Keratin Associations with Synthetic, Biosynthetic and Natural Polymers: An Extensive Review



Abstract
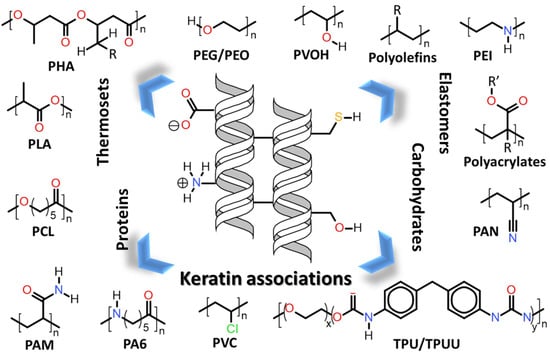
:
Contents
| 1. | A brief historical context of knowledge and use of keratin based-materials |
| 2. | Keratin’s structure and chemical toolset |
| 3. | Sustainability and safety assessment |
| 4. | Keratins extraction |
| 4.1. | Oxidative and reductive extraction |
| 4.2. | Steam explosion extraction |
| 4.3. | Extraction with Ionic liquids and deep eutectic solvents |
| 5. | Keratin sources and their distinctions |
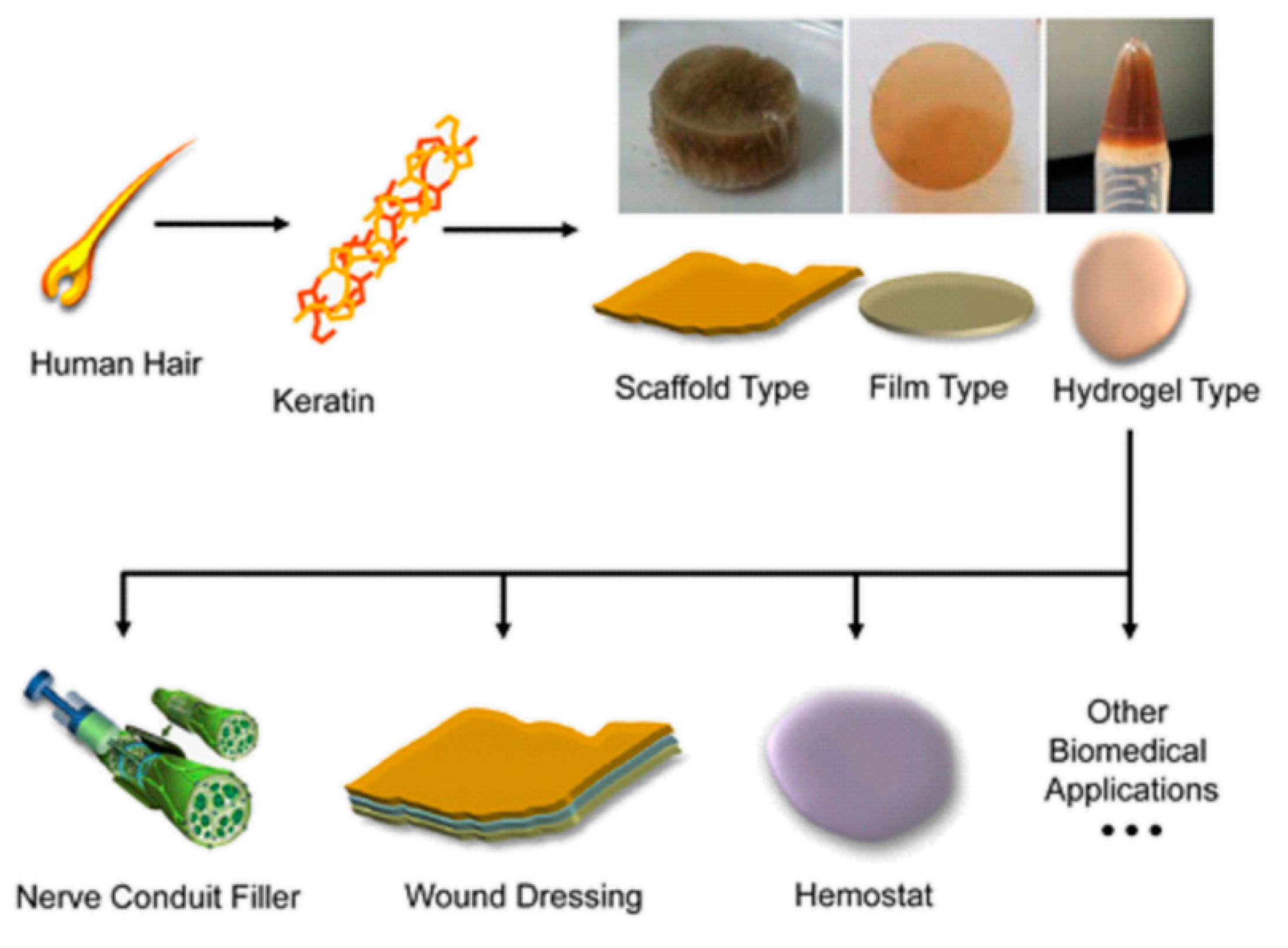
| 6. | Keratin-based biomaterials |
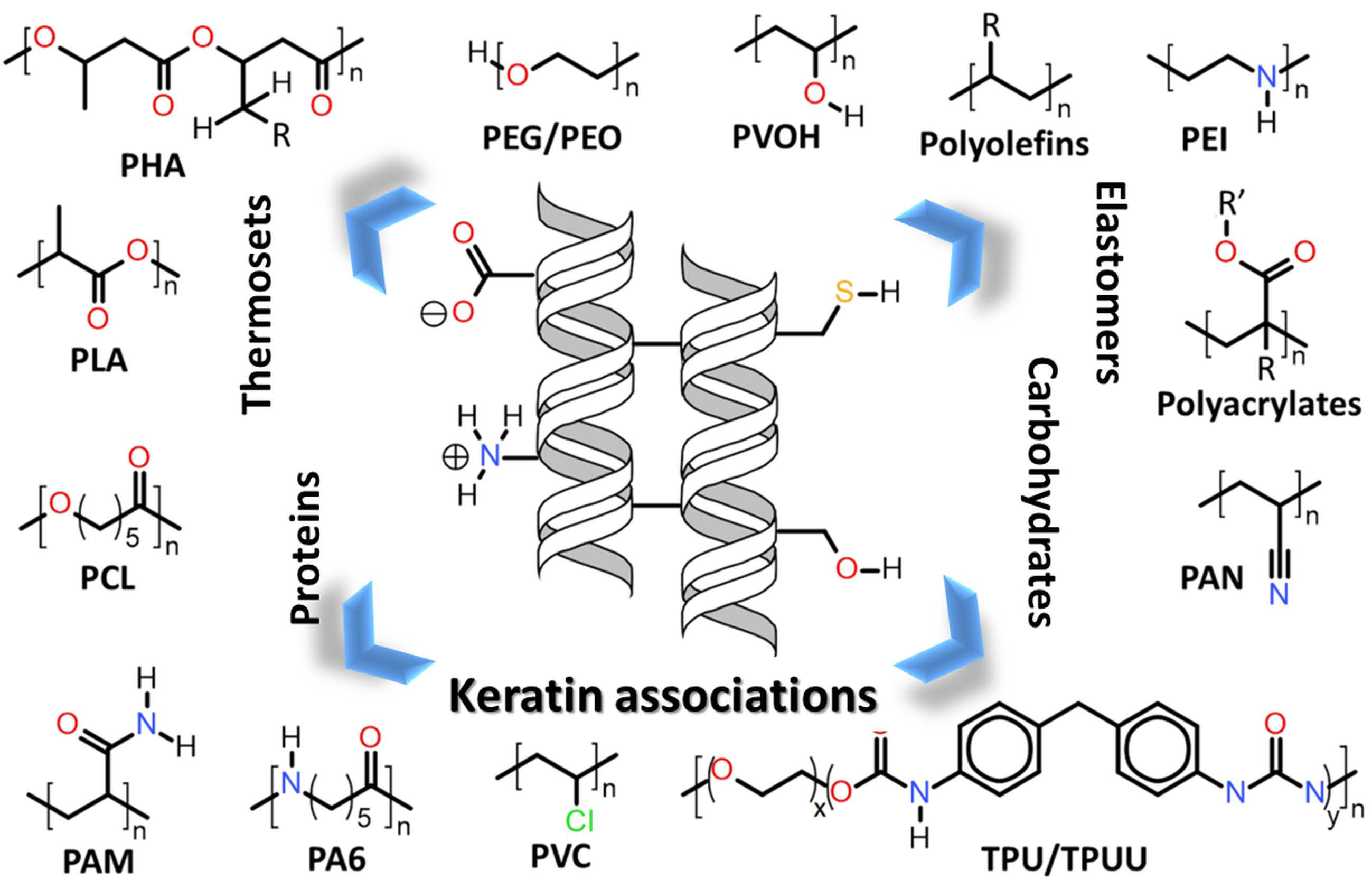
| 7. | Keratin associations with other polymers |
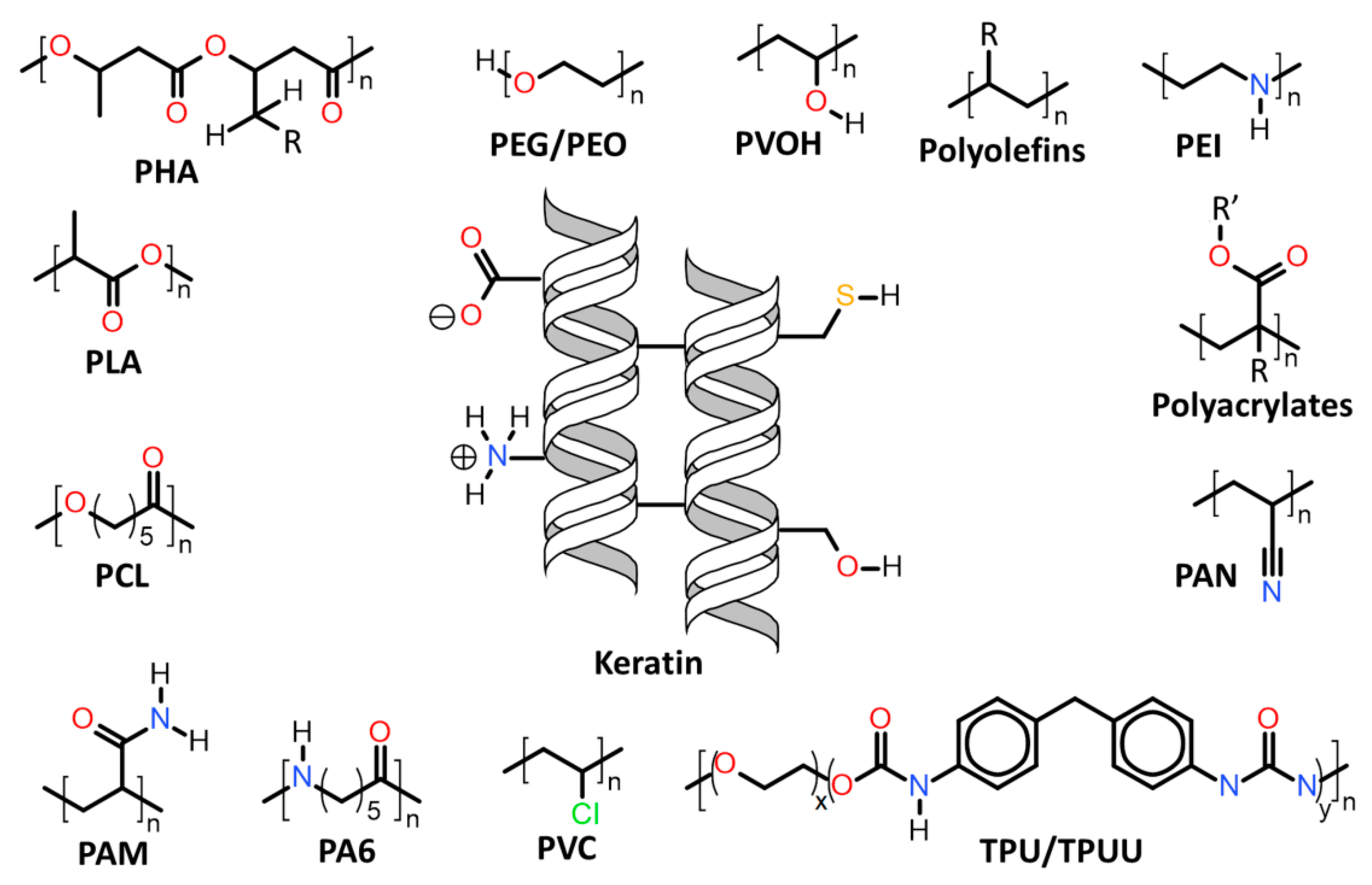
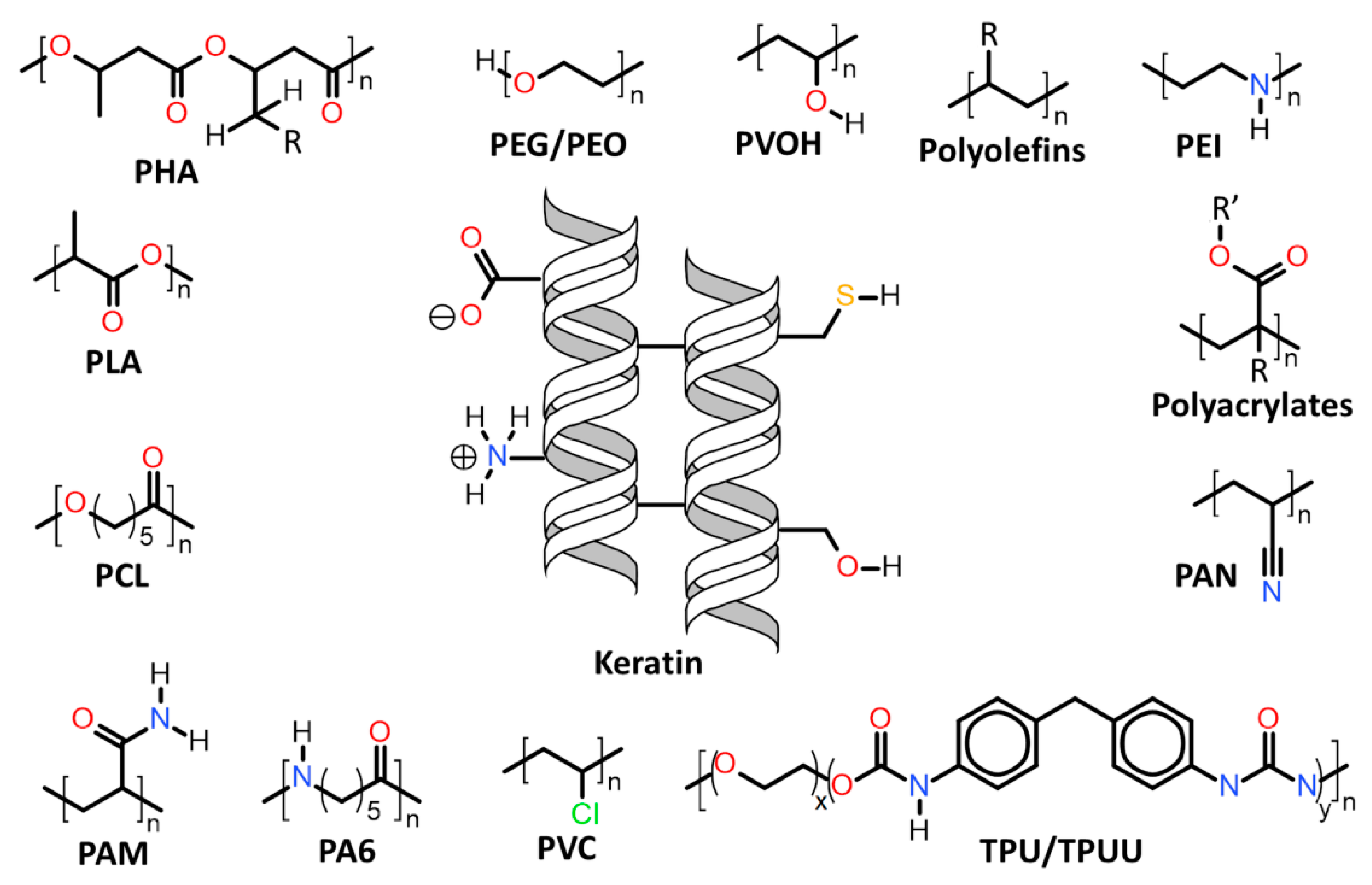
| 7.1. | Keratin associations with synthetic and biosynthetic thermoplastics |
| 7.1.1. | Polyolefins |
| 7.1.2. | Polyethylene glycol (PEG) and Polyethylene oxide (PEO) |
| 7.1.3. | Poly(ethylene imide) (PEI) |
| 7.1.4. | Polyacrylates, polyacrylonitrile (PAN) and polyacrylamide (PAM) |
| 7.1.5. | Polyvinyl chloride (PVC) |
| 7.1.6. | Polyvinyl alcohol (PVOH) |
| 7.1.7. | Polyamide-6 (PA6) |
| 7.1.8. | ε-Polycaprolactone (PCL) |
| 7.1.9. | Polylactic acid (PLA) |
| 7.1.10. | Polyhydroxyalkanoates (PHA) |
| 7.1.11. | Thermoplastic polyurethanes (TPU) and polyurea-urethanes (TPUU) |
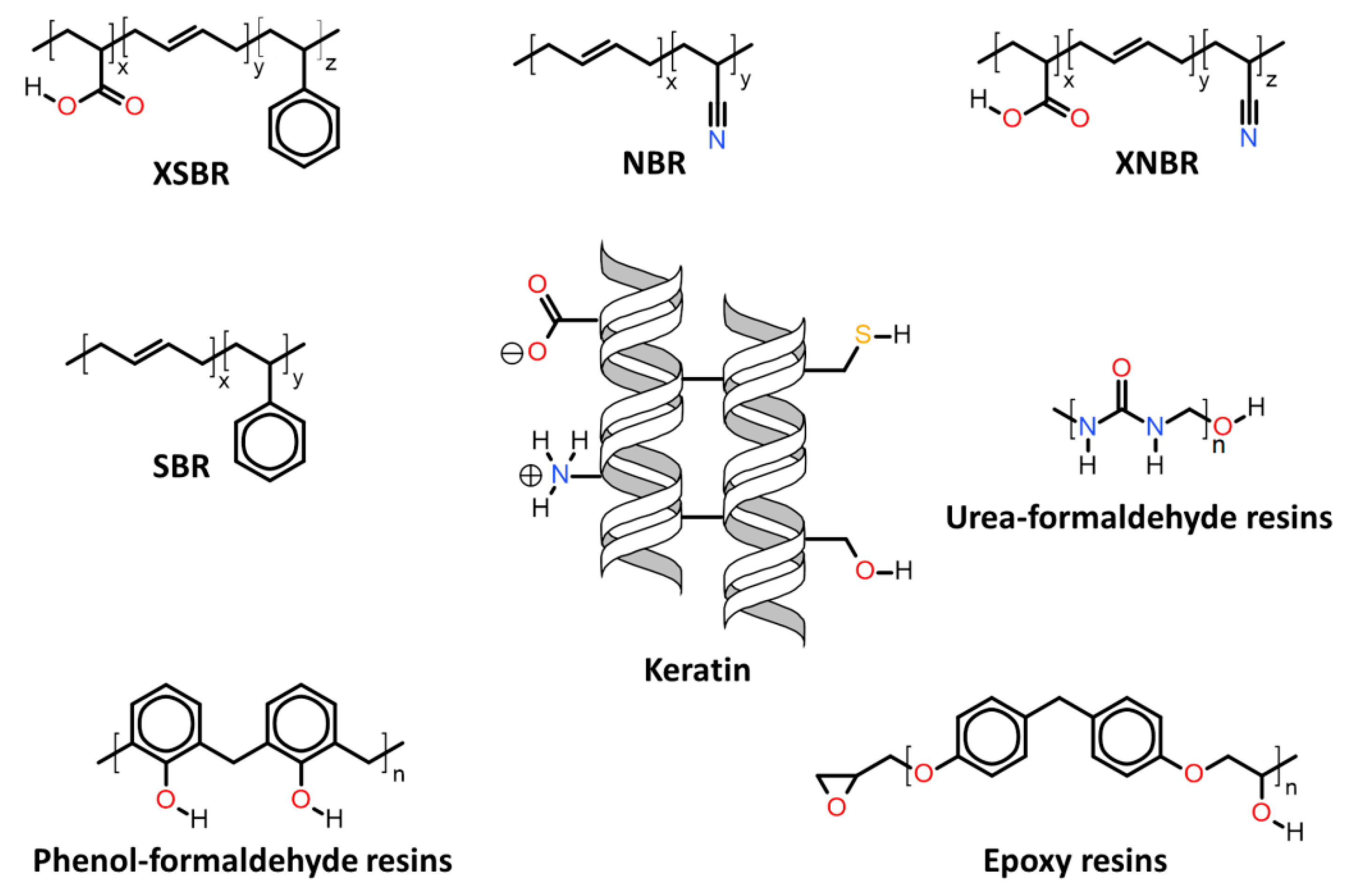
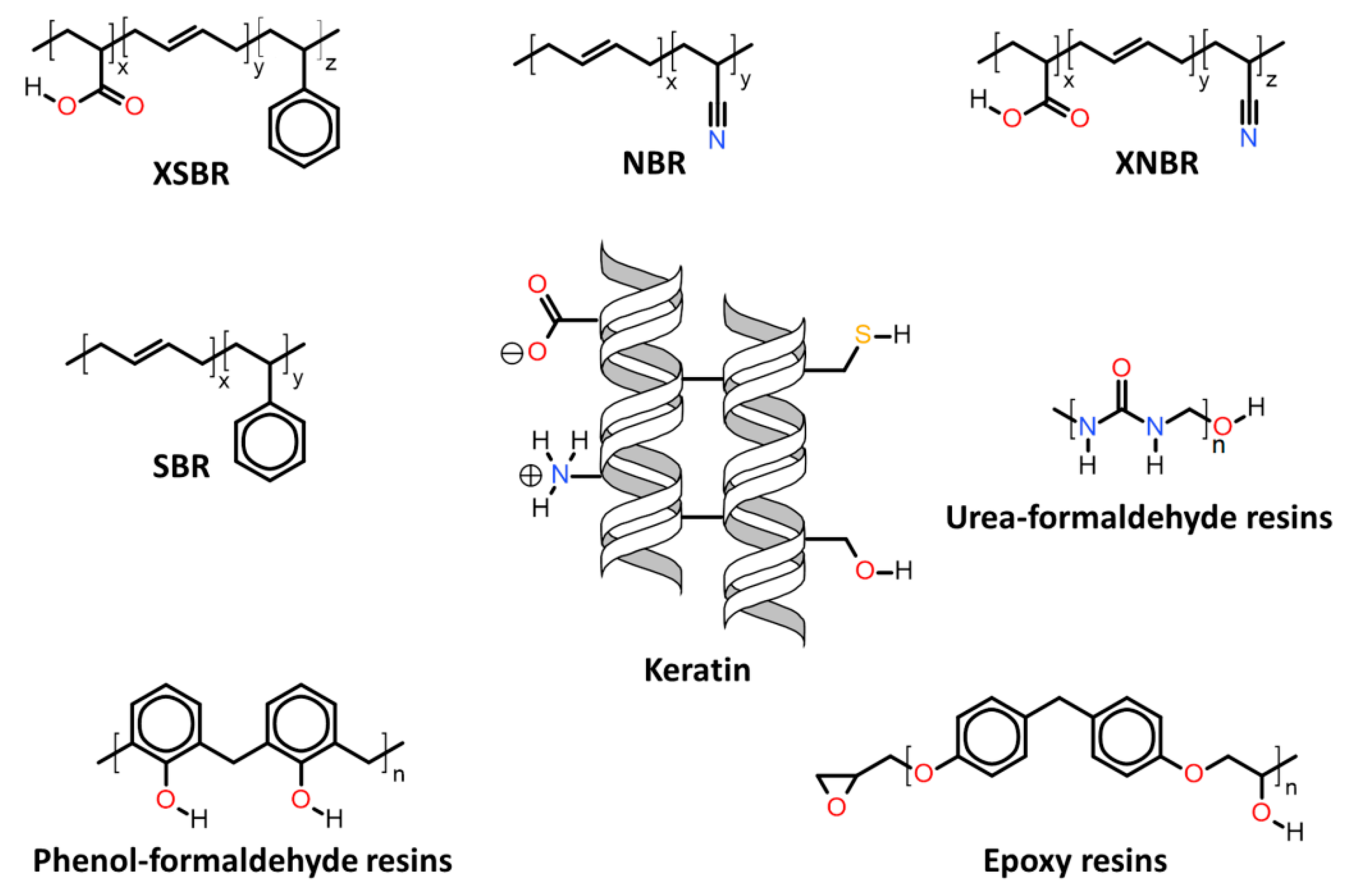
