New Monomer Based on Eugenol Methacrylate, Synthesis, Polymerization and Copolymerization with Methyl Methacrylate–Characterization and Thermal Properties

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Analysis
2.3. Synthesis
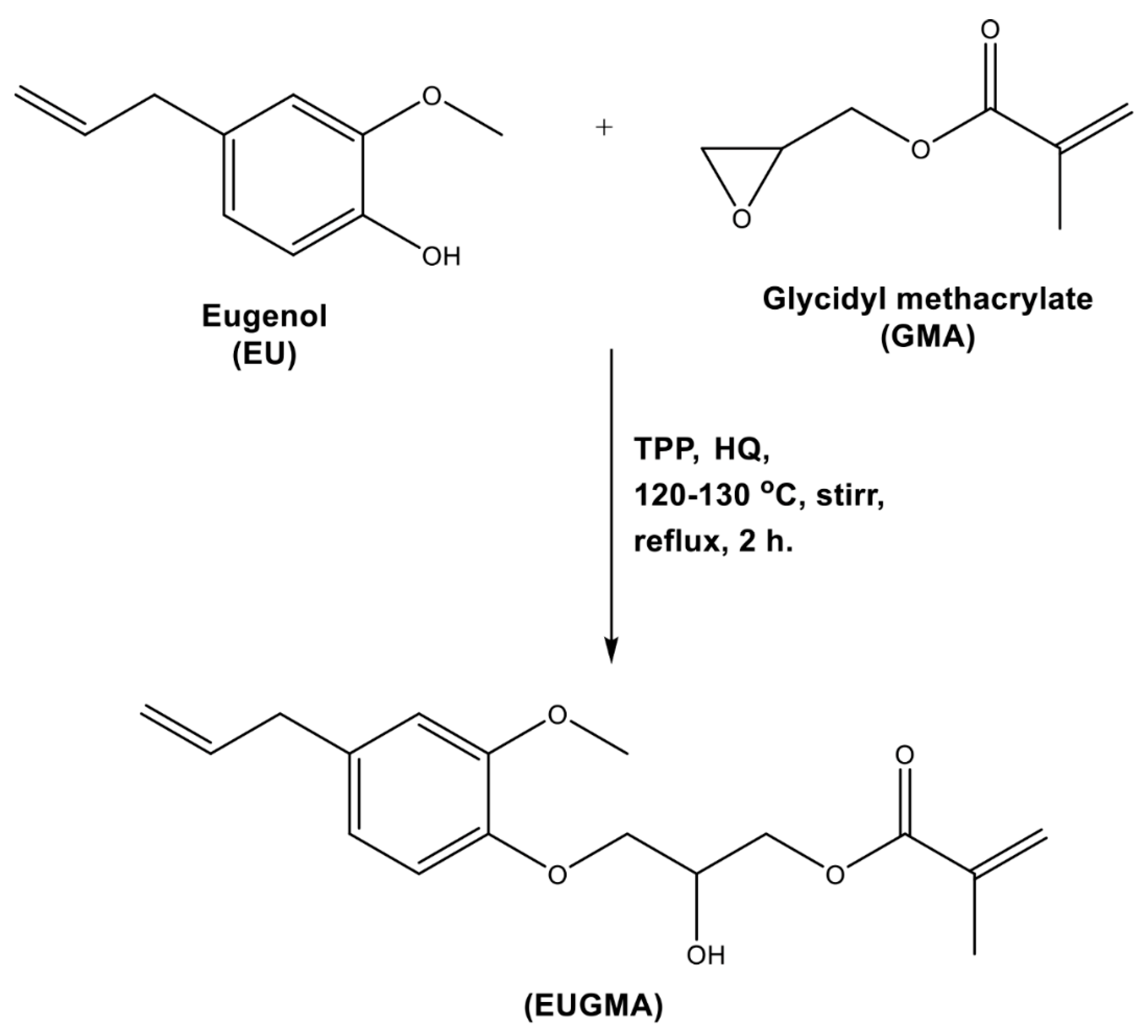
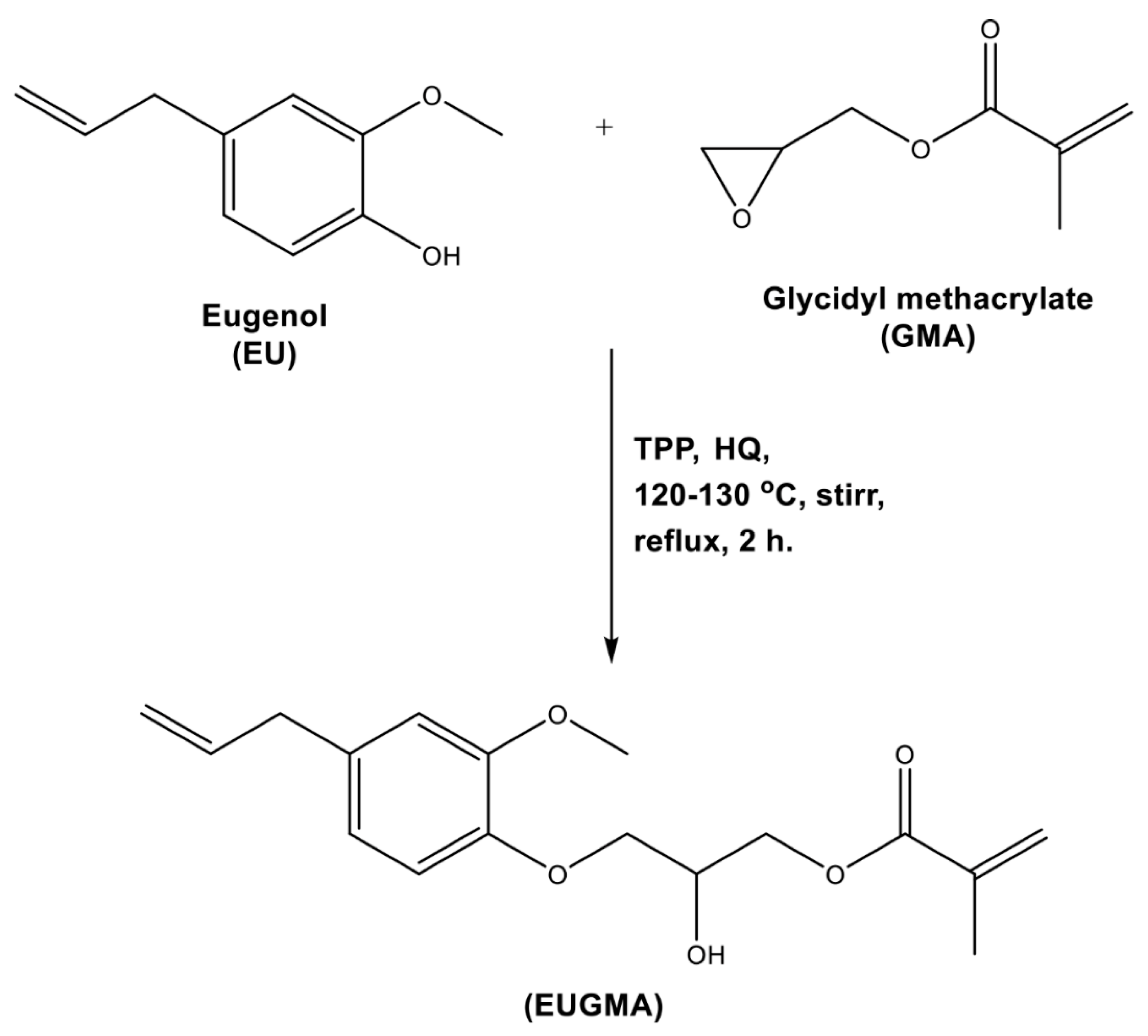
2.3.1. Eugenyl-2-Hydroxypropyl Methacrylate (EUGMA)
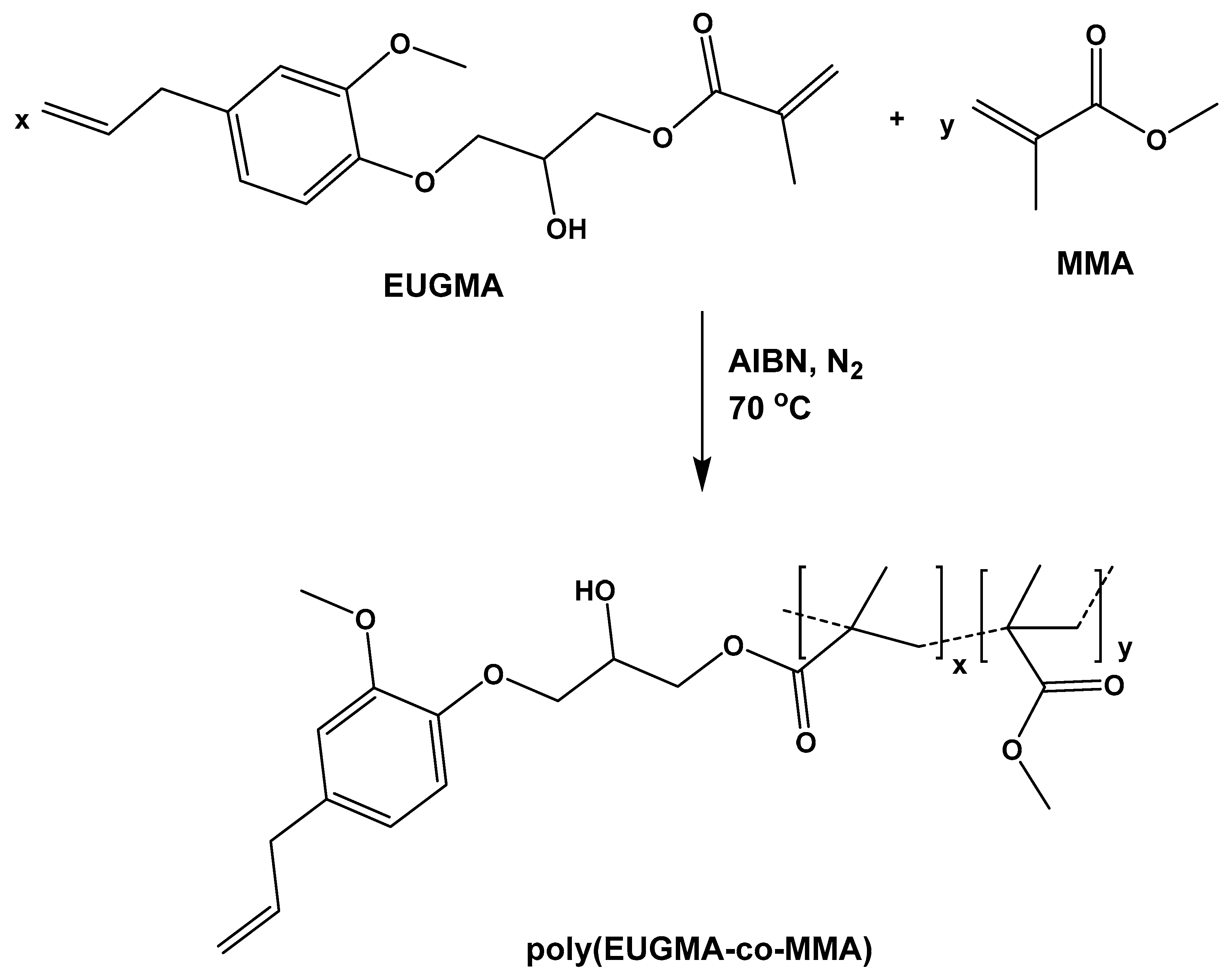
2.3.2. Synthesis of Polymers and Copolymers
3. Results and Discussion
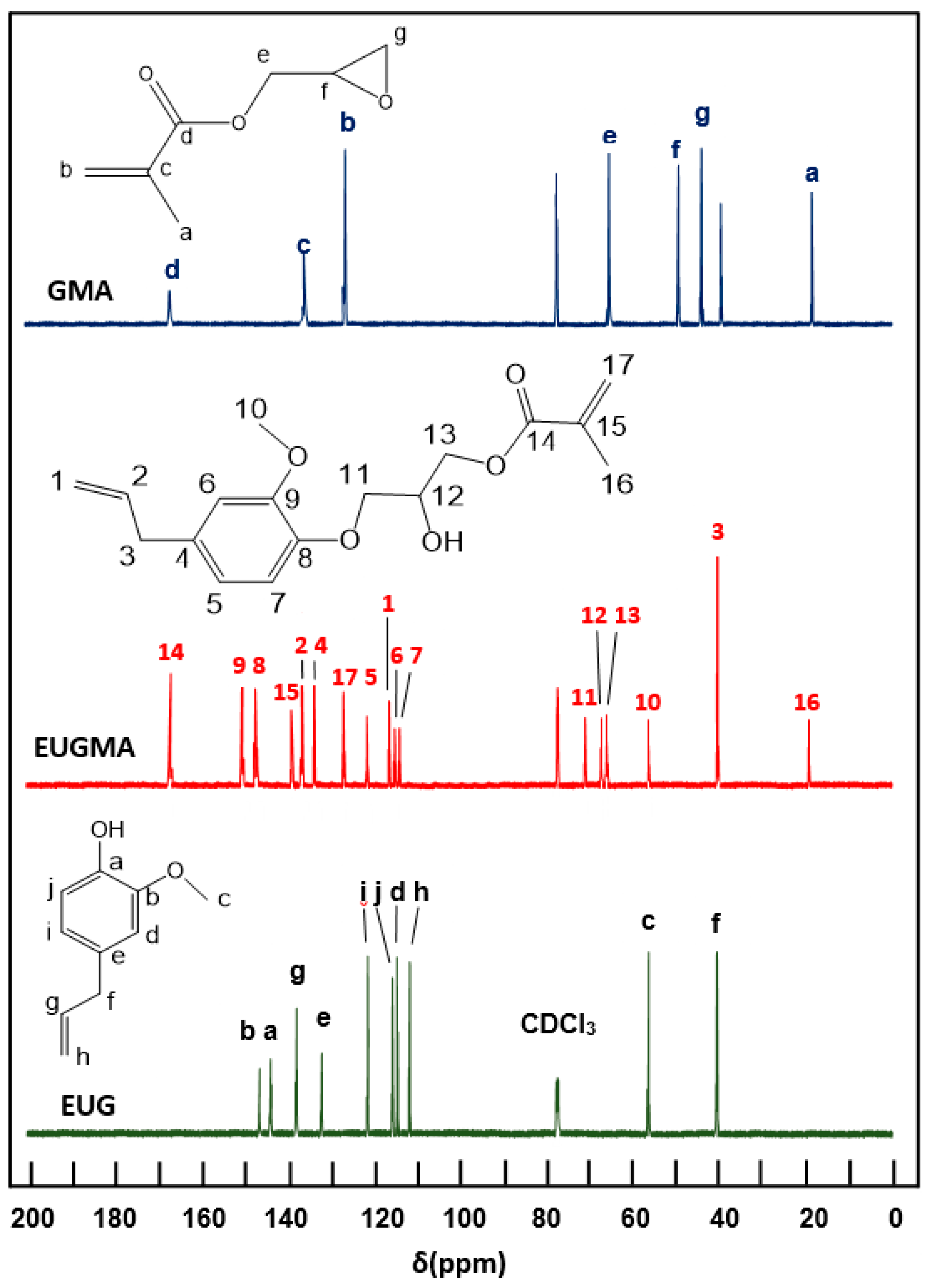
3.1. Synthesis and Characterization of EUGMA
3.2. Synthesis of Homopolymers and Copolymers
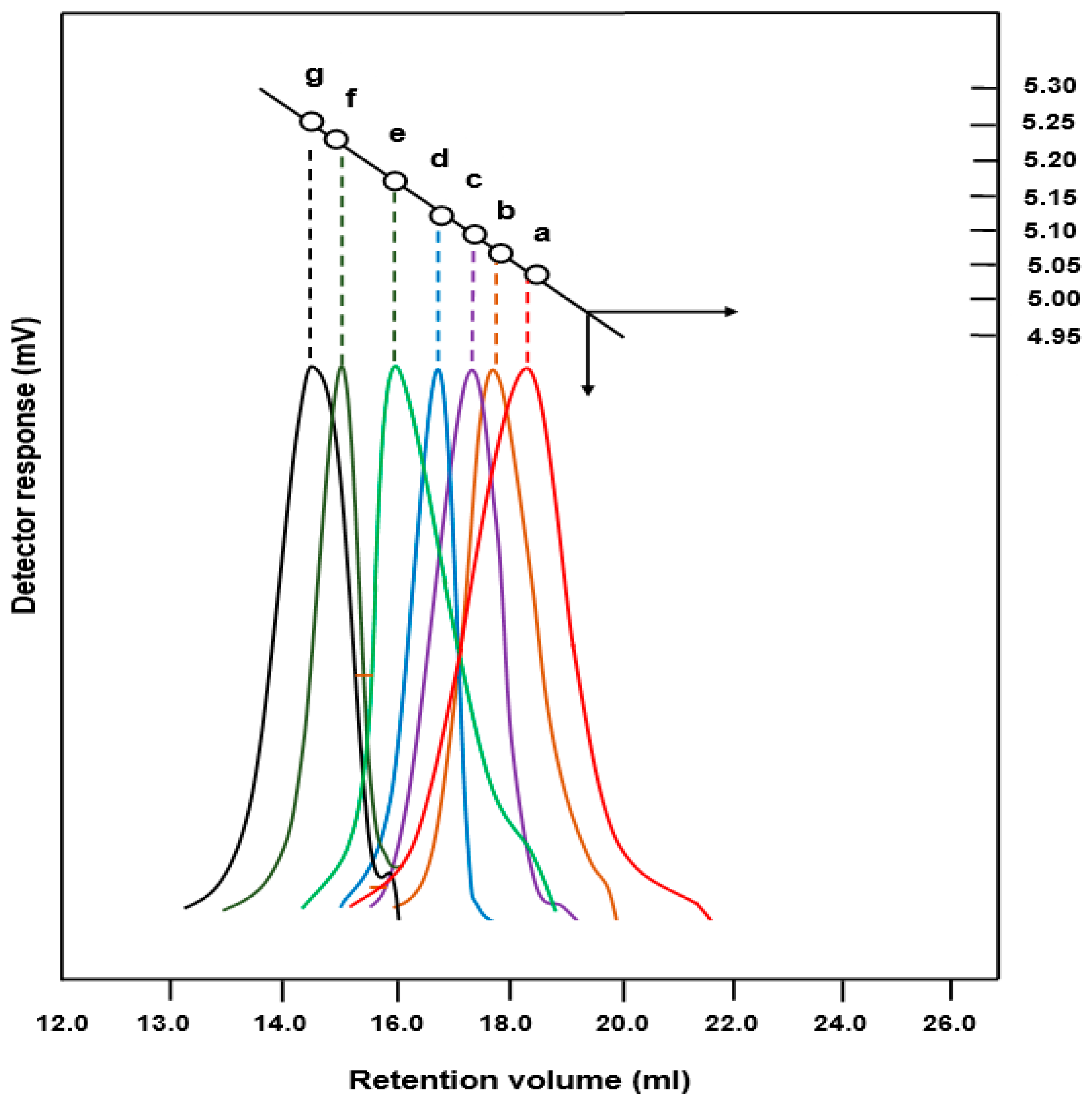
3.2.1. SEC Analysis
3.2.2. Structure
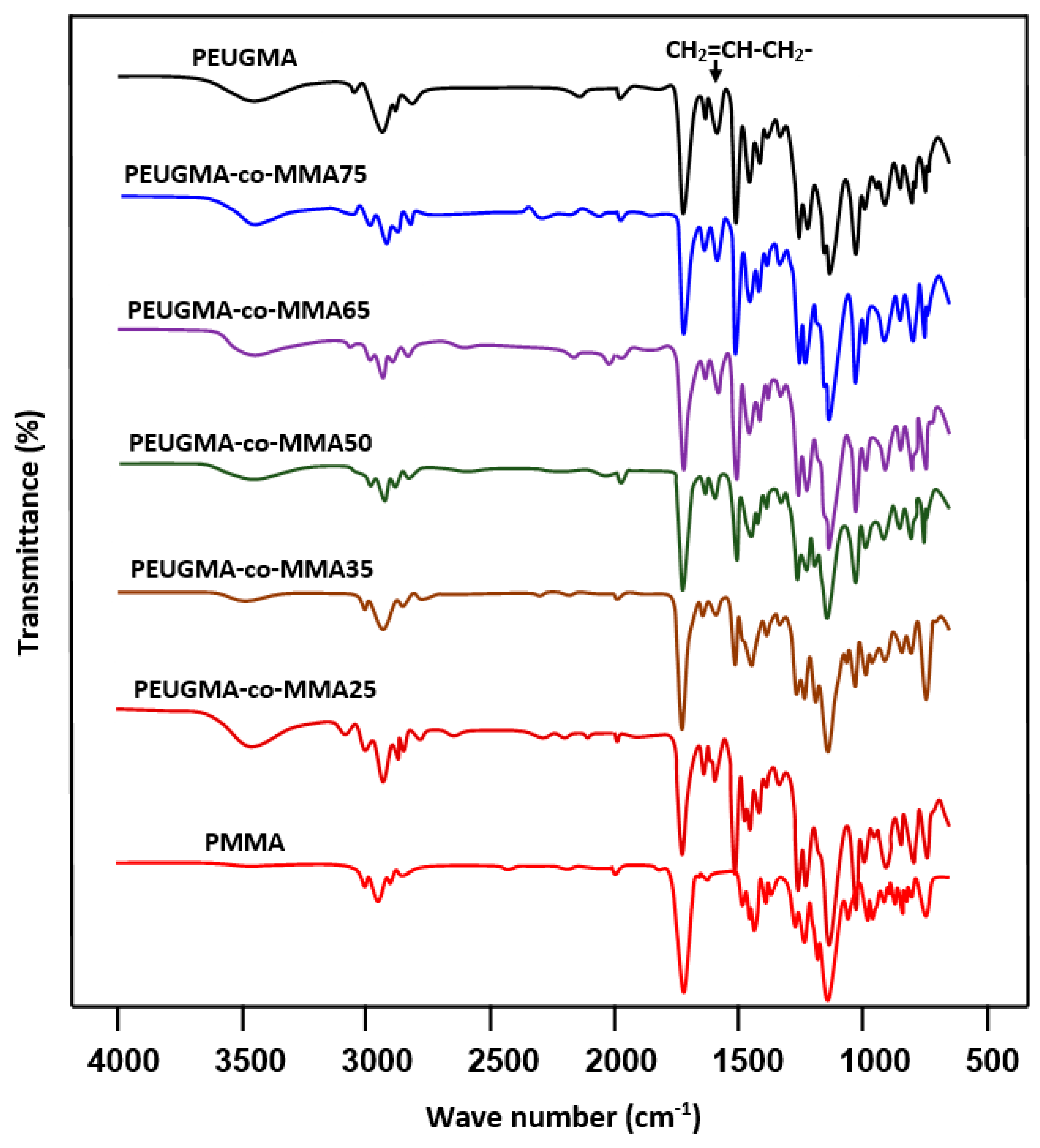
FTIR Analysis
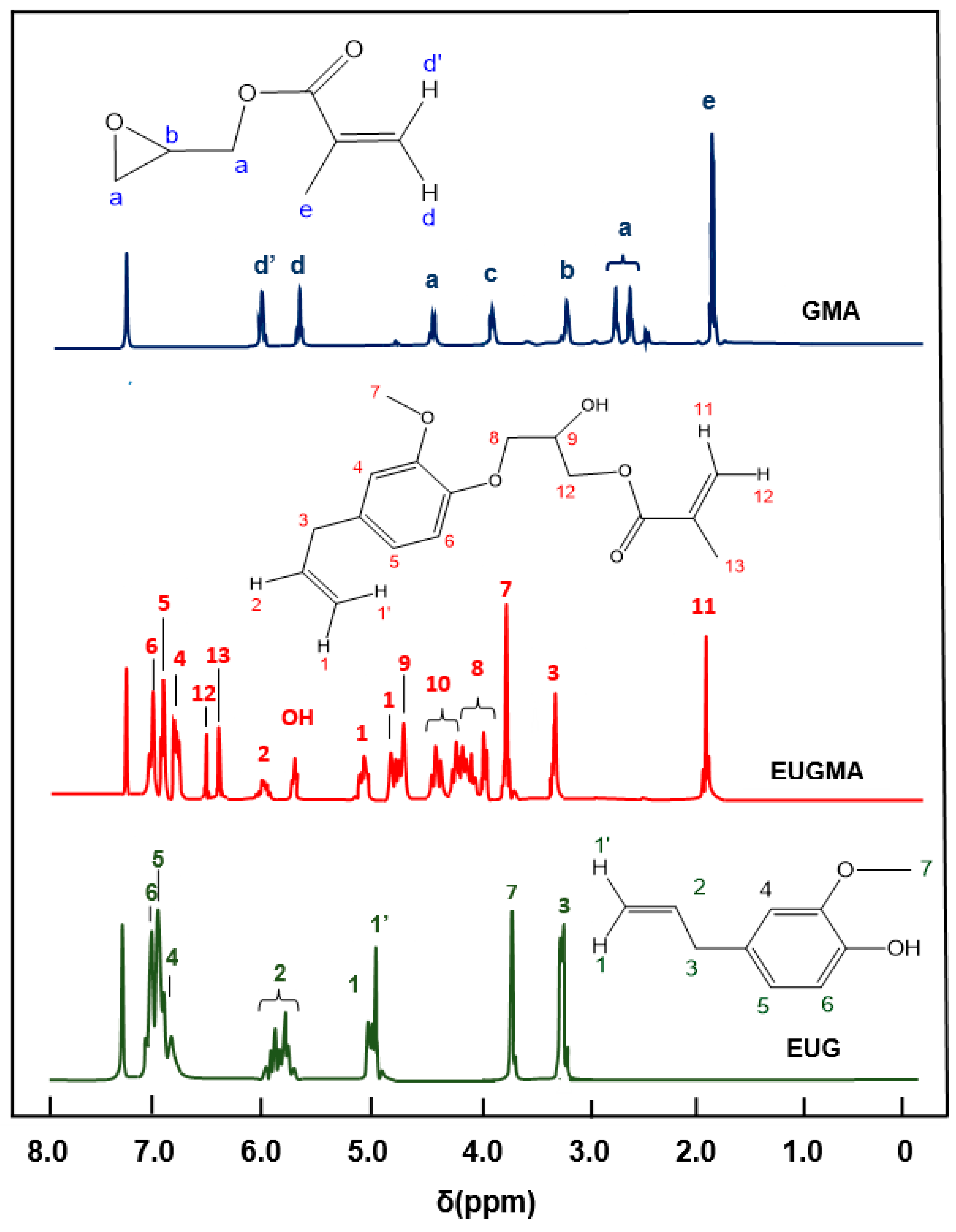
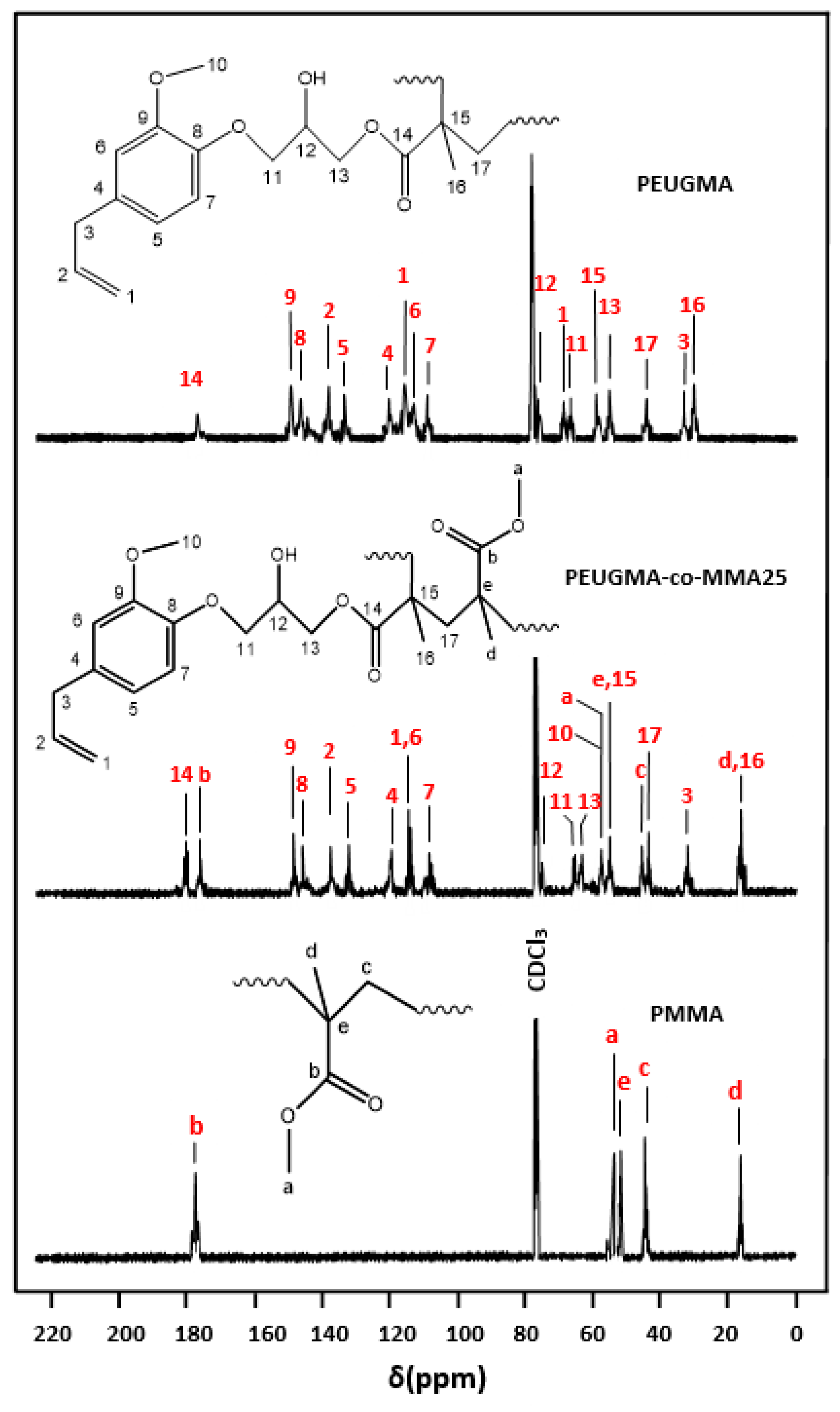
NMR Analysis
UV Analysis
Comonomer Distribution
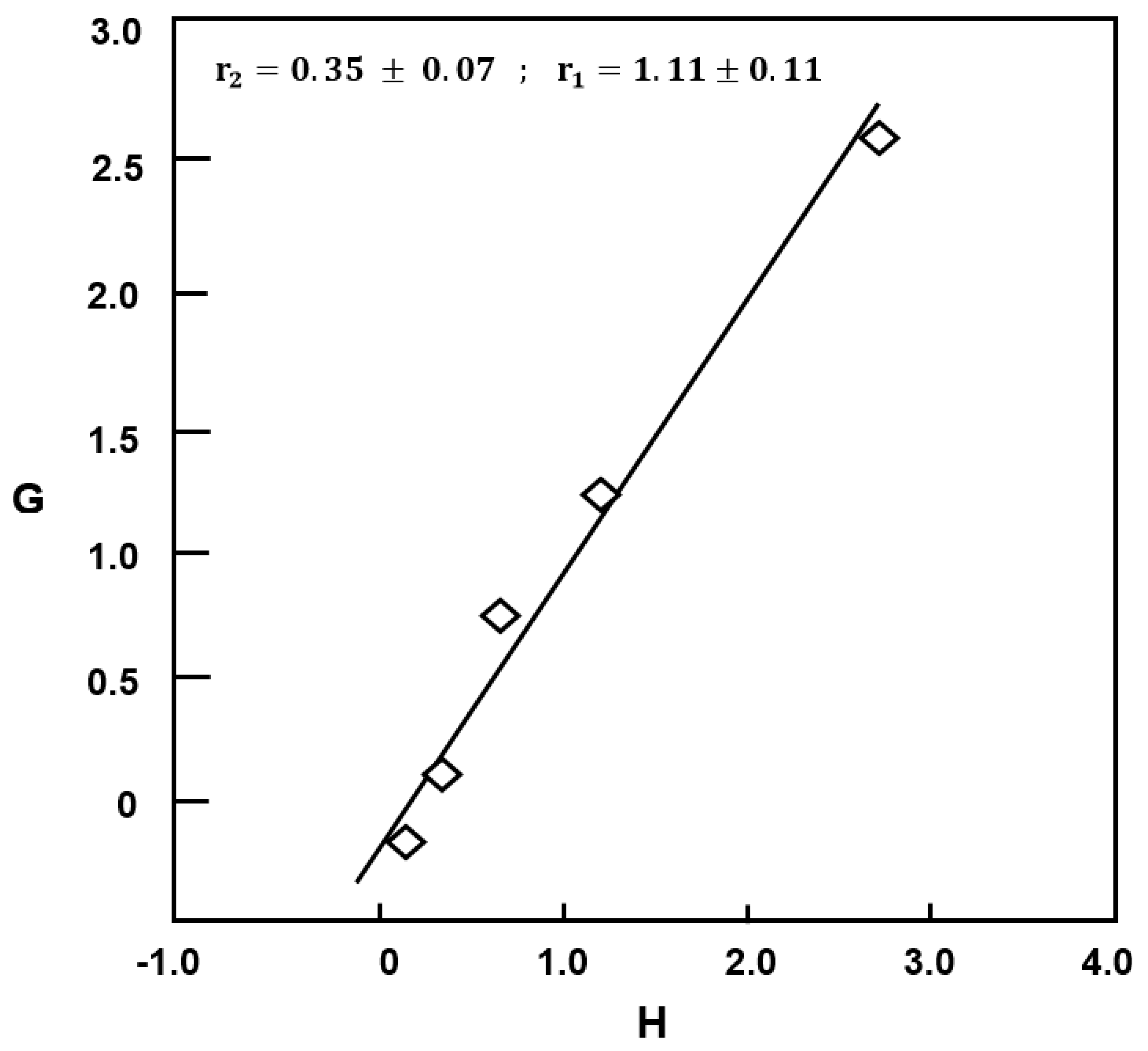
3.2.3. Distribution of the Dyad Monomer Sequences
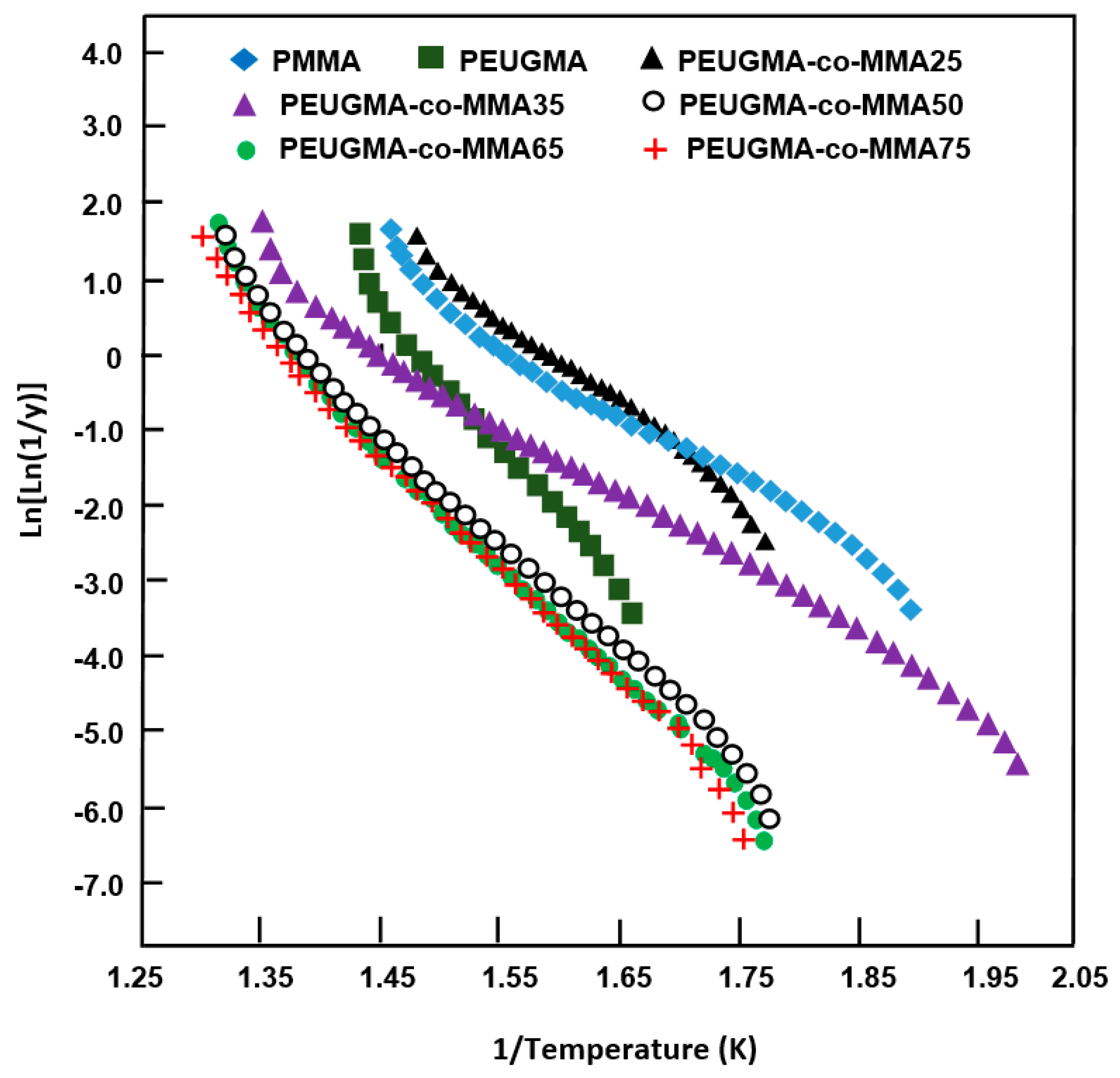
3.3. Thermal Analysis
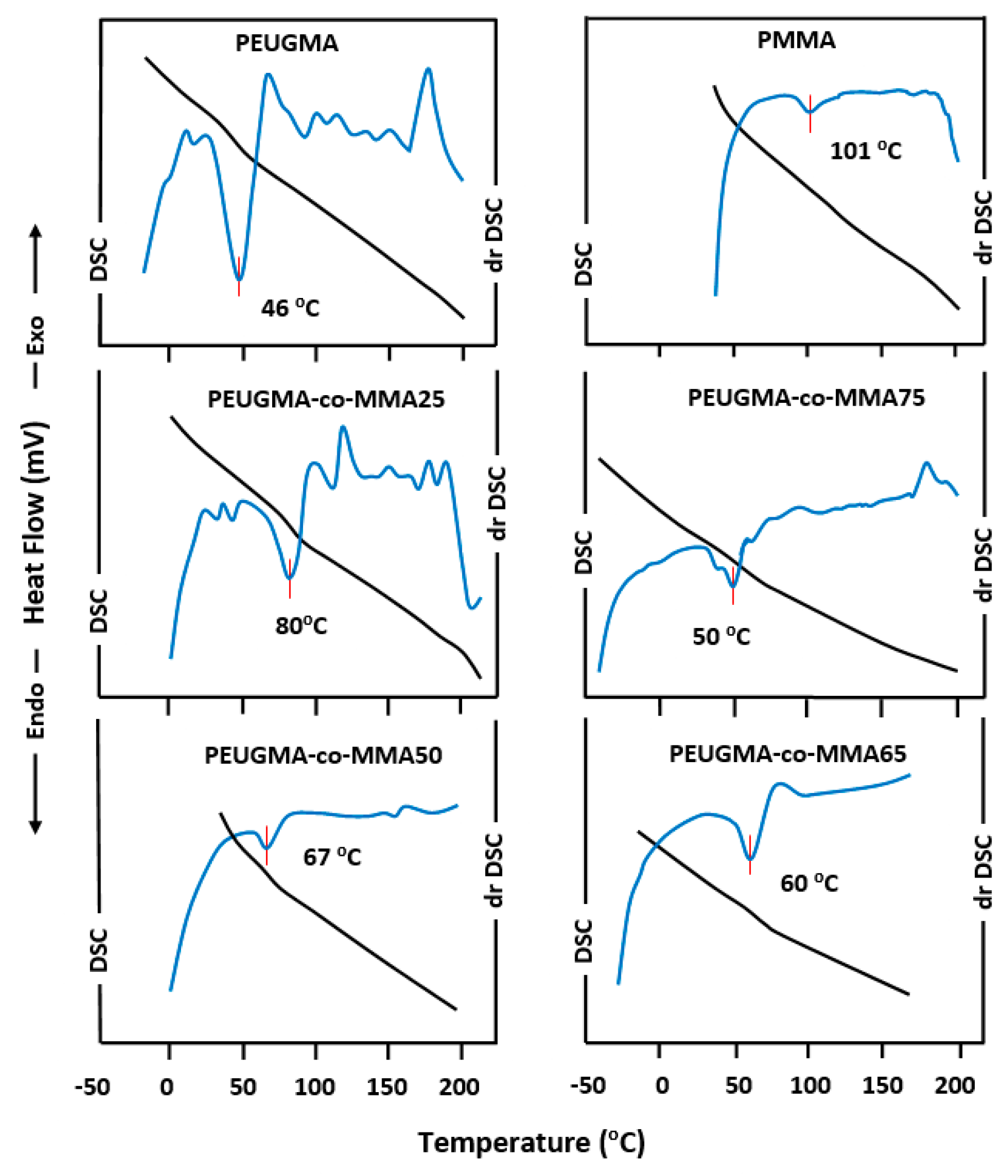
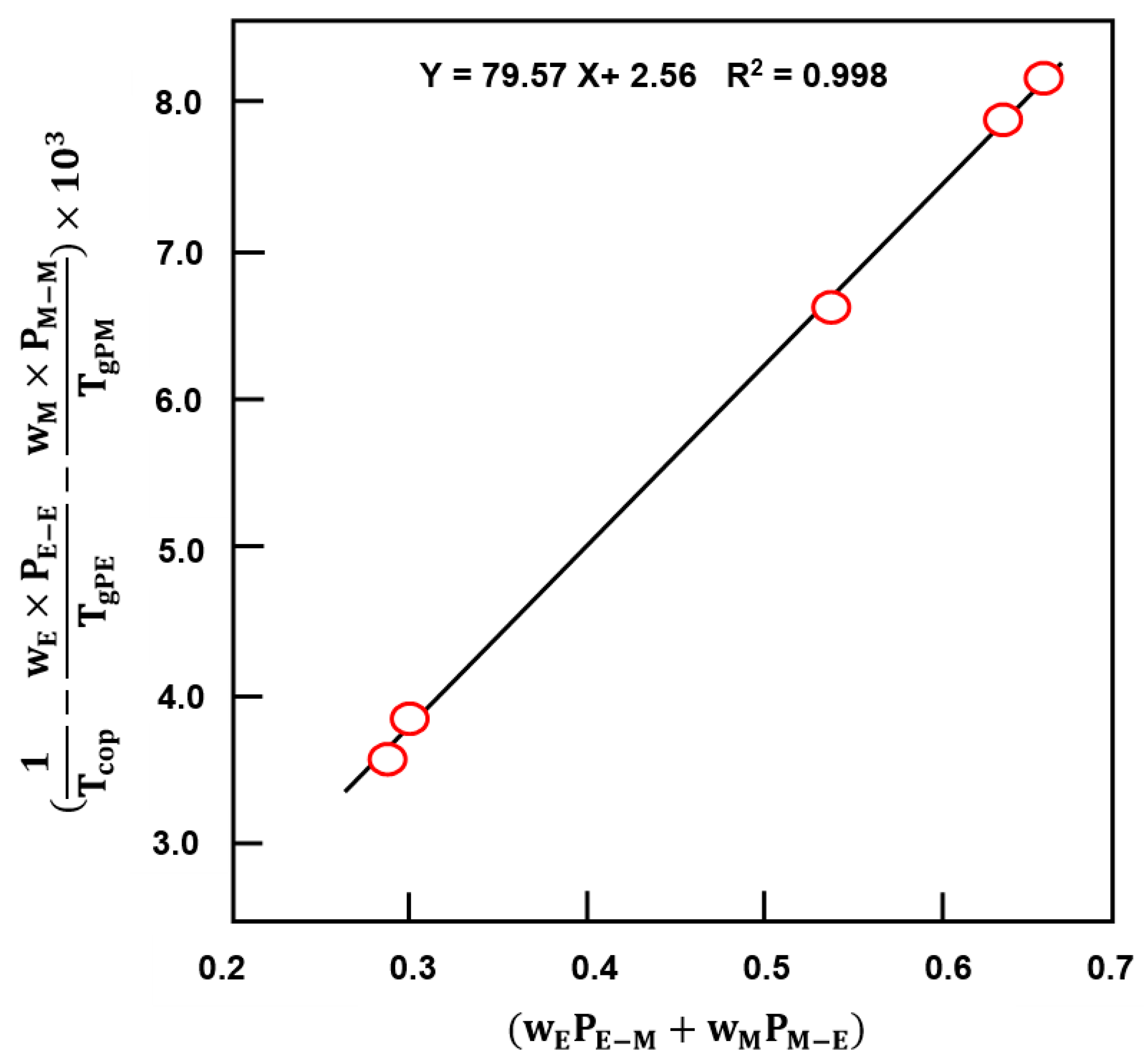
3.3.1. DSC Analysis
3.3.2. TGA Analysis
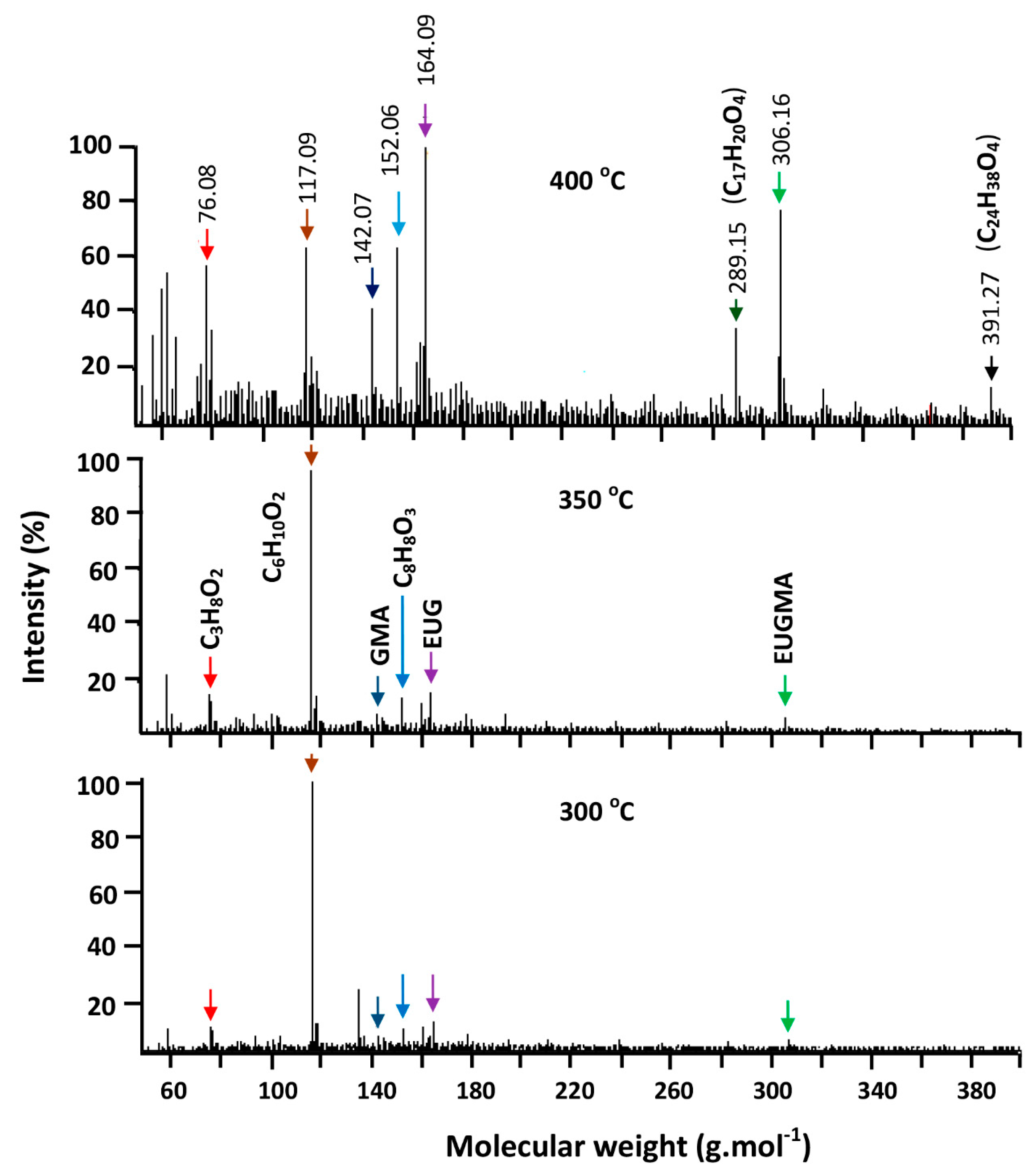
3.3.3. DART-ToF-MS Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, H.Y.; Jeong, U.; Kim, J.K. Reaction kinetics and morphological changes of reactive polymer-polymer interface. Macromolecules 2003, 36, 1594–1602. [Google Scholar] [CrossRef]
- Kang, E.T.; Zhang, Y. Surface modification of fluoropolymers via molecular design. Adv. Mater. 2000, 12, 1481–1494. [Google Scholar] [CrossRef]
- Grubbs, R.B.; Dean, J.M.; Broz, M.E.; Bates, F.S. Reactive block copolymers for modification of thermosetting epoxy. Macromolecules 2000, 33, 9522–9534. [Google Scholar] [CrossRef]
- Tarducci, C.; Kinmond, E.; Badyal, J.; Brewer, S.; Willis, C. Epoxide-functionalized solid surfaces. Chem. Mater. 2000, 12, 1884–1889. [Google Scholar] [CrossRef]
