Urner, M.; Hasler, M.; Roth-Z’Graggen, B.; Aemisegger, C.; Baulig, W.; Athanassiou, E.K.; Regenass, S.; Stark, W.J.; Beck-Schimmer, B. Iron core/shell nanoparticles as magnetic drug carriers: Possible interactions with the vascular compartment Research Article. Nanomedicine 2011, 6, 1199–1213. [Google Scholar] [CrossRef]
- Fornaguera, C.; Calderó, G.; Mitjans, M.; Vinardell, M.P.; Solans, C.; Vauthier, C. Interactions of PLGA nanoparticles with blood components: Protein adsorption, coagulation, activation of the complement system and hemolysis studies. Nanoscale 2015, 7, 6045–6058. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Clogston, J.D.; Neun, B.W.; Hall, J.B.; Patri, A.K.; McNeil, S.E. Method for Analysis of Nanoparticle Hemolytic Properties In Vitro. Nano Lett. 2008, 8, 2180–2187. [Google Scholar] [CrossRef]
- Simberg, D.; Zhang, W.-M.; Merkulov, S.; McCrae, K.; Park, J.-H.; Sailor, M.J.; Ruoslahti, E. Contact activation of kallikrein-kinin system by superparamagnetic iron oxide nanoparticles in vitro and in vivo. J. Control. Release 2009, 140, 301–305. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, H.; Liu, Z.; Chen, B. Effects of major parameters of nanoparticles on their physical and chemical properties and recent application of nanodrug delivery system in targeted chemotherapy. Int. J. Nanomed. 2017, 12, 8483–8493. [Google Scholar] [CrossRef]
- Comănescu, M.V.; Mocanu, M.A.; Anghelache, L.; Marinescu, B.; Dumitrache, F.; Bădoi, A.D.; Manda, G. Toxicity of L-DOPA coated iron oxide nanoparticles in intraperitoneal delivery setting—Preliminary preclinical study. Romanian J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2015, 56, 691–696. [Google Scholar]
- Cooper, D.L.; Harirforoosh, S. Effect of Formulation Variables on Preparation of Celecoxib Loaded. PLoS ONE 2014, 12, 1135–1158. [Google Scholar]
- Yamagishi, Y.; Watari, A.; Hayata, Y.; Li, X.; Kondoh, M.; Yoshioka, Y.; Tsutsumi, Y.; Yagi, K. Acute and chronic nephrotoxicity of platinum nanoparticles in mice. Nanoscale Res. Lett. 2013, 8, 395. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Yan, H.-M.; Ramachandran, A.; Murray, G.J.; Rollins, D.E.; Jaeschke, H. HepaRG cells: A human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 2011, 53, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Al-Salam, S.; Zia, S.; Marzouqi, F.; Al-Dhaheri, A.; Subramaniyan, D.; Dhanasekaran, S.; Yasin, J.; Ali, B.H.; E Kazzam, E. Contrasting actions of diesel exhaust particles on the pulmonary and cardiovascular systems and the effects of thymoquinone. Br. J. Pharmacol. 2011, 164, 1871–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manke, A.; Wang, L.; Rojanasakul, Y. Mechanisms of Nanoparticle-Induced Oxidative Stress and Toxicity. BioMed Res. Int. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostan, H.B.; Rezaee, R.; Valokala, M.G.; Tsarouhas, K.; Golokhvast, K.; Tsatsakis, A.M.; Karimi, G. Cardiotoxicity of nano-particles. Life Sci. 2016, 165, 91–99. [Google Scholar] [CrossRef] [PubMed]
- A Kang, K.; Wang, J.; Jasinski, J.B.; Achilefu, S. Fluorescence Manipulation by Gold Nanoparticles: From Complete Quenching to Extensive Enhancement. J. Nanobiotechnol. 2011, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Grosse, S.; Rokstad, A.M.; Ali, S.; Lambris, J.D.; E Mollnes, T.; Nilsen, A.M.; Stenvik, J.; Mollnes, T. Iron oxide nanoparticles induce cytokine secretion in a complement-dependent manner in a human whole blood model. Int. J. Nanomed. 2017, 12, 3927–3940. [Google Scholar] [CrossRef]
- Mousavi, N.; Nohria, A. Radiation-Induced Cardiovascular Disease. Curr. Treat. Options Cardiovasc. Med. 2013, 15, 507–517. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novoselova, M.V.; German, S.V.; Sindeeva, O.A.; Kulikov, O.A.; Minaeva, O.V.; Brodovskaya, E.P.; Ageev, V.P.; Zharkov, M.N.; Pyataev, N.A.; Sukhorukov, G.B.; et al. Submicron-Sized Nanocomposite Magnetic-Sensitive Carriers: Controllable Organ Distribution and Biological Effects. Polymers 2019, 11, 1082. https://doi.org/10.3390/polym11061082
Novoselova MV, German SV, Sindeeva OA, Kulikov OA, Minaeva OV, Brodovskaya EP, Ageev VP, Zharkov MN, Pyataev NA, Sukhorukov GB, et al. Submicron-Sized Nanocomposite Magnetic-Sensitive Carriers: Controllable Organ Distribution and Biological Effects. Polymers. 2019; 11(6):1082. https://doi.org/10.3390/polym11061082
Chicago/Turabian StyleNovoselova, Marina V., Sergey V. German, Olga A. Sindeeva, Oleg A. Kulikov, Olga V. Minaeva, Ekaterina P. Brodovskaya, Valentin P. Ageev, Mikhail N. Zharkov, Nikolay A. Pyataev, Gleb B. Sukhorukov, and et al. 2019. "Submicron-Sized Nanocomposite Magnetic-Sensitive Carriers: Controllable Organ Distribution and Biological Effects" Polymers 11, no. 6: 1082. https://doi.org/10.3390/polym11061082