| 7.2. | Keratin associations with elastomers and thermosets |
| 7.2.1. | Butadiene copolymer rubbers |
| 7.2.2. | Epoxy resins |
| 7.2.3. | Urea-formaldehyde resins |
| 7.2.4. | Phenol-formaldehyde resins |
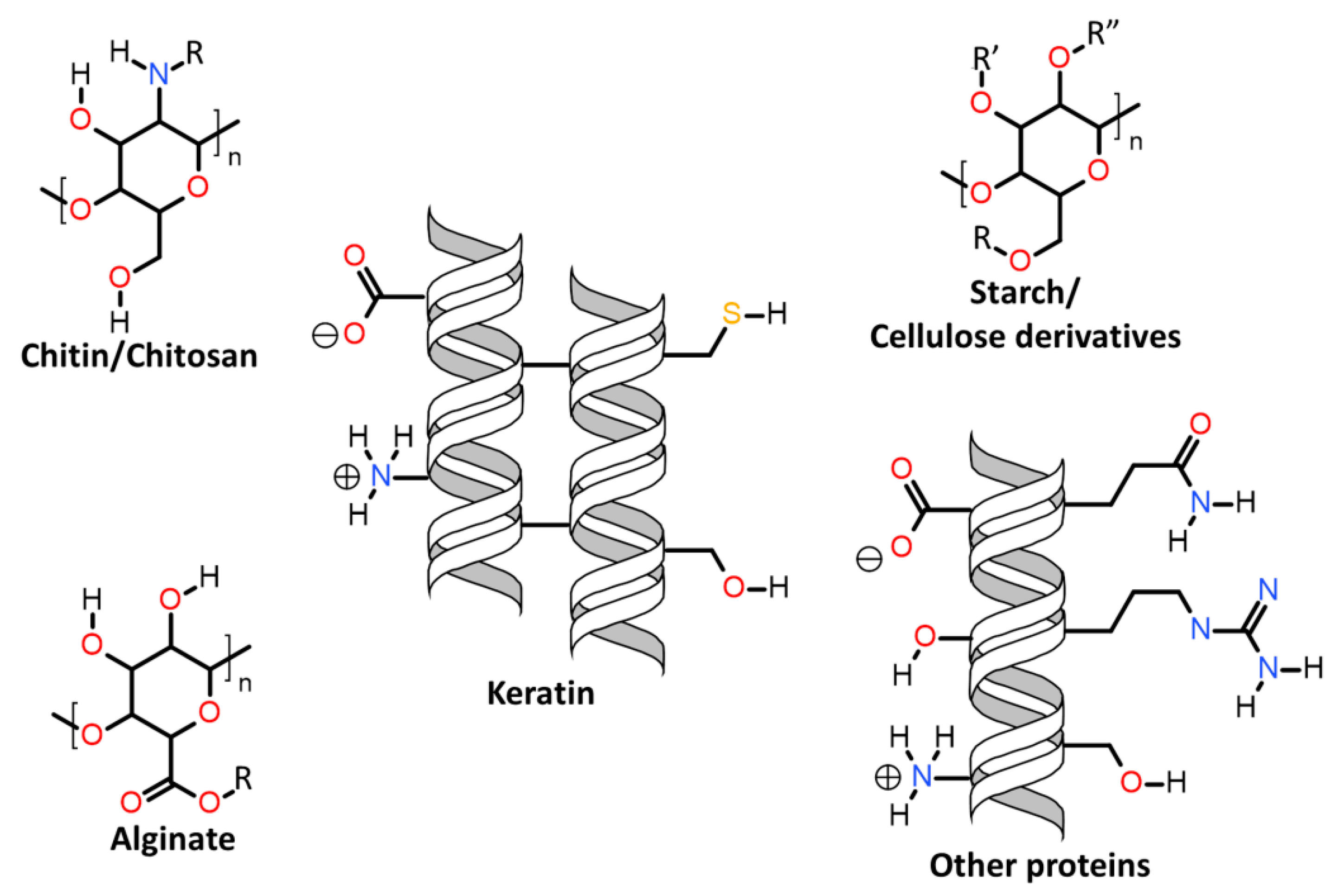
| 7.3. | Keratin associations with natural polymers and fibres |
| 7.3.1. | Keratin associations with carbohydrates |
| 7.3.1.1. | Cellulose |
| 7.3.1.2. | Chitosan |
| 7.3.1.3. | Alginate |
| 7.3.1.4. | Starch |
| 7.3.2. | Keratin association with other proteins |
| 7.3.2.1. | Collagen and gelatine |
| 7.3.2.2. | Soy and wheat protein |
| 7.3.2.3. | Silk fibroin |
| 7.3.2.4. | Associations between different keratin sources |
| 8. | Conclusions and outlook |
| Author Contributions | |
| Conflicts of Interest | |
| References | |
1. Brief Historical Context of Knowledge and Use of Keratin-Based Materials
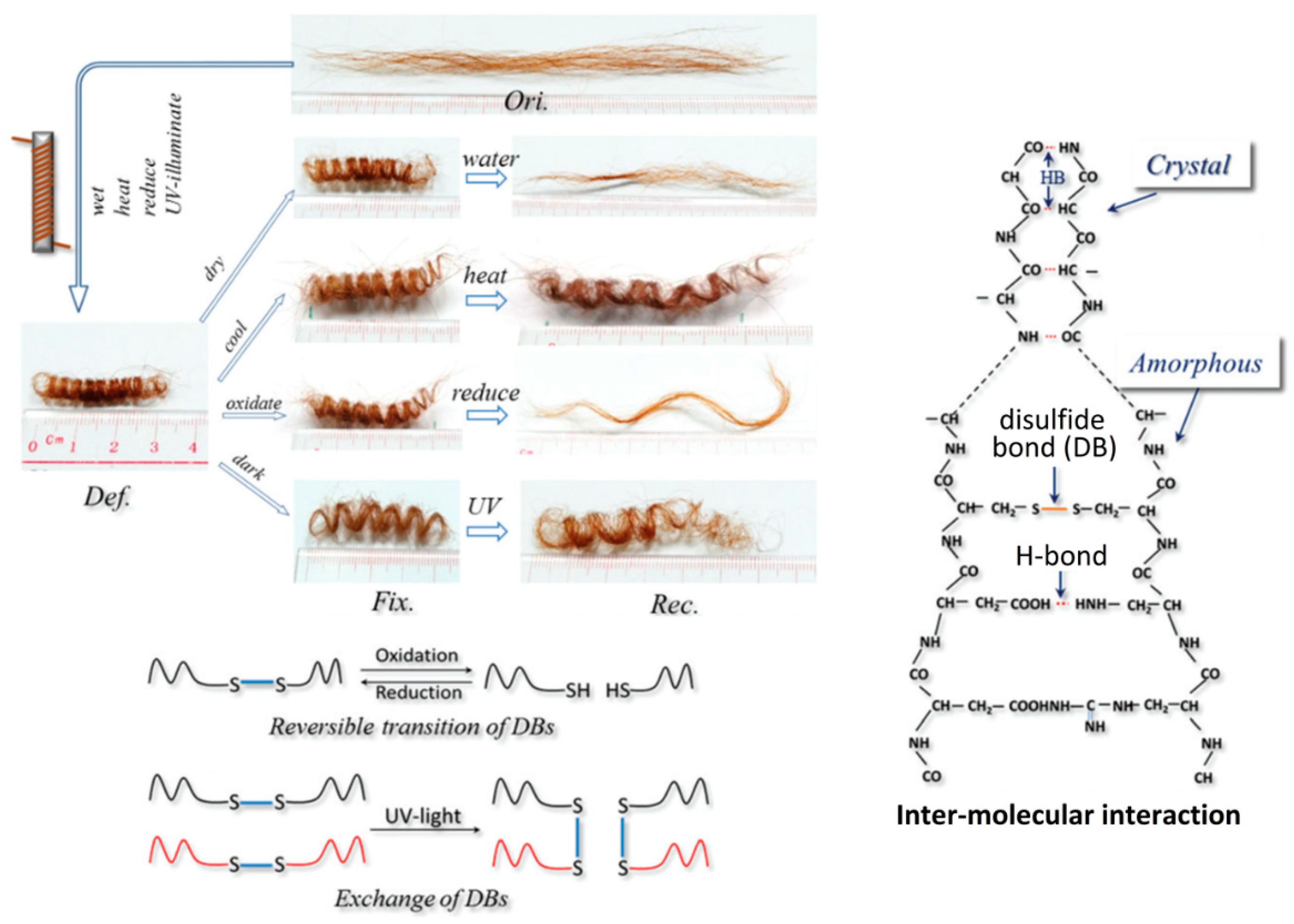
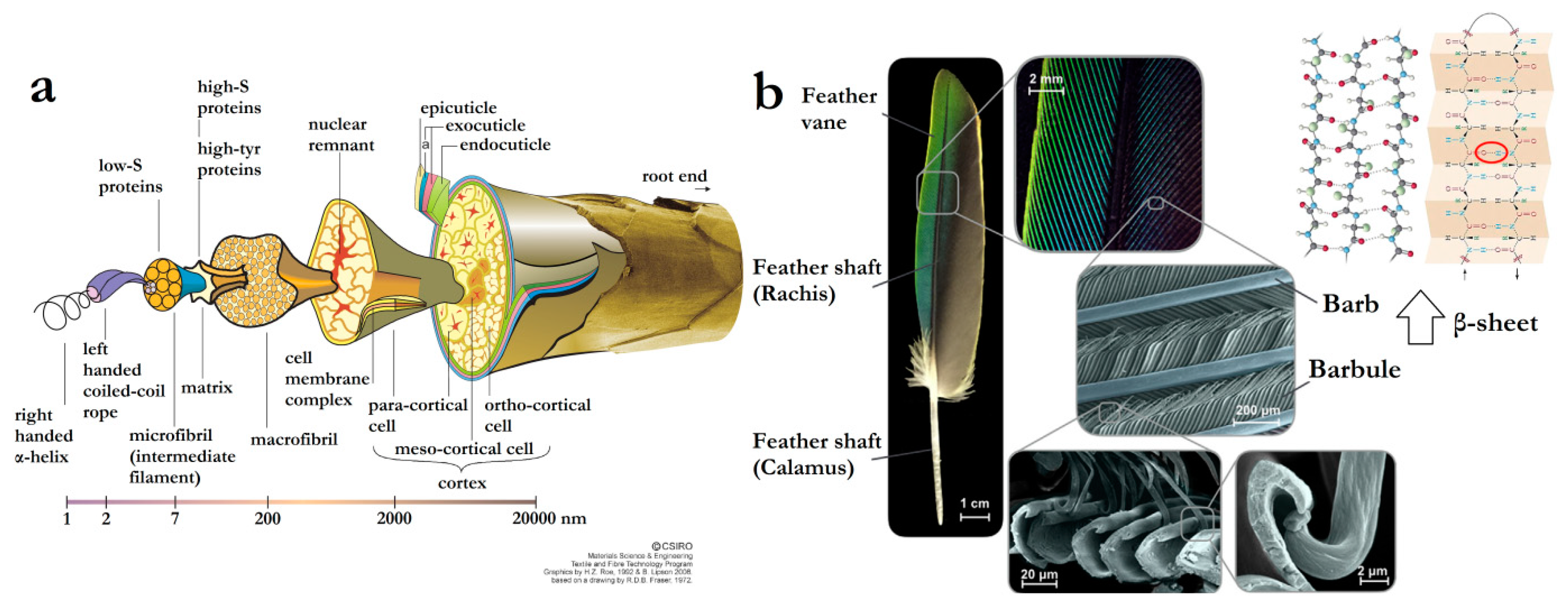
2. Keratin’s Structure and Chemical Toolset
3. Sustainability and Safety Assessment
4. Keratins Extraction
4.1. Oxidative and Reductive Extraction
4.2. Steam Explosion Extraction
4.3. Extraction with Ionic Liquids and Deep Eutectic Solvents
5. Keratin Sources and Their Distinctions
6. Keratin-Based Biomaterials
7. Keratin Associations with Other Polymers
7.1. Keratin Associations with Synthetic and Biosynthetic Thermoplastics
7.1.1. Polyolefins
7.1.2. Polyethylene Glycol (PEG) and Polyethylene Oxide (PEO)