- Murugan, R.; Ramakrishna, S. Modification of demineralized bone matrix by a chemical route. J. Mater. Chem. 2004, 14, 2041–2045. [Google Scholar] [CrossRef]
- Liu, Y.; Klep, V.; Zdyrko, B.; Luzinov, I. Polymer grafting via ATRP initiated from macroinitiator synthesized on surface. Langmuir 2004, 20, 6710–6718. [Google Scholar] [CrossRef]
- Zou, X.; Kang, E.; Neoh, K.; Zhang, Y.; Tan, K.; Cui, C.; Lim, T. Plasma polymerization and deposition of glycidyl methacrylate on Si (100) surface for adhesion improvement with polyimide. Polym. Adv. Technol. 2001, 12, 583–595. [Google Scholar] [CrossRef]
- Ghosh, S.; Krishnamurti, N. Use of glycidyl methacrylate monomers for developing cross-linkable pressure sensitive adhesives. Eur. Polym. J. 2000, 36, 2125–2131. [Google Scholar] [CrossRef]
- Lee, M.S.; Park, W.H. Compatibility and thermal properties of poly (3-hydroxybutyrate)/poly (glycidyl methacrylate) blends. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 351–358. [Google Scholar] [CrossRef]
- Arostegui, A.; Nazabal, J. Compatibilization of a poly (butylene terephthalate)/poly (ethylene octene) copolymer blends with different amounts of an epoxy resin. J. Appl. Polym. Sci. 2004, 91, 260–269. [Google Scholar] [CrossRef]
- Sailaja, R.R.N.; Reddy, A.P.; Chanda, M. Effect of epoxy functionalized compatibilizer on the mechanical properties of low-density polyethylene/plasticized tapioca starch blends. Polym. Int. 2001, 50, 1352–1359. [Google Scholar] [CrossRef]
- Martin, P.; Maquet, C.; Legras, R.; Bailly, C.; Leemans, L.; Van Gurp, M.; Van Duin, M. Conjugated effects of the compatibilization and the dynamic vulcanization on the phase inversion behavior in poly (butylene terephthalate)/epoxide-containing rubber reactive polymer blends. Polymer 2004, 45, 5111–5125. [Google Scholar] [CrossRef]
- Zukowska, G.Z.; Robertson, V.J.; Marcinek, M.L.; Jeffrey, K.R.; Stevens, J.R. Structure of proton-conducting anhydrous gel electrolytes based on poly (glycidyl methacrylate). J. Phys. Chem. B 2003, 107, 5797–5805. [Google Scholar] [CrossRef]
- Ulusoy, A.; Onur, M.A. Measurement of in vitro phagocytic activity using functional groups carrying monodisperse poly (glycidyl methacrylate) microspheres in rat blood. J. Biomater. Sci. Polym. Ed. 2003, 14, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Bedair, M.; El Rassi, Z. Capillary electrochromatography with monolithic stationary phases: II. Preparation of cationic stearyl-acrylate monoliths and their electrochromatographic characterization. J. Chromatogr. A 2003, 1013, 35–45. [Google Scholar] [CrossRef]
- Malmsten, M.; Larsson, A. Immobilization of trypsin on porous glycidyl methacrylate beads: Effects of polymer hydrophilization. Colloids Surf. B Biointerfaces 2000, 18, 277–284. [Google Scholar] [CrossRef]
- Mao, Y.; Gleason, K.K. Hot filament chemical vapor deposition of poly (glycidyl methacrylate) thin films using tert-butyl peroxide as an initiator. Langmuir 2004, 20, 2484–2488. [Google Scholar] [CrossRef]
- Zhang, M.; Kang, E.; Neoh, K.; Tan, K. Consecutive graft copolymerization of glycidyl methacrylate and aniline on poly (tetrafluoroethylene) films. Langmuir 2000, 16, 9666–9672. [Google Scholar] [CrossRef]
- Minegishi, S.; Otsuka, T.; Kameyama, A.; Nishikubo, T. Synthesis of phosphorus-containing vinyl ether monomers and oligomers and their photoinitiated polymerization. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 2031–2042. [Google Scholar] [CrossRef]
- Ochiai, B.; Iwamoto, T.; Miyagawa, T.; Nagai, D.; Endo, T. Direct incorporation of gaseous carbon dioxide into solid-state copolymer containing oxirane and quaternary ammonium halide structure as self-catalytic function. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 4941–4947. [Google Scholar] [CrossRef]