7.1.3. Poly(ethylene imide) (PEI)
7.1.4. Polyacrylates, Polyacrylonitrile (PAN) and Polyacrylamide (PAM)
7.1.5. Polyvinyl Chloride (PVC)
7.1.6. Polyvinyl Alcohol (PVOH)
7.1.7. Polyamide-6 (PA6)
7.1.8. ε-Polycaprolactone (PCL)
7.1.9. Polylactic Acid (PLA)
7.1.10. Polyhydroxyalkanoates (PHA)
7.1.11. Thermoplastic Polyurethanes (TPU) and Polyurea-Uretanes (TPUU)
7.2. Keratin Associations with Elastomers and Thermosets
7.2.1. Butadiene Copolymer Rubbers
7.2.2. Epoxy Resins
7.2.3. Urea-Formaldehyde Resin
7.2.4. Phenol-Formaldehyde Resins
7.3. Keratin Associations with Natural Polymers and Fibres
7.3.1. Keratin Associations with Carbohydrates
7.3.1.1. Cellulose
7.3.1.2. Chitosan
7.3.1.3. Alginate
7.3.1.4. Starch
7.3.2. Keratin Association with Other Proteins
7.3.2.1. Collagen and Gelatine
7.3.2.2. Soy and Wheat Protein
7.3.2.3. Silk Fibroin
7.3.2.4. Associations between Different Keratin Sources
8. Summary and Outlook
Author Contributions
Conflicts of Interest
References
- Gupta, R.; Ramnani, P. Microbial keratinases and their prospective applications: An overview. Appl. Microbiol. Biotechnol. 2006, 70, 21. [Google Scholar] [CrossRef] [PubMed]
- Thys, R.C.S.; Brandelli, A. Purification and properties of a keratinolytic metalloprotease from Microbacterium sp. J. Appl. Microbiol. 2006, 101, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Unesco Memory Of The World Register. In Ben Cao Gang Mu; Compendium of Materia Medica (P.R. China); 2010.