- Li, W.H.; Stöver, H.D. Mono-or narrow disperse poly (methacrylate-co-divinylbenzene) microspheres by precipitation polymerization. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 2899–2907. [Google Scholar] [CrossRef]
- Bao, H.; Chen, Z.; Liu, J. Fabrication of nanoscale latex arrays based on hydroxylated poly (butyl methacrylate-b-glycidyl methacrylate). Colloid Polym. Sci. 2003, 282, 92–95. [Google Scholar] [CrossRef]
- Du, Y.-Z.; Tomohiro, T.; Kodaka, M. Synthesis of hemispherical poly (2-hydroxylethyl methacrylate-co-methyl methacrylate)/poly (styrene-co-glycidyl methacrylate) composite particles with heterobifunctional groups by soap-free seeded emulsion polymerization. Macromolecules 2004, 37, 803–812. [Google Scholar] [CrossRef]
- Mouaziz, H.; Larsson, A.; Sherrington, D. One-step batch synthesis of high solids monodisperse styrene/glycidyl methacrylate and styrene/methacrylic acid emulsion copolymers. Macromolecules 2004, 37, 1319–1323. [Google Scholar] [CrossRef]
- Czech, Z. Solvent-based pressure-sensitive adhesives for removable products. Int. J. Adhes. Adhes. 2006, 26, 414–418. [Google Scholar] [CrossRef]
- Grassie, N.; MacCallum, J. Thermal and photochemical degradation of poly (n-butyl methacrylate). J. Polym. Sci. Part A Gen. Pap. 1964, 2, 983–1000. [Google Scholar] [CrossRef] [Green Version]
- Czech, Z.; Agnieszka, K.; Ragańska, P.; Antosik, A. Thermal stability and degradation of selected poly (alkyl methacrylates) used in the polymer industry. J. Therm. Anal. Calorim. 2015, 119, 1157–1161. [Google Scholar] [CrossRef] [Green Version]
- Zulfiqar, S.; Zulfiqar, M.; Nawaz, M.; McNeill, I.; Gorman, J. Thermal degradation of poly (glycidyl methacrylate). Polym. Degrad. Stab. 1990, 30, 195–203. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Tate, F.A. The Polymerization of Allyl Compounds. VI. The Polymerization of Allyl-1-d2 Acetate and the Mechanism of its Chain Termination. J. Am. Chem. Soc. 1953, 75, 91–95. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Altschul, R. The Polymerization of Allyl Compounds. I. Factors Governing the Acyl Peroxide-Induced Polymerization of Allyl Acetate, and the Fate of the Peroxide. J. Am. Chem. Soc. 1945, 67, 812–816. [Google Scholar] [CrossRef]