- Bragulla, H.H.; Homberger, D.G. Structure and functions of keratin proteins in simple, stratified, keratinized and cornified epithelia. J. Anat. 2009, 214, 516–559. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.D.B.; MacRae, T.P.; Rogers, G.E. Keratins: Their Composition, Structure, and Biosyntahesis; Charles, C., Ed.; Thomas: Springfield, IL, USA, 1972; ISBN 0398022836. [Google Scholar]
- Moll, R.; Franke, W.W.; Schiller, D.L.; Geiger, B.; Krepler, R. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982, 31, 11–24. [Google Scholar] [CrossRef]
- Schweizer, J.; Bowden, P.E.; Coulombe, P.A.; Langbein, L.; Lane, E.B.; Magin, T.M.; Maltais, L.; Omary, M.B.; Parry, D.A.D.; Rogers, M.A.; et al. New consensus nomenclature for mammalian keratins. J. Cell Biol. 2006, 174, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D. The discovery of the structural features of proteins alpha-helix and beta-sheet, the principal. Proc. Natl. Acad. Sci. USA 2003, 100, 11207–11210. [Google Scholar] [CrossRef] [Green Version]
- Rafik, M.E.; Doucet, J.; Briki, F. The intermediate filament architecture as determined by X-ray diffraction modeling of hard α-keratin. Biophys. J. 2004, 86, 3893–3904. [Google Scholar] [CrossRef] [Green Version]
- Goddard, D.R.; Michaelis, L.J. Derivatives of Keratin. J. Biol. Chem. 1935, 112, 361–371. [Google Scholar]
- Rivett, D.E.; Ward, S.W.; Belkin, L.M.; Ramshaw, J.A.M.; Wilshire, J.F.K. Keratin and Wool Research. In The Lennox Legacy; CSIRO Publishing: Collingwood, Australia, 1996. [Google Scholar]
- Crewther, W.G.; Fraser, R.D.B.; Lennox, F.G.; Lindley, H. The Chemistry of Keratins. In Advances in Protein Chemistry; Anfinsen, C.B., Anson, M.L., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Cambridge, MA, USA, 1965; Volume 20, pp. 191–346. [Google Scholar]
- Jarman, T.; Light, J. Prospects for novel biomaterials development. In World Biotech Report; 1985; p. 505. [Google Scholar]
- Brown, C.H. Keratins in Invertebrates. Nature 1950, 166, 439. [Google Scholar] [CrossRef]
- Makar, I.A.; Havryliak, V.V.; Sedilo, H.M. Genetic and biochemical aspects of keratin synthesis by hair follicles. Tsitologiia I Genetika 2007, 41, 75–79. [Google Scholar] [CrossRef]
- Fuchs, E. Evolution and Complexity of the Genes Encoding the Keratins of Human Epidermal Cells. J. Investig. Dermatol. 1983, 81, S141–S144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinert, P.M.; Parry, D.A.D.; Racoosin, E.L.; Idler, W.W.; Steven, A.C.; Trus, B.L.; Roop, D.R. The complete cDNA and deduced amino acid sequence of a type II mouse epidermal keratin of 60,000 Da: Analysis of sequence differences between type I and type II keratins. Proc. Natl. Acad. Sci. USA 1984, 81, 5709–5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulombe, P.A.; Omary, M.B. Hard and soft principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 2002, 14, 110–122. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Bousquet, O.; Ma, L.; Yamada, S.; Wirtz, D. The ins and outs of intermediate filament organization. Trends Cell Biol. 2000, 10, 420–428. [Google Scholar] [CrossRef]
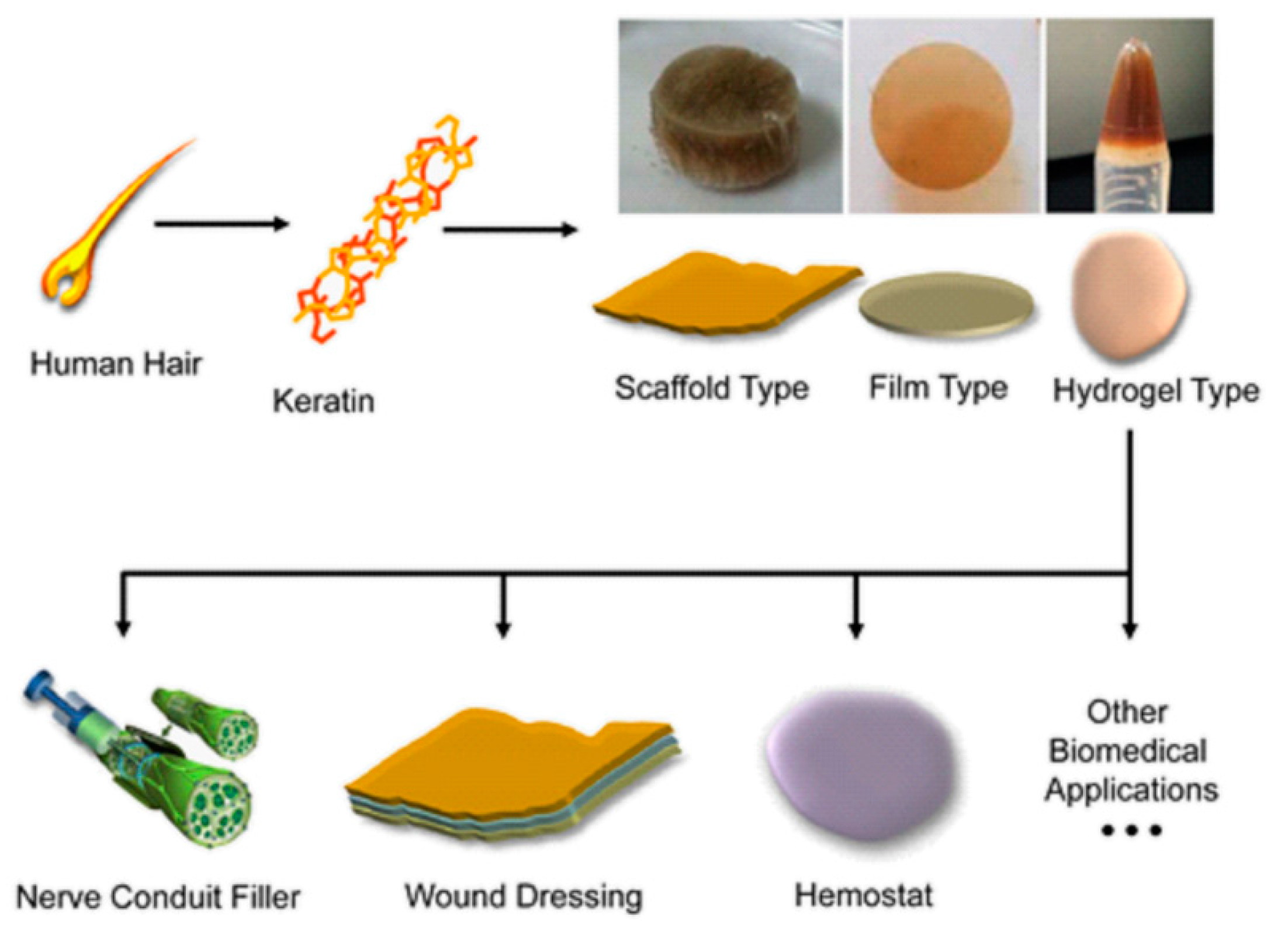
- Rouse, J.G.; Van Dyke, M.E. A review of keratin-based biomaterials for biomedical applications. Materials 2010, 3, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Mizuguchi, R.; Tanaka, K.; Dorman, M. Palmoplantar keratoderma Voerner with composite keratohyalin granules Studies on keratinization parameters and ultrastructures. J. Dermatol. 2000, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Strasser, B.; Mlitz, V.; Hermann, M.; Tschachler, E.; Eckhart, L. Convergent evolution of cysteine-rich proteins in feathers and hair. BMC Evol. Biol. 2015, 15, 82. [Google Scholar] [CrossRef] [Green Version]
- Strelkov, S.V.; Herrmann, H.; Aebi, U. Molecular architecture of intermediate filaments. BioEssays 2003, 25, 243–251. [Google Scholar] [CrossRef]
- Kreplak, L.; Bär, H.; Leterrier, J.F.; Herrmann, H.; Aebi, U. Exploring the mechanical behavior of single intermediate filaments. J. Mol. Biol. 2005, 354, 569–577. [Google Scholar] [CrossRef]
- Fudge, D.S.; Gosline, J.M. Molecular design of the α–keratin composite: Insights from a matrix–free model, hagfish slime threads. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2004, 271, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Guthold, M.; Liu, W.; Sparks, E.A.; Jawerth, L.M.; Peng, L.; Falvo, M.; Superfine, R.; Hantgan, R.R.; Lord, S.T. A Comparison of the Mechanical and Structural Properties of Fibrin Fibers with Other Protein Fibers. Cell Biochem. Biophys. 2007, 49, 165–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hearle, J.W.S. A critical review of the structural mechanics of wool and hair fibres. Int. J. Biol. Macromol. 2000, 27, 123–138. [Google Scholar] [CrossRef]
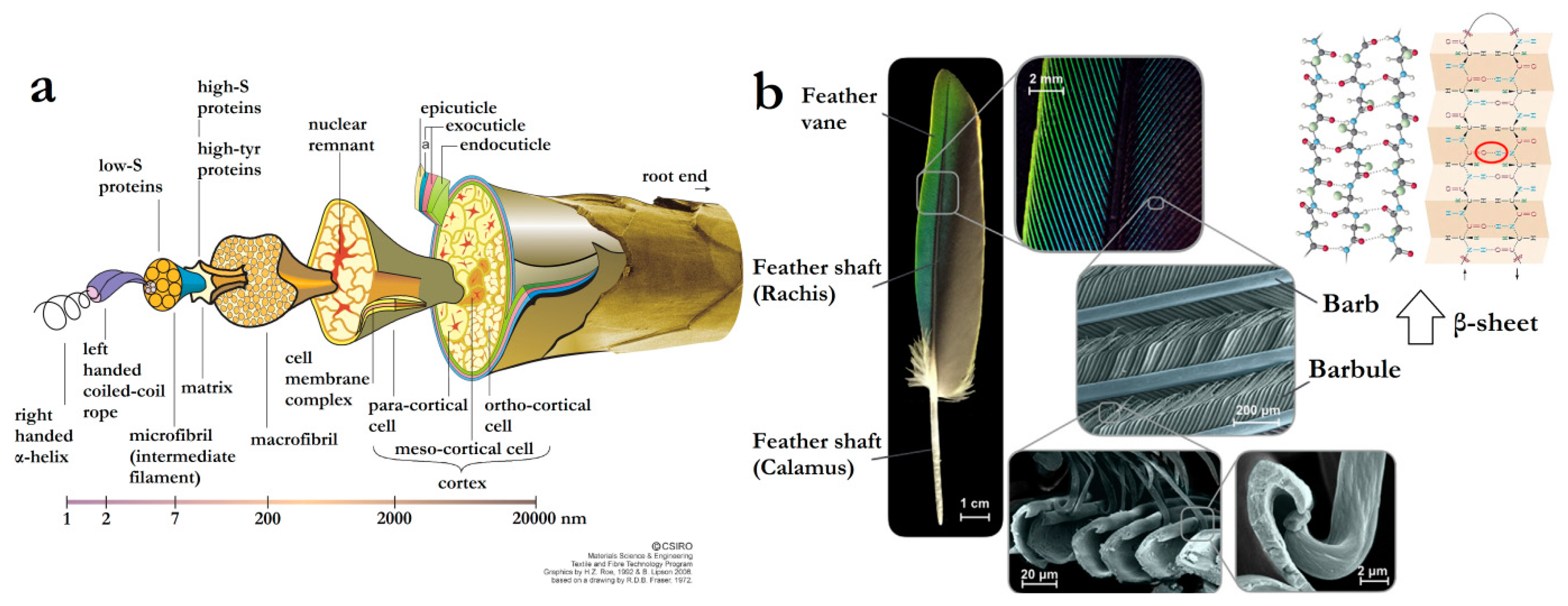
- Fraser, R.D.B.; Parry, D.A.D. Molecular packing in the feather keratin filament. J. Struct. Biol. 2008, 162, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Kim, M.-S.; Chung, B.M.; Leahy, D.J.; Coulombe, P.A. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 2012, 19, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Hu, J.; Gui, X.; Lu, J.; Luo, H. Is biopolymer hair a multi-responsive smart material? Polym. Chem. 2017, 8, 283–294. [Google Scholar] [CrossRef]
- Zhan, M.; Wool, R.P. Mechanical properties of chicken feather fibers. Polym. Compos. 2011, 32, 937–944. [Google Scholar] [CrossRef]
- Europe, P. An Analysis of European Plastics Production, Demand and Waste Data. Plast. Fact 2018, 18–57. [Google Scholar]
- Neufeld, L.; Stassen, F.; Sheppard, R.; Gilman, T. The New Plastics Economy: Rethinking the Future of Plastics; In World Economic Forum: Colony, Switzerland, 2016. [Google Scholar]
- Jambeck, J.R.; Geyer, R.; Wilcox, C.; Siegler, T.R.; Perryman, M.; Andrady, A.; Narayan, R.; Law, K.L. Plastic waste inputs from land into the ocean. Science 2015, 347, 768–771. [Google Scholar] [CrossRef]
- Cole, M.; Lindeque, P.; Halsband, C.; Galloway, T.S. Microplastics as contaminants in the marine environment: A review. Mar. Pollut. Bull. 2011, 62, 2588–2597. [Google Scholar] [CrossRef]
- Weithmann, N.; Möller, J.N.; Löder, M.G.J.; Piehl, S.; Laforsch, C.; Freitag, R. Organic fertilizer as a vehicle for the entry of microplastic into the environment. Sci. Adv. 2018, 4, 8060. [Google Scholar] [CrossRef] [Green Version]
- Van Cauwenberghe, L.; Janssen, C.R. Microplastics in bivalves cultured for human consumption. Environ. Pollut. 2014, 193, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Werlang, P.O.; Brandelli, A. Characteriztion of a novel feather-degrading Bacillus sp. strain. Appl. Biochem. Biotechnol. 2005, 120, 71–79. [Google Scholar] [CrossRef]
- Kanoksilapatham, W.; Intagun, W. A Review: Biodegradation and Applications of Keratin Degrading Microorganisms and Keratinolytic Enzymes, Focusing on Thermophiles and Thermostable Serine Proteases. Am. J. Appl. Sci. 2017, 14, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Korniłłowicz-Kowalska, T.; Bohacz, J. Biodegradation of keratin waste: Theory and practical aspects. Waste Manag. 2011, 31, 1689–1701. [Google Scholar] [CrossRef]
- Bohacz, J. Biodegradation of feather waste keratin by a keratinolytic soil fungus of the genus Chrysosporium and statistical optimization of feather mass loss. World J. Microbiol. Biotechnol. 2016, 33, 13. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, B.; Nagarajan, R.; Senthilkumar, P.; Thangamani, R.; Noorthen, A. Field study of Chicken feather waste open dumping on road sides of Tuticorin city, Tamilnadu, India. Int. J. Curr. Sci. Res. 2016, 2, 960–966. [Google Scholar]
- Joardar, J.C.; Rahman, M.M. Poultry feather waste management and effects on plant growth. Int. J. Recycl. Org. Waste Agric. 2018, 7, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Khosa, M.; Ullah, A. A sustainable role of keratin biopolymer in green chemistry: A review. J Food Process. Beverages 2013, 1, 8. [Google Scholar]
- Scarfato, P.; Di Maio, L.; Incarnato, L. Recent advances and migration issues in biodegradable polymers from renewable sources for food packaging. J. Appl. Polym. Sci. 2015, 132, 42597. [Google Scholar] [CrossRef]
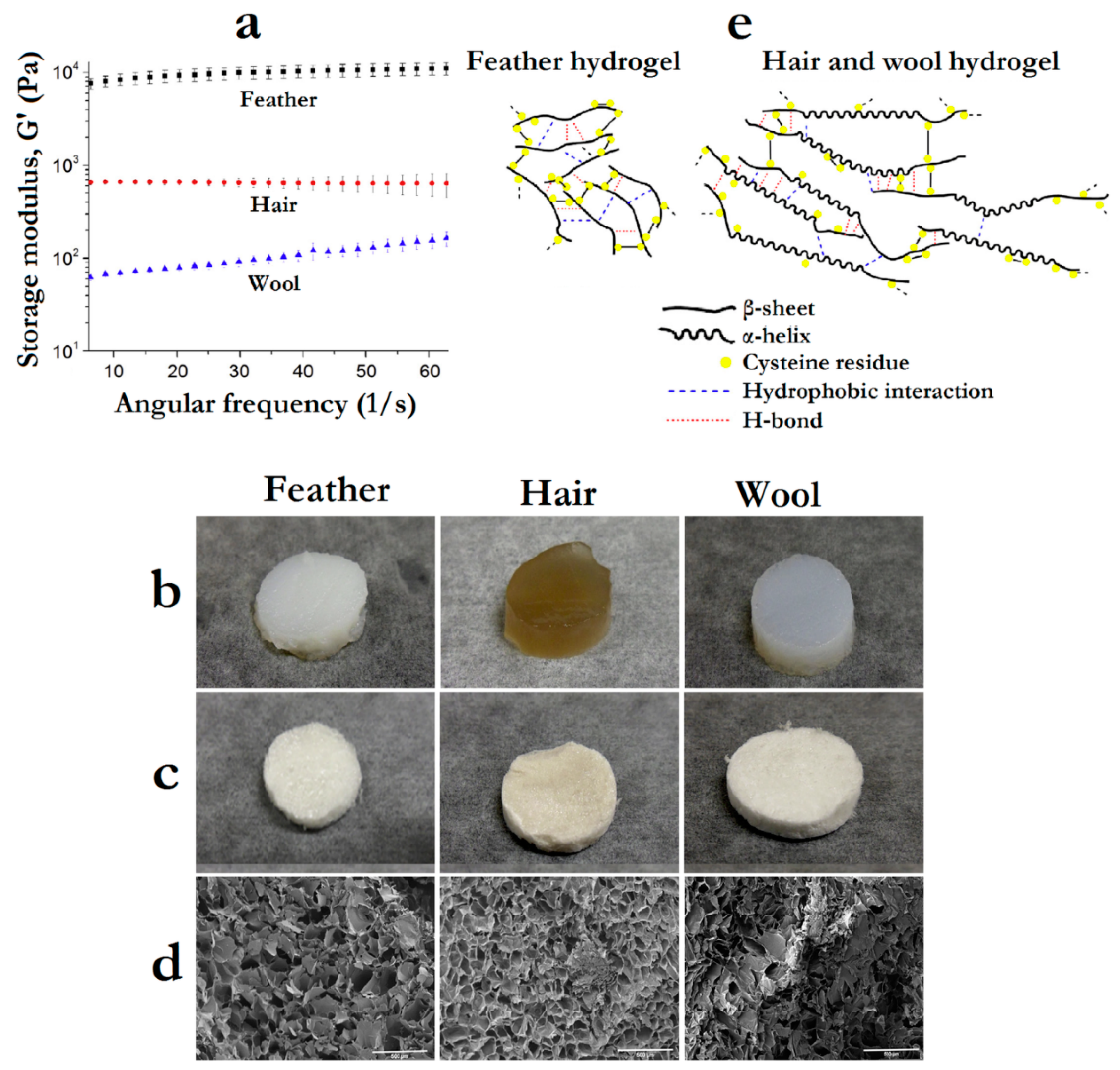
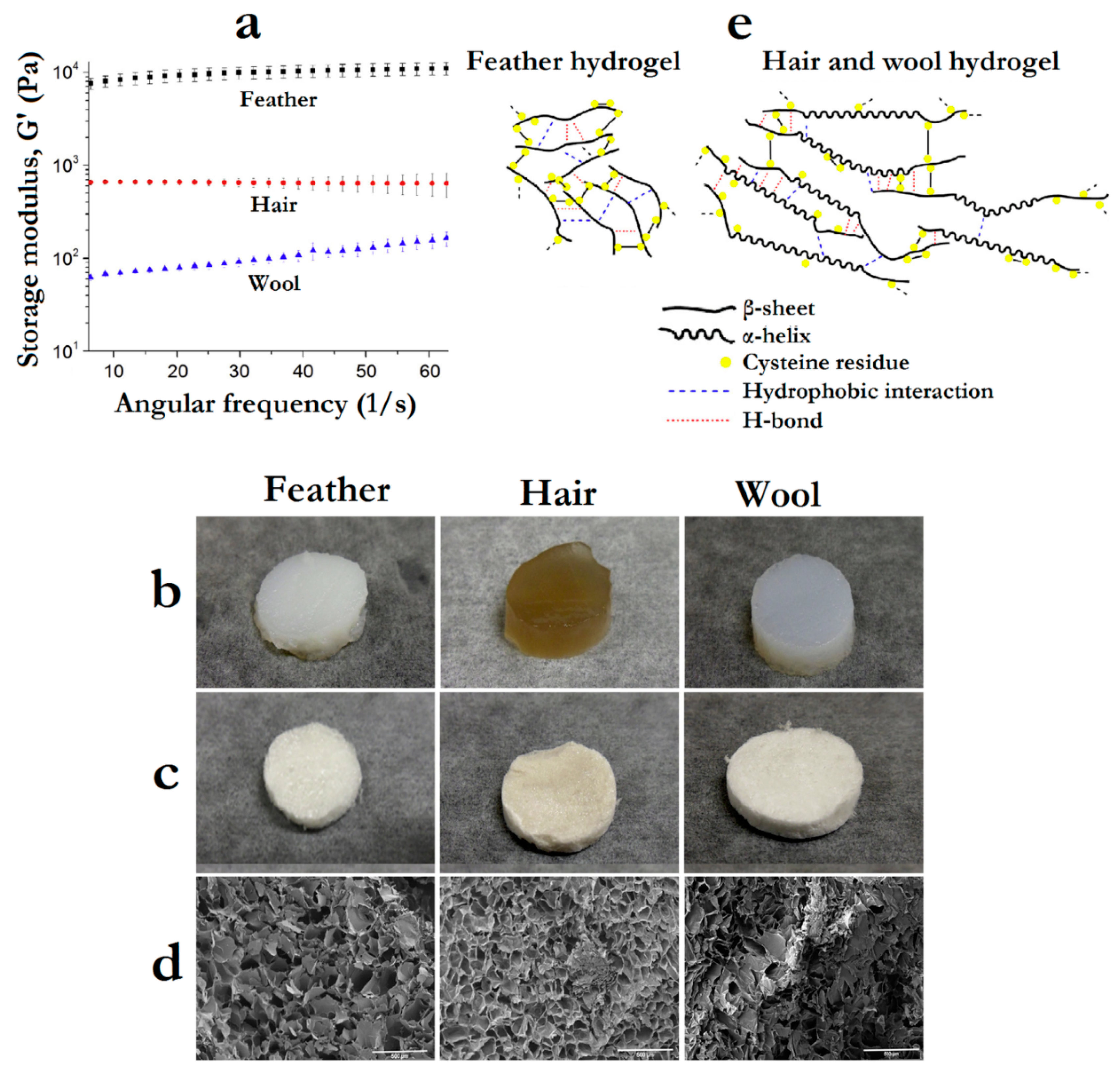
- Esparza, Y.; Bandara, N.; Ullah, A.; Wu, J. Hydrogels from feather keratin show higher viscoelastic properties and cell proliferation than those from hair and wool keratins. Mater. Sci. Eng. C 2018, 90, 446–453. [Google Scholar] [CrossRef]
- Shavandi, A.; Silva, T.H.; Bekhit, A.A.; Bekhit, A.E.D.A. Keratin: Dissolution, extraction and biomedical application. Biomater. Sci. 2017, 5, 1699–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmeier, J. Horn-lime plastic masses from keratin substances. Ger. Pat. 1905, 18, DE184915. [Google Scholar]
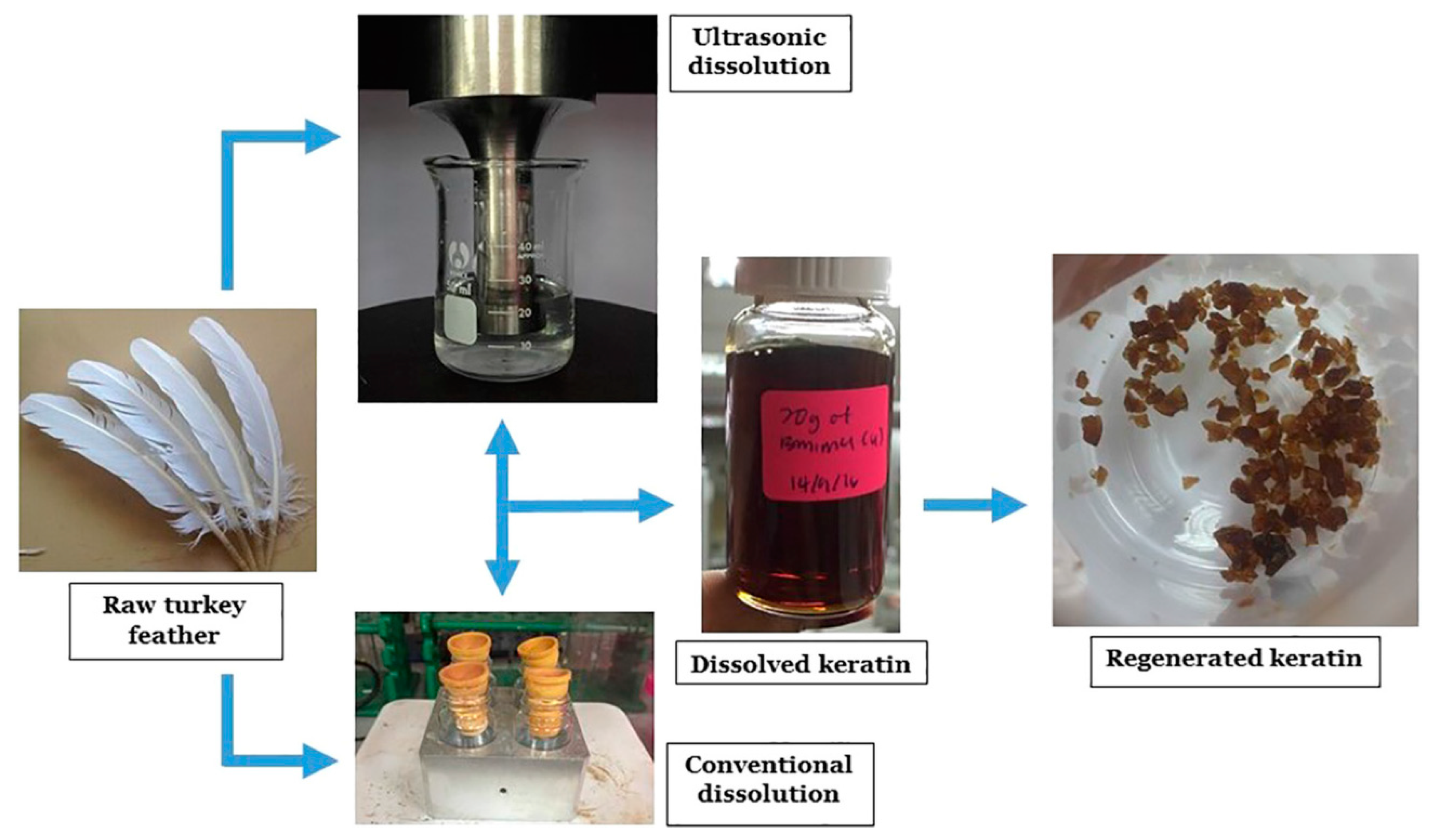
- Shavandi, A.; Carne, A.; Bekhit, A.A.; Bekhit, A.E.D.A. An improved method for solubilisation of wool keratin using peracetic acid. J. Environ. Chem. Eng. 2017, 5, 1977–1984. [Google Scholar] [CrossRef]
- Sinkiewicz, I.; Śliwińska, A.; Staroszczyk, H.; Kołodziejska, I. Alternative Methods of Preparation of Soluble Keratin from Chicken Feathers. Waste Biomass Valoriz. 2017, 8, 1043–1048. [Google Scholar] [CrossRef]
- Gupta, A.; Kamarudin, N.B.; Kee, C.Y.G.; Yunus, R.B.M. Extraction of Keratin Protein from Chicken Feather. J. Chem. Chem. Eng. 2012, 732–737. [Google Scholar]
- Fujii, T.; Li, D. Preparation and properties of protein films and particles from chicken feather. J. Biol. Macromol. 2008, 8, 48–55. [Google Scholar]
- Xu, W.; Ke, G.; Wu, J.; Wang, X. Modification of wool fiber using steam explosion. Eur. Polym. J. 2006, 42, 2168–2173. [Google Scholar] [CrossRef] [Green Version]
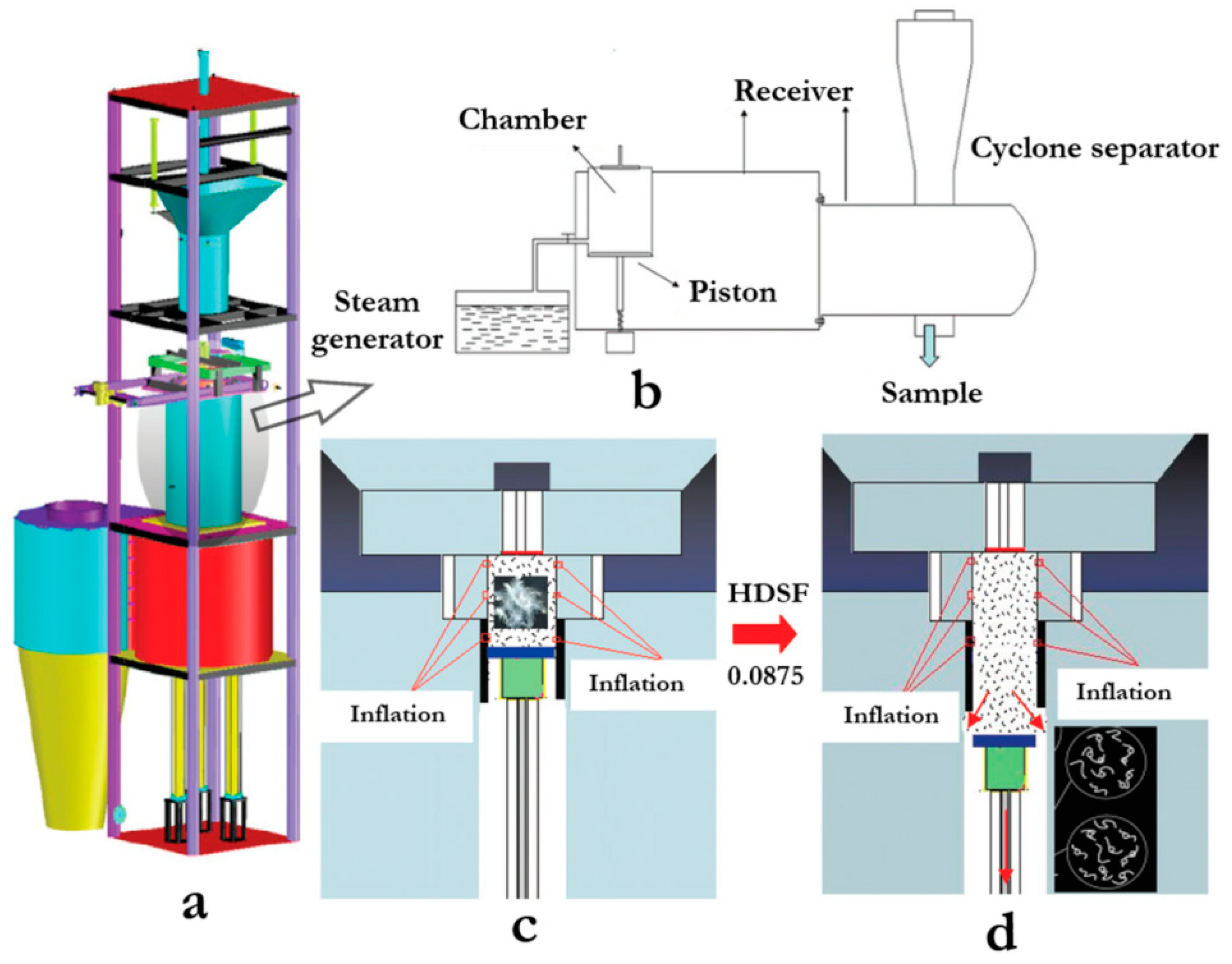
- Zhao, W.; Yang, R.; Zhang, Y.; Wu, L. Sustainable and practical utilization of feather keratin by an innovative physicochemical pretreatment: High density steam flash-explosion. Green Chem. 2012, 14, 3352–3360. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, B.; Yu, F.; Xu, G.; Song, A. A real explosion: The requirement of steam explosion pretreatment. Bioresour. Technol. 2012, 121, 335–341. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Yang, R. Steam Flash Explosion Assisted Dissolution of Keratin from Feathers. ACS Sustain. Chem. Eng. 2015, 3, 2036–2042. [Google Scholar] [CrossRef]
- Dupont, J. On the solid, liquid and solution structural organization of imidazolium ionic liquids. J. Braz. Chem. Soc. 2004, 15, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, H.; Moniruzzaman, M.; Yusup, S.; Welton, T. Ionic liquids assisted processing of renewable resources for the fabrication of biodegradable composite materials. Green Chem. 2017, 19, 2051–2075. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, D. Preparation of regenerated wool keratin films from wool keratin-ionic liquid solutions. J. Appl. Polym. Sci. 2013, 127, 2648–2653. [Google Scholar] [CrossRef]
- Chen, J.; Vongsanga, K.; Wang, X.; Byrne, N. What happens during natural protein fibre dissolution in ionic liquids. Materials 2014, 7, 6158–6168. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Cao, X.J. Extracting keratin from chicken feathers by using a hydrophobic ionic liquid. Process Biochem. 2012, 47, 896–899. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, J.; Lv, J.; Li, Z.; Xing, L.; Ding, S. Extraction of keratin with ionic liquids from poultry feather. Sep. Purif. Technol. 2014, 132, 577–583. [Google Scholar] [CrossRef]
- Sun, P.; Liu, Z.T.; Liu, Z.W. Particles from bird feather: A novel application of an ionic liquid and waste resource. J. Hazard. Mater. 2009, 170, 786–790. [Google Scholar] [CrossRef]
- Zheng, S.; Nie, Y.; Zhang, S.; Zhang, X.; Wang, L. Highly Efficient Dissolution of Wool Keratin by Dimethylphosphate Ionic Liquids. ACS Sustain. Chem. Eng. 2015, 3, 2925–2932. [Google Scholar] [CrossRef]
- Idris, A.; Vijayaraghavan, R.; Rana, U.A.; Fredericks, D.; Patti, A.F.; MacFarlane, D.R. Dissolution of feather keratin in ionic liquids. Green Chem. 2013, 15, 525–534. [Google Scholar] [CrossRef]
- Liu, X.; Nie, Y.; Meng, X.; Zhang, Z.; Zhang, X.; Zhang, S. DBN-based ionic liquids with high capability for the dissolution of wool keratin. RSC Adv. 2017, 7, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Azmi, N.A.; Idris, A.; Yusof, N.S.M. Ultrasonic technology for value added products from feather keratin. Ultrason. Sonochem. 2018, 47, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yuan, J.; Wang, P.; Fan, X.; Xu, J.; Wang, Q.; Zhang, L. Dissolution and regeneration of wool keratin in the deep eutectic solvent of choline chloride-urea. Int. J. Biol. Macromol. 2018, 119, 423–430. [Google Scholar] [CrossRef]
- Moore, K.E.; Mangos, D.N.; Slattery, A.D.; Raston, C.L.; Boulos, R.A. Wool deconstruction using a benign eutectic melt. RSC Adv. 2016, 6, 20095–20101. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tang, R.C. Dissolution of wool in the choline chloride/oxalic acid deep eutectic solvent. Mater. Lett. 2018, 231, 217–220. [Google Scholar] [CrossRef]
- Wang, D.; Yang, X.H.; Tang, R.C.; Yao, F. Extraction of keratin from rabbit hair by a deep eutectic solvent and its characterization. Polymers 2018, 10, 993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashim, U.; Arshad, M.K.M.; Lakshmipriya, T.; Chen, Y.; Tang, T.-H.; Gopinath, S.C.B.; Ruslinda, A.R.; Anbu, P. Biotechnological Aspects and Perspective of Microbial Keratinase Production. Biomed Res. Int. 2015, 2015, 1–10. [Google Scholar]
- Martinez-Hernandez, A.L.; Velasco-Santos, C. 17-Keratin Fibers from Chicken Feathers: Structure and Advances in Polymer Composites. In Keratin: Structure, Properties and Applications; Nova Science Publishers: Hauppauge, NY, USA, 2012; ISBN 9789174158991. Available online: http://www.novapublishers.org/catalog/product_info.php?products_id=32840 (accessed on 21 December 2019).
- Karthikeyan, R.; Balaji, S.; Sehgal, P.K. Industrial applications of keratins—A review. J. Sci. Ind. Res India 2007, 66, 710–715. [Google Scholar]
- Norell, M.; Ji, Q.; Gao, K.; Yuan, C.; Zhao, Y.; Wang, L. Modern feathers on a non-avian dinosaur. Nature 2002, 416, 36–37. [Google Scholar] [CrossRef]
- Ullah, A.; Vasanthan, T.; Bressler, D.; Elias, A.L.; Wu, J. Bioplastics from feather quill. Biomacromolecules 2011, 12, 3826–3832. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; McKittrick, J.; Meyers, M.A. Keratin: Structure, mechanical properties, occurrence in biological organisms, and efforts at bioinspiration. Prog. Mater. Sci. 2016, 76, 229–318. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, T.N.; Pissarenko, A.; Herrera, S.A.; Kisailus, D.; Lubarda, V.A.; Meyers, M.A. A lightweight, biological structure with tailored stiffness: The feather vane. Acta Biomater. 2016, 41, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.N.; Wang, B.; Espinosa, H.D.; Meyers, M.A. Extreme lightweight structures: Avian feathers and bones. Mater. Today 2017, 20, 377–391. [Google Scholar] [CrossRef]
- Fraser, R.D.; Roe, H.Z.; Lipson, B. The Structure of a Merino Wool Fibre. Available online: http://www.scienceimage.csiro.au/library/textile/i/7663/the-structure-of-a-merino-wool-fibre/ (accessed on 6 February 2019).
- DeFrates, K.; Moore, R.; Borgesi, J.; Lin, G.; Mulderig, T.; Beachley, V.; Hu, X. Protein-Based Fiber Materials in Medicine: A Review. Nanomaterials 2018, 8, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jao, D.; Xue, Y.; Medina, J.; Hu, X. Protein-based drug-delivery materials. Materials 2017, 10, 517. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-M.; LI, F.-Y.; Wang, X.-J.; LI, Q.-F.; He, Y.-F.; Wang, Y.-B. The Application of Feather Keratin and Its Derivatives In Treatment Of Potato Starch Wastewater. Funct. Mater. Lett. 2010, 3, 213–216. [Google Scholar] [CrossRef]
- Khosa, M.A.; Wu, J.; Ullah, A. Chemical modification, characterization, and application of chicken feathers as novel biosorbents. RSC Adv. 2013, 3, 20800–20810. [Google Scholar] [CrossRef] [Green Version]
- Latha, P.P.; Singh, R.K.; Kukrety, A.; Saxena, R.C.; Bhatt, M.; Jain, S.L. Poultry Chicken Feather Derived Biodegradable Multifunctional Additives for Lubricating Formulations. ACS Sustain. Chem. Eng. 2016, 4, 999–1005. [Google Scholar] [CrossRef]
- Torculas, M.; Medina, J.; Xue, W.; Hu, X. Protein-Based Bioelectronics. ACS Biomater. Sci. Eng. 2016, 2, 1211–1223. [Google Scholar] [CrossRef]
- Ikkai, F.; Naito, S. Dynamic light scattering and circular dichroism studies on heat-induced gelation of hard-keratin protein aqueous solutions. Biomacromolecules 2002, 3, 482–487. [Google Scholar] [CrossRef]