- Volodina, V.I.; Tarasov, A.I.; Spasskii, S.S. Kinetics of copolymerization of allyl and fumaric oligoesters. Polym. Sci. USSR 1976, 18, 2658–2662. [Google Scholar] [CrossRef]
- Fujisawa, S.; Kadoma, Y. Action of eugenol as a retarder against polymerization of methyl methacrylate by benzoyl peroxide. Biomaterials 1997, 18, 701–703. [Google Scholar] [CrossRef]
- Mayo, F.R.; Lewis, F.M. Copolymerization. I. A basis for comparing the behavior of monomers in copolymerization: The copolymerization of styrene and methyl methacrylate. J. Am. Chem. Soc. 1944, 66, 1594–1601. [Google Scholar] [CrossRef]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdös, F.; Turcsányi, B. Confidence intervals for copolymerization reactivity ratios determined by the Kelen-Tüdös method. Polym. Bull. 1980, 2, 71–76. [Google Scholar] [CrossRef]
- Deshpande, A.; Deshpande, D.D.; Babu, G.N. Effect of temperature on the Monomer Reactivity Ratios of 2-Hydroxypropyl Methacrylate with Methyl Acrylate and Methyl Methacrylate. Polym. Bull. 1981, 6, 1–6. [Google Scholar] [CrossRef]
- Igarashi, S. Representation of composition and blockiness of the copolymer by a triangular coordinate system. J. Polym. Sci. Part B Polym. Lett. 1963, 1, 359–363. [Google Scholar] [CrossRef]
- Teng, H.; Koike, K.; Zhou, D.; Satoh, Z.; Koike, Y.; Okamoto, Y. High Glass Transition Temperatures of Poly(methyl methacrylate)Prepared by Free Radical Initiator. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 315–317. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergnt, E.H. Polymer Hand-book, 3rd ed.; Wiley: New York, NY, USA, 1989; p. V78. [Google Scholar]
- Fox, T.G. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull. Am. Phys. Soc. 1956, 1, 123–125. [Google Scholar]
- Brostowa, W.; Chiua, R.; Kalogerasb, I.M.; Vassilikou-Dova, A. Prediction of glass transition temperatures: Binary blends and copolymers. Mater. Lett. 2008, 62, 3152–3155. [Google Scholar] [CrossRef]
- Bonardelli, P.G.; Moggi, G.; Turturro, A. Glass transition temperatures of copolymer and terpolymer fluoroelastomers. Polymer 1986, 27, 905–909. [Google Scholar] [CrossRef]
- Azhar Juhari, A. Structure Property Relations in Complex Copolymer Systems. Ph.D. Thesis, Johannes Gutenberg Univeristy, Mainz, Germany, 2010. [Google Scholar]
- Liu, T.; Geng, X.; Nie, Y.; Chen, R.; Meng, Y.; Li, X. Hyperbranched polyethers with tunable glass transition temperature: Controlled synthesis and mixing rules. RSC Adv. 2014, 4, 30250–30258. [Google Scholar] [CrossRef]
- San Roman, J.; Madruga, E.L.; Guzman, J. Glass transition temperatures of acrylic polymers and copolymers containing phenyl side groups. Polym. Commun. 1984, 25, 373–377. [Google Scholar]
- Johnston, N.W. Sequence distribution-glass transition effects. J. Macromol. Sci. Rev. Macromol. Chem. 1976, 14, 215–250. [Google Scholar] [CrossRef]
- Barton, J.M. Relation of glass transition temperature to molecular structure of addition copolymers. J. Polym. Sci. Part C 1970, 30, 573–597. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Inaba, A.; Brown, J.E.; Hatada, K.; Kitayama, T.; Masuda, E. Effects of weak linkages on the thermal and oxidative degradation of poly (methyl methacrylates). Macromolecules 1986, 19, 2160–2168. [Google Scholar] [CrossRef]
- Saunders, K.J. Organic Polymer Chemistry: An Introduction to the Organic Chemistry of Adhesives, Fibres, Paints, Plastics and Rubbers; Springer Science & Business Media: New York, NY, USA, 2012. [Google Scholar]
- Broido, A. A simple, sensitive graphical method of treating thermogravimetric analysis data. J. Polym. Sci. Part A-2 Polym. Phys. 1969, 7, 1761–1773. [Google Scholar] [CrossRef]