- Xu, H.; Cai, S.; Xu, L.; Yang, Y. Water-stable three-dimensional ultrafine fibrous scaffolds from keratin for cartilage tissue engineering. Langmuir 2014, 30, 8461–8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Noh, K.; Lee, S.C.; Kwon, I.K.; Han, D.W.; Lee, I.S.; Hwang, Y.S. Human hair keratin and its-based biomaterials for biomedical applications. Tissue Eng. Regen. Med. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Barone, J.R.; Schmidt, W.F.; Gregoire, N.T. Extrusion of feather keratin. J. Appl. Polym. Sci. 2006, 100, 1432–1442. [Google Scholar] [CrossRef]
- Drack, M.; Wimmer, R. Woolrock-a material for technical use consisting of keratin. J. Mater. Sci. 2007, 42, 6183–6187. [Google Scholar] [CrossRef]
- Tanabe, T.; Okitsu, N.; Tachibana, A.; Yamauchi, K. Preparation and characterization of keratin–chitosan composite film. Biomaterials 2002, 23, 817–825. [Google Scholar] [CrossRef]
- Vasconcelos, A.; Freddi, G.; Cavaco-Paulo, A. Biodegradable Materials Based on Silk Fibroin and Keratin. Biomacromolecules 2008, 9, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Tonin, C.; Aluigi, A.; Vineis, C.; Varesano, A.; Montarsolo, A.; Ferrero, F. Thermal and structural characterization of poly(ethylene-oxide)/keratin blend films. J. Therm. Anal. Calorim. 2007, 89, 601–608. [Google Scholar] [CrossRef]
- Zoccola, M.; Montarsolo, A.; Aluigi, A.; Varesano, A.; Vineis, C.; Tonin, C. Electrospinning of polyamide 6/modified-keratin blends. E-Polymers 2007, 7, 1–19. [Google Scholar] [CrossRef]
- Council, N.R. Utilization of Chicken Feathers as Filling Materials; Kennedy, S.J., Schubert, A., Weiner, L.I., Eds.; National Academies Press: Washington, DC, USA, 1956; ISBN 978-0-309-31032-1. [Google Scholar]
- Du Pisani, J.A. Sustainable development—Historical roots of the concept. Environ. Sci. 2006, 3, 83–96. [Google Scholar] [CrossRef]
- Utracki, L.A.; Mukhopadhyay, P.; Gupta, R.K. Polymer Blends: Introduction. In Polymer Blends Handbook; Utracki, L.A., Wilkie, C.A., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 3–170. ISBN 978-94-007-6064-6. [Google Scholar]
- Posch, W. 3-Polyolefins. In Applied Plastics Engineering Handbook; Kutz, M., Ed.; Plastics Design Library; William Andrew Publishing: Oxford, UK, 2011; pp. 23–48. ISBN 978-1-4377-3514-7. [Google Scholar]
- Sauter, D.W.; Taoufik, M.; Boisson, C. Polyolefins, a success story. Polymers 2017, 9, 1–13. [Google Scholar]
- Sam, S.T.; Nuradibah, M.A.; Ismail, H.; Noriman, N.Z.; Ragunathan, S. Recent Advances in Polyolefins/Natural Polymer Blends Used for Packaging Application. Polym. Plast. Technol. Eng. 2014, 53, 631–644. [Google Scholar] [CrossRef]
- Shavandi, A.; Ali, M.A. Keratin based thermoplastic biocomposites: A review. Rev. Environ. Sci. Biotechnol. 2019, 18, 299–316. [Google Scholar] [CrossRef] [Green Version]
- Bullions, T.A.; Gillespie, R.A.; Price-O’Brien, J.; Loos, A.C. The effect of maleic anhydride modified polypropylene on the mechanical properties of feather fiber, kraft pulp, polypropylene composites. J. Appl. Polym. Sci. 2004, 92, 3771–3783. [Google Scholar] [CrossRef]
- Barone, J.R.; Gregoire, N.T. Characterisation of fibre–polymer interactions and transcrystallinity in short keratin fibre–polypropylene composites. Plast. Rubber Compos. 2014, 35, 287–293. [Google Scholar] [CrossRef]
- Barone, J.R.; Schmidt, W.F. Polyethylene reinforced with keratin fibers obtained from chicken feathers. Compos. Sci. Technol. 2005, 65, 173–181. [Google Scholar] [CrossRef]
- Kim, N.K.; Bhattacharyya, D. Development of fire resistant wool polymer composites: Mechanical performance and fire simulation with design perspectives. Mater. Des. 2016, 106, 391–403. [Google Scholar] [CrossRef]
- Huda, M.S.; Schmidt, W.F.; Misra, M.; Drzal, L.T. Effect of fiber surface treatment of poultry feather fibers on the properties of their polymer matrix composites. J. Appl. Polym. Sci. 2013, 128, 1117–1124. [Google Scholar] [CrossRef]
- Wang, H.; Jin, X.-Y.; Wu, H.-B. Adsorption and desorption properties of modified feather and feather/PP melt-blown filter cartridge of lead ion (Pb 2+). J. Appl. Polym. Sci. 2014, 132, 41555. [Google Scholar]
- Amieva, E.J.-C.; Velasco-Santos, C.; Martínez-Hernández, A.L.; Rivera-Armenta, J.L.; Mendoza-Martínez, A.M.; Castaño, V.M. Composites from chicken feathers quill and recycled polypropylene. J. Compos. Mater. 2015, 49, 275–283. [Google Scholar] [CrossRef]
- Patnam, P.L.; Ray, S.S.; Chatterjee, A.K.; Jain, S.L. Self-driven graft polymerization of vinyl monomers on poultry chicken feathers in the absence of initiator/catalyst. J. Appl. Polym. Sci. 2017, 134, 1–9. [Google Scholar] [CrossRef]
- Bailey, F.E.; Koleske, J.V. Properties of Poly Ethylene Oxide. In Poly Ethylene Oxide; Bailey, F.E., Koleske, J.V., Eds.; Academic Press: Cambridge, MA, USA, 1976; Chapter 6; pp. 105–149. ISBN 978-0-12-073250-0. [Google Scholar]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Hutanu, D.; Frishberg, M.D.; Guo, L.; Darie, C.C. Recent Applications of Polyethylene Glycols (PEGs) and PEG Derivatives. Mod. Chem. Appl. 2014, 2, 1000132. [Google Scholar] [CrossRef] [Green Version]
- Varesano, A.; Aluigi, A.; Vineis, C.; Tonin, C. Study on the shear viscosity behavior of keratin/PEO blends for nanofibre electrospinning. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 1193–1201. [Google Scholar] [CrossRef]
- Aluigi, A.; Vineis, C.; Varesano, A.; Mazzuchetti, G.; Ferrero, F.; Tonin, C. Structure and properties of keratin/PEO blend nanofibres. Eur. Polym. J. 2008, 44, 2465–2475. [Google Scholar] [CrossRef]
- Aluigi, A.; Varesano, A.; Montarsolo, A.; Vineis, C.; Ferrero, F.; Mazzuchetti, G.; Tonin, C. Electrospinning of keratin/poly (ethylene oxide) blend nanofibers. J. Appl. Polym. Sci. 2007, 104, 863–870. [Google Scholar] [CrossRef]
- Ma, H.; Shen, J.; Cao, J.; Wang, D.; Yue, B.; Mao, Z.; Wu, W.; Zhang, H. Fabrication of wool keratin/polyethylene oxide nano-membrane from wool fabric waste. J. Clean. Prod. 2017, 161, 357–361. [Google Scholar] [CrossRef]
- Fan, J.; Lei, T.-D.; Li, J.; Zhai, P.-Y.; Wang, Y.-H.; Cao, F.-Y.; Liu, Y. High protein content keratin/poly (ethylene oxide) nanofibers crosslinked in oxygen atmosphere and its cell culture. Mater. Des. 2016, 104, 60–67. [Google Scholar] [CrossRef]
- Grkovic, M.; Stojanovic, D.B.; Kojovic, A.; Strnad, S.; Kreze, T.; Aleksic, R.; Uskokovic, P.S. Keratin-polyethylene oxide bio-nanocomposites reinforced with ultrasonically functionalized graphene. RSC Adv. 2015, 5, 91280–91287. [Google Scholar] [CrossRef]
- Yue, K.; Liu, Y.; Byambaa, B.; Singh, V.; Liu, W.; Li, X.; Sun, Y.; Zhang, Y.S.; Tamayol, A.; Zhang, P.; et al. Visible light crosslinkable human hair keratin hydrogels. Bioeng. Transl. Med. 2018, 3, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Yemul, O.; Imae, T. Synthesis and characterization of poly (ethyleneimine) dendrimers. Colloid Polym. Sci. 2008, 286, 747–752. [Google Scholar] [CrossRef]
- Kuzuhara, A.; Hori, T. New Method of Dyeing Keratin Fibers Using Poly(ethylene imine) and Its Coloring Mechanism. J. Appl. Polym. Sci. 2003, 90, 3806–3810. [Google Scholar] [CrossRef]
- Kuzuhara, A. Influence of urea on the coloring ability of a low-temperature coloring method of keratin fibers using polyethyleneimine. J. Appl. Polym. Sci. 2004, 91, 3827–3834. [Google Scholar] [CrossRef]
- Kuzuhara, A.; Hori, T. Diffusion behavior of poly(ethylene imine) into keratin fibers using microspectrophotometry. J. Appl. Polym. Sci. 2005, 97, 65–71. [Google Scholar] [CrossRef]
- Penzel, E.; Ballard, N.; Asua, J.M. Polyacrylates. In Ullmann’s Encyclopedia of Industrial Chemistry; American Cancer Society: New York, NY, USA, 2018; pp. 1–20. ISBN 9783527306732. [Google Scholar]
- Wichterle, O.; LÍM, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Paul, R.; Genescà, E. 8-The use of enzymatic techniques in the finishing of technical textiles. In Advances in the Dyeing and Finishing of Technical Textiles; Gulrajani, M.L., Ed.; Woodhead Publishing Series in Textiles; Woodhead Publishing: Cambridge, UK, 2013; pp. 177–198. ISBN 978-0-85709-433-9. [Google Scholar]
- Hearle, J.W.S. Textile Fibers: A Comparative Overview. In Encyclopedia of Materials: Science and Technology; Buschow, K.H.J., Cahn, R.W., Flemings, M.C., Ilschner, B., Kramer, E.J., Mahajan, S., Veyssière, P., Eds.; Elsevier: Oxford, UK, 2001; pp. 9100–9116. ISBN 978-0-08-043152-9. [Google Scholar]