- El-Sonbati, A.; Diab, M. Thermal stability of poly (acryloyl chloride) homopolymer and copolymers of acryloyl chloride with methyl methacrylate. J. Therm. Anal. Calorim. 1988, 34, 769–776. [Google Scholar] [CrossRef]
- Hirata, T.; Kashiwagi, T.; Brown, J.E. Thermal and oxidative degradation of poly (methyl methacrylate): Weight loss. Macromolecules 1985, 18, 1410–1418. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer and Copolymer | Monomer (g) | Monomer (Mole Fraction) | ||
|---|---|---|---|---|
| EUGMA | MMA | EUGMA | MMA | |
| PEUGMA | 3.06 | - | 1.00 | - |
| PEUGMA-co-MMA25 | 0.51 | 0.49 | 0.25 | 0.75 |
| PEUGMA-co-MMA35 | 0.62 | 0.38 | 0.35 | 0.65 |
| PEUGMA-co-MMA50 | 0.75 | 0.25 | 0.50 | 0.50 |
| PEUGMA-co-MMA65 | 0.85 | 0.15 | 0.65 | 0.35 |
| PEUGMA-co-MMA75 | 0.90 | 0.10 | 0.75 | 0.25 |
| PMMA | - | 3.00 | - | 1.00 |
| Copolymer | Yield (wt %) | (g·mol−1) × 105 | (g·mol−1) × 105 | I |
|---|---|---|---|---|
| PEUGMA | - | 1.08 | 3.21 | 2.96 |
| PEUGMA-co-MMA25 | 10.0 | 1.22 | 2.99 | 2.45 |
| PEUGMA-co-MMA35 | 10.6 | 1.28 | 2.46 | 1.92 |
| PEUGMA-co-MMA50 | 11.4 | 1.32 | 2.81 | 2.12 |
| PEUGMA-co-MMA65 | 11.2 | 1.36 | 2.94 | 2.16 |
| PEUGMA-co-MMA75 | 12.0 | 1.48 | 3.32 | 2.25 |
| PMMA | - | 1.78 | 4.40 | 2.47 |
| Copolymer | Composition (mol %) | |||||
|---|---|---|---|---|---|---|
| 1H NMR | UV-Visible | Average | ||||
| EUGMA | MMA | EUGMA | MMA | EUGMA | MMA | |
| PEUGMA-co-MMA25 | 38.52 | 64.42 | 41.06 | 58.94 | 39.79 | 60.21 |
| PEUGMA-co-MMA35 | 47.22 | 52.78 | 50.18 | 49.82 | 48.70 | 51.30 |
| PEUGMA-co-MMA50 | 58.80 | 44.20 | 64.82 | 35.18 | 61.81 | 38.19 |
| PEUGMA-co-MMA65 | 73.18 | 26.82 | 74.82 | 25.18 | 74.00 | 26.00 |
| PEUGMA-co-MMA75 | 78.18 | 12.82 | 75.92 | 24.08 | 77.05 | 22.95 |
| Reactivity | Mayo–Lewis | Fineman–Ross | Kelen–Tüdős | Average |
|---|---|---|---|---|
| r1 | 1.05 ± 0.07 | 1.11 ± 0.11 | 0.87 ± 0.08 | 1.01 ± 0.09 |
| r2 | 0.32 ± 0.10 | 0.35 ± 0.07 | 0.38 ± 0.05 | 0.35 ± 0.07 |
| Polymer and Copolymer | WE * | WM * | (°C) | (°C) | (°C) | (°C) | (°C) |
|---|---|---|---|---|---|---|---|
| PMMA | 0 | 1.0 | 101 | 101 | - | ||
| PEUGMA-co-MMA25 | 0.67 | 0.33 | 80 | 56.0 | 24.0 | 74.0 | 6 |
| PEUGMA-co-MMA35 | 0.75 | 0.25 | 73 | 53.2 | 19.8 | 68.0 | 5 |
| PEUGMA-co-MMA50 | 0.83 | 0.17 | 67 | 51.0 | 16.0 | 61.0 | 6 |
| PEUGMA-co-MMA65 | 0.90 | 0.10 | 60 | 48.6 | 16.4 | 55.0 | 5 |
| PEUGMA-co-MMA75 | 0.91 | 0.09 | 50 | 48.3 | 1.7 | 53.0 | 3 |
| PEUGMA | 1.0 | 0 | 46 | 46 | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Odayni, A.-B.; Saeed, W.S.; Ahmed, A.Y.B.H.; Alrahlah, A.; Al-Kahtani, A.; Aouak, T. New Monomer Based on Eugenol Methacrylate, Synthesis, Polymerization and Copolymerization with Methyl Methacrylate–Characterization and Thermal Properties. Polymers 2020, 12, 160. https://doi.org/10.3390/polym12010160
Al-Odayni A-B, Saeed WS, Ahmed AYBH, Alrahlah A, Al-Kahtani A, Aouak T. New Monomer Based on Eugenol Methacrylate, Synthesis, Polymerization and Copolymerization with Methyl Methacrylate–Characterization and Thermal Properties. Polymers. 2020; 12(1):160. https://doi.org/10.3390/polym12010160
Chicago/Turabian StyleAl-Odayni, Abdel-Basit, Waseem Sharaf Saeed, Ahmed Yacine Badjah Hadj Ahmed, Ali Alrahlah, Abdullah Al-Kahtani, and Taieb Aouak. 2020. "New Monomer Based on Eugenol Methacrylate, Synthesis, Polymerization and Copolymerization with Methyl Methacrylate–Characterization and Thermal Properties" Polymers 12, no. 1: 160. https://doi.org/10.3390/polym12010160
APA StyleAl-Odayni, A.-B., Saeed, W. S., Ahmed, A. Y. B. H., Alrahlah, A., Al-Kahtani, A., & Aouak, T. (2020). New Monomer Based on Eugenol Methacrylate, Synthesis, Polymerization and Copolymerization with Methyl Methacrylate–Characterization and Thermal Properties. Polymers, 12(1), 160. https://doi.org/10.3390/polym12010160