- Yan, Y. 2-Developments in fibers for technical nonwovens. In Advances in Technical Nonwovens; Kellie, G., Ed.; Woodhead Publishing Series in Textiles; Woodhead Publishing: Sawston, UK, 2016; pp. 19–96. ISBN 978-0-08-100575-0. [Google Scholar]
- Hennecke, D.; Bauer, A.; Herrchen, M.; Wischerhoff, E.; Gores, F. Cationic polyacrylamide copolymers (PAMs): Environmental half life determination in sludge-treated soil. Environ. Sci. Eur. 2018, 30, 16. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Negishi, M.; Okabe, T. Infrared spectroscopy of graft polymers separated from graft copolymers of wool and silk with methyl methacrylate. J. Appl. Polym. Sci. 1968, 12, 2585–2596. [Google Scholar] [CrossRef]
- Leeder, J.D.; Pratt, A.J.; Watt, I.C. Wool–polymer systems: Effect of vinyl polymers on water absorption. J. Appl. Polym. Sci. 1967, 11, 1649–1659. [Google Scholar] [CrossRef]
- Varma, D.S.; Sadhir, R.K. Radiation-Induced graft copolymerization of methyl methacrylate on natural and modified wool. II. Sorption behavior. J. Appl. Polym. Sci. 1979, 23, 393–400. [Google Scholar] [CrossRef]
- Niezette, J. Stress-strain behaviour of graft copolymers of methyl methacrylate and natural wool fibres. Eur. Polym. J. 1981, 17, 281–283. [Google Scholar] [CrossRef]
- Bresee, R.R.; Annis, P.A.; Reagan, B.M. Localization of methyl acrylate graft copolymerization in exposed cortex of abraided wool fibers. J. Appl. Polym. Sci. 1986, 31, 2839–2843. [Google Scholar] [CrossRef]
- Arai, K.; Negishi, M.; Suda, T.; Arai, S. Grafting onto wool. III. Relationship between alpha- and beta-forms in wool keratin of grafted fibers. J. Appl. Polym. Sci. 1973, 17, 483–502. [Google Scholar] [CrossRef]
- Varma, D.S.; Sadhir, R.K. Radiation-induced graft copolymerization of methyl methacrylate on natural and modified wool. V. Crystalline and morphological structure. J. Appl. Polym. Sci. 1980, 25, 487–498. [Google Scholar] [CrossRef]
- Elangovan, V.J.; Saccubai, S. Chemical and mechanical properties of methyl methacrylate-grafted wool fiber. J. Appl. Polym. Sci. 1992, 44, 2179–2183. [Google Scholar] [CrossRef]
- Elangovan, V.J.; Saccubai, S. Thermal properties of wool-g-poly (methyl methacrylate) copolymers. J. Appl. Polym. Sci. 1992, 45, 1823–1830. [Google Scholar] [CrossRef]
- Xu, W.; Bao, J.; Zhang, J.; Shi, M. Microwave irradiation graft copolymerization of hydroxyethyl methacrylate onto wool fabrics. J. Appl. Polym. Sci. 1998, 70, 2343–2347. [Google Scholar] [CrossRef]
- Meng, X.Y. Chemical and mechanical properties of butyl methacrylate grafted wool fiber. J. Appl. Polym. Sci. 2004, 91, 3813–3817. [Google Scholar] [CrossRef]
- Tsukada, M.; Shiozaki, H.; Freddi, G.; Crighton, J.S. Graft copolymerization of benzyl methacrylate onto wool fibers. J. Appl. Polym. Sci. 1997, 64, 343–350. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, B.; Shan, Z. Preparation of sound-insulating material based on discarded cow hair. J. Appl. Polym. Sci. 2018, 135, 46332. [Google Scholar] [CrossRef]
- Martínez-Hernández, A.L.; Velasco-Santos, C.; De Icaza, M.; Castaño, V.M. Grafting of methyl methacrylate onto natural keratin. E-Polymers 2003, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Reddy, N.; Hou, X.; Yang, Y. Tensile properties of thermoplastic feather films grafted with different methacrylates. ACS Sustain. Chem. Eng. 2014, 2, 1849–1856. [Google Scholar] [CrossRef]
- Schaller, J.; Miyamoto, T.; Shimamura, K.; Inagaki, H. Membranes prepared from keratin–polyacrylonitrile graft copolymers. J. Appl. Polym. Sci. 1980, 25, 783–794. [Google Scholar] [CrossRef]
- Giri, G.; Sahoo, P.K.; Samal, R.K. Graft copolymerization onto wool fibers: Grafting of acrylamide onto wool fibers initiated by potassium monopersulphate/Fe (II) redox system. J. Appl. Polym. Sci. 1990, 40, 471–483. [Google Scholar] [CrossRef]
- Shavandi, A.; Ali, M.A. Graft polymerization onto wool fibre for improved functionality. Prog. Org. Coat. 2019, 130, 182–199. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Sun, L.; Ma, C.; Qiao, Y.; Yao, H. Thermal degradation of PVC: A review. Waste Manag. 2016, 48, 300–314. [Google Scholar] [CrossRef]
- Wagoner, J.K. Toxicity of vinyl chloride and poly (vinyl chloride): A critical review. Environ. Health Perspect. 1983, 52, 61–66. [Google Scholar] [CrossRef]
- Folarin, O.M.; Sadiku, E.R. Thermal stabilizers for poly (vinyl chloride): A review. Int. J. Phys. Sci. 2011, 6, 4323–4330. [Google Scholar]
- Yngve, V. Vinyl Resin Phonograph Record. U.S. Patent No. 2,307,180, 7 December 1943. [Google Scholar]
- Lucio, D.S.V.; Rivera-Armenta, J.L.; Rivas-Orta, V.; Díaz-Zavala, N.P.; Páramo-García, U.; Rivas, N.V.G.; Cinco, M.Y.C. Manufacturing of Composites from Chicken Feathers and Polyvinyl Chloride (PVC). In Handbook of Composites from Renewable Materials; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 159–174. ISBN 9781119441632. [Google Scholar]
- Akhlaghi, S.; Sharif, A.; Kalaee, M.; Manafi, M. Miscibility and thermal behavior of poly (vinyl chloride)/feather keratin blends. J. Appl. Polym. Sci. 2011, 121, 3252–3261. [Google Scholar] [CrossRef]
- Aslam, M.; Kalyar, M.A.; Raza, Z.A. Polyvinyl alcohol: A review of research status and use of polyvinyl alcohol based nanocomposites. Polym. Eng. Sci. 2018, 58, 2119–2132. [Google Scholar] [CrossRef]
- Baker, M.I.; Walsh, S.P.; Schwartz, Z.; Boyan, B.D. A review of polyvinyl alcohol and its uses in cartilage and orthopedic applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100, 1451–1457. [Google Scholar] [CrossRef]
- DeMerlis, C.; Schoneker, D. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef]
- Katoh, K.; Shibayama, M.; Tanabe, T.; Yamauchi, K. Preparation and properties of keratin-poly (vinyl alcohol) blend fiber. J. Appl. Polym. Sci. 2004, 91, 756–762. [Google Scholar] [CrossRef]
- Liu, R.; Li, L.; Liu, S.; Li, S.; Zhu, X.; Yi, M.; Liao, X. Structure and properties of wool keratin/poly (vinyl alcohol) blended fiber. Adv. Polym. Technol. 2018, 37, 2756–2762. [Google Scholar] [CrossRef]
- Esparza, Y.; Ullah, A.; Boluk, Y.; Wu, J. Preparation and characterization of thermally crosslinked poly (vinyl alcohol)/feather keratin nanofiber scaffolds. Mater. Des. 2017, 133, 1–9. [Google Scholar] [CrossRef]
- Ding, J.; Chen, M.; Chen, W.; He, M.; Zhou, X.; Yin, G. Vapor-Assisted Crosslinking of a FK/PVA/PEO Nanofiber Membrane. Polymers 2018, 10, 747. [Google Scholar] [CrossRef] [Green Version]
- Kadirvelu, K.; Fathima, N.N. Self-assembly of keratin peptides: Its implication on the performance of electrospun PVA nanofibers. Sci. Rep. 2016, 6, 36558. [Google Scholar] [CrossRef] [Green Version]
- El-Kheir, A.A.; Mowafi, S.; Taleb, M.A.; El-Sayed, H. Preparation and Characterization of Keratin-Polyvinyl Alcohol Composite Film. Egypt. J. Chem. 2012, 55, 491–507. [Google Scholar]
- Dou, Y.; Zhang, B.; He, M.; Yin, G.; Cui, Y. Preparation and Physicochemical Properties of Dialdehyde Starch Crosslinked Feather Keratin/PVA Composite Films. J. Macromol. Sci. Part A 2014, 51, 1009–1015. [Google Scholar] [CrossRef]
- Dou, Y.; Zhang, B.; He, M.; Yin, G.; Cui, Y.; Savina, I. Keratin/Polyvinyl Alcohol Blend Films Cross-Linked by Dialdehyde Starch and Their Potential Application for Drug Release. Polymers 2015, 7, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, S.; Yi, M.; Ge, J.; Yin, G.; Li, X. Preparation and Physicochemical Properties of Blend Films of Feather Keratin and Poly (vinyl alcohol) Compatibilized by Tris (hydroxymethyl) aminomethane. Polymers 2018, 10, 1054. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, S.; Maass, T.A. Ueber ɛ-Amidocapronsäure. Berichte der Dtsch. Chem. Gesellschaft 1899, 32, 1266–1272. [Google Scholar] [CrossRef] [Green Version]
- Matthies, P.; Seydl, W.F. History and Development of Nylon 6. In High Performance Polymers: Their Origin and Development; Seymour, R.B., Kirshenbaum, G.S., Eds.; Springer: Dordrecht, The Netherlands, 1986; pp. 39–53. ISBN 978-94-011-7073-4. [Google Scholar]
- Vlasveld, D.P.N.; Groenewold, J.; Bersee, H.E.N.; Picken, S.J. Moisture absorption in polyamide-6 silicate nanocomposites and its influence on the mechanical properties. Polymer 2005, 46, 12567–12576. [Google Scholar] [CrossRef]
- Aluigi, A.; Tonetti, C.; Vineis, C.; Tonin, C.; Mazzuchetti, G. Adsorption of copper (II) ions by keratin/PA6 blend nanofibres. Eur. Polym. J. 2011, 47, 1756–1764. [Google Scholar] [CrossRef]
- Aluigi, A.; Varesano, A.; Vineis, C.; Del Rio, A. Electrospinning of immiscible systems: The wool keratin/polyamide-6 case study. Mater. Des. 2017, 127, 144–153. [Google Scholar] [CrossRef]
- Akhlaghi, S.; Sharif, A.; Kalaee, M.; Nouri, A.; Manafi, M. Morphology, nanomechanical and thermodynamic surface characteristics of nylon 6/feather keratin blend films: An atomic force microscopy investigation. Polym. Int. 2012, 61, 646–656. [Google Scholar] [CrossRef]
- Natta, F.J.; van Hill, J.W.; Carothers, W.H. Studies of Polymerization and Ring Formation. XXIII. 1 ε-Caprolactone and its Polymers. J. Am. Chem. Soc. 1934, 56, 455–457. [Google Scholar] [CrossRef]
- Guarino, V.; Gentile, G.; Sorrentino, L.; Ambrosio, L. Polycaprolactone: Synthesis, Properties, and Applications. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–36. ISBN 0471440264. [Google Scholar]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Li, R.; Wu, X.; Chen, K.; Che, J. Controllable fabrication and characterization of hydrophilic PCL/wool keratin nanonets by electronetting. Eur. Polym. J. 2017, 86, 154–161. [Google Scholar] [CrossRef]
- Wu, P.; Dai, X.; Chen, K.; Li, R.; Xing, Y. Fabrication of regenerated wool keratin/polycaprolactone nanofiber membranes for cell culture. Int. J. Biol. Macromol. 2018, 114, 1168–1173. [Google Scholar] [CrossRef]
- Edwards, A.; Jarvis, D.; Hopkins, T.; Pixley, S.; Bhattarai, N. Poly (ε-caprolactone)/keratin-based composite nanofibers for biomedical applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 103, 21–30. [Google Scholar] [CrossRef]
- Boakye, M.; Rijal, N.; Adhikari, U.; Bhattarai, N. Fabrication and Characterization of Electrospun PCL-MgO-Keratin-Based Composite Nanofibers for Biomedical Applications. Materials 2015, 8, 4080–4095. [Google Scholar] [CrossRef]
- Zhao, X.; Lui, Y.S.; Choo, C.K.C.; Sow, W.T.; Huang, C.L.; Ng, K.W.; Tan, L.P.; Loo, J.S.C. Calcium phosphate coated Keratin–PCL scaffolds for potential bone tissue regeneration. Mater. Sci. Eng. C 2015, 49, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Jamshidian, M.; Tehrany, E.A.; Imran, M.; Jacquot, M.; Desobry, S. Poly-Lactic Acid: Production, Applications, Nanocomposites, and Release Studies. Compr. Rev. Food Sci. Food Saf. 2010, 9, 552–571. [Google Scholar] [CrossRef]
- Södergård, A.; Stolt, M. Industrial Production of High Molecular Weight Poly (Lactic Acid). In Poly Lactic Acid; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 27–41. ISBN 9780470649848. [Google Scholar]
- Liu, Z.; Wang, Y.; Wu, B.; Cui, C.; Guo, Y.; Yan, C. A critical review of fused deposition modeling 3D printing technology in manufacturing polylactic acid parts. Int. J. Adv. Manuf. Technol. 2019, 102, 2877–2889. [Google Scholar] [CrossRef]
- Fortunati, E.; Aluigi, A.; Armentano, I.; Morena, F.; Emiliani, C.; Martino, S.; Santulli, C.; Torre, L.; Kenny, J.M.; Puglia, D. Keratins extracted from Merino wool and Brown Alpaca fibres: Thermal, mechanical and biological properties of PLLA based biocomposites. Mater. Sci. Eng. C 2015, 47, 394–406. [Google Scholar] [CrossRef]
- Puglia, D.; Ceccolini, R.; Fortunati, E.; Armentano, I.; Morena, F.; Martino, S.; Aluigi, A.; Torre, L.; Kenny, J.M. Effect of processing techniques on the 3D microstructure of poly (l-lactic acid) scaffolds reinforced with wool keratin from different sources. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Li, J.-S.; Li, Y.; Liu, X.; Zhang, J.; Zhang, Y. Strategy to introduce an hydroxyapatite–keratin nanocomposite into a fibrous membrane for bone tissue engineering. J. Mater. Chem. B 2013, 1, 432–437. [Google Scholar] [CrossRef]
- Spiridon, I.; Paduraru, O.M.; Zaltariov, M.F.; Darie, R.N. Influence of Keratin on Polylactic Acid/Chitosan Composite Properties. Behavior upon Accelerated Weathering. Ind. Eng. Chem. Res. 2013, 52, 9822–9833. [Google Scholar] [CrossRef]
- Aranberri, I.; Montes, S.; Azcune, I.; Rekondo, A.; Grande, H.-J. Fully Biodegradable Biocomposites with High Chicken Feather Content. Polymers 2017, 9, 593. [Google Scholar] [CrossRef] [Green Version]
- Cañavate, J.; Aymerich, J.; Garrido, N.; Colom, X.; Macanás, J.; Molins, G.; Álvarez, M.; Carrillo, F. Properties and optimal manufacturing conditions of chicken feathers/poly (lactic acid) biocomposites. J. Compos. Mater. 2016, 50, 1671–1683. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Olivares, G.; Sanchez-Solis, A.; Calderas, F.; Alongi, J. Keratin fibres derived from tannery industry wastes for flame retarded PLA composites. Polym. Degrad. Stab. 2017, 140, 42–54. [Google Scholar] [CrossRef]
- Koller, M.; Atlić, A.; Dias, M.; Reiterer, A.; Braunegg, G. Microbial PHA Production from Waste Raw Materials. In Plastics from Bacteria: Natural Functions and Applications; Chen, G.G.-Q., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 85–119. ISBN 978-3-642-03287-5. [Google Scholar]
- Raza, Z.A.; Abid, S.; Banat, I.M. Polyhydroxyalkanoates: Characteristics, production, recent developments and applications. Int. Biodeterior. Biodegrad. 2018, 126, 45–56. [Google Scholar] [CrossRef]
- Li, Z.; Yang, J.; Loh, X.J. Polyhydroxyalkanoates: Opening doors for a sustainable future. NPG Asia Mater. 2016, 8, e265. [Google Scholar] [CrossRef]
- Yuan, J.; Xing, Z.-C.; Park, S.-W.; Geng, J.; Kang, I.-K.; Yuan, J.; Shen, J.; Meng, W.; Shim, K.-J.; Han, I.-S.; et al. Fabrication of PHBV/keratin composite nanofibrous mats for biomedical applications. Macromol. Res. 2009, 17, 850–855. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Yuan, J.; Shen, J. Differences in cytocompatibility between collagen, gelatin and keratin. Mater. Sci. Eng. C 2016, 59, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Ibáñez, P.; Lopez-Rubio, A.; Martínez-Sanz, M.; Cabedo, L.; Lagaron, J.M. Keratin-polyhydroxyalkanoate melt-compounded composites with improved barrier properties of interest in food packaging applications. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Fabra, M.J.; Pardo, P.; Martinez-Sanz, M.; Lopez-Rubio, A. Combining polyhydroxyalkanoates with nanokeratin to develop novel biopackaging structures. J. Appl. Polym. Sci. 2016, 133, 42695. [Google Scholar] [CrossRef]
- Datta, J.; Kasprzyk, P. Thermoplastic polyurethanes derived from petrochemical or renewable resources: A comprehensive review. Polym. Eng. Sci. 2018, 58, E14–E35. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Gao, Y. The manufacture of 3D printing of medical grade TPU. Prog. Addit. Manuf. 2017, 2, 117–123. [Google Scholar] [CrossRef]
- Tang, D.; Noordover, B.A.J.; Sablong, R.J.; Koning, C.E. Thermoplastic Poly(urethane urea)s From Novel, Bio-based Amorphous Polyester Diols. Macromol. Chem. Phys. 2012, 213, 2541–2549. [Google Scholar] [CrossRef]
- Mattia, J.; Painter, P. A Comparison of Hydrogen Bonding and Order in a Polyurethane and Poly (urethane−urea) and Their Blends with Poly (ethylene glycol). Macromolecules 2007, 40, 1546–1554. [Google Scholar] [CrossRef]
- Ban, J.-L.; Li, S.-Q.; Yi, C.-F.; Zhao, J.-B.; Zhang, Z.-Y.; Zhang, J.-Y. Amorphous and Crystallizable Thermoplastic Polyureas Synthesized through a One-pot Non-isocyanate Route. Chin. J. Polym. Sci. 2019, 37, 43–51. [Google Scholar] [CrossRef]
- Saucedo-Rivalcoba, V.; Martínez-Hernández, A.L.; Martínez-Barrera, G.; Velasco-Santos, C.; Castaño, V.M. (Chicken feathers keratin)/polyurethane membranes. Appl. Phys. A 2011, 104, 219–228. [Google Scholar] [CrossRef]
- Wrześniewska-Tosik, K.; Zajchowski, S.; Bryśkiewicz, A.; Ryszkowska, J. Feathers as a flame-retardant in elastic polyurethane foam. Fibres Text. East. Eur. 2014, 103, 119–128. [Google Scholar]
- Gokce, O.; Kasap, M.; Akpinar, G.; Ozkoc, G. Preparation, characterization, and in vitro evaluation of chicken feather fiber-thermoplastic polyurethane composites. J. Appl. Polym. Sci. 2017, 134, 45338. [Google Scholar] [CrossRef] [Green Version]
- Pourjavaheri, F.; Jones, O.A.H.; Czajka, M.; Martinez-Pardo, I.; Blanch, E.W.; Shanks, R.A. Design and characterization of sustainable bio-composites from waste chicken feather keratin and thermoplastic polyurethane. Polym. Compos. 2018, 39, E620–E632. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Xiang, P.; Lu, J.; Yuan, J.; Shen, J. Electrospun polyurethane/keratin/AgNP biocomposite mats for biocompatible and antibacterial wound dressings. J. Mater. Chem. B 2016, 4, 635–648. [Google Scholar] [CrossRef]
- Li, H.; Oh, J.S.; Sinha, T.K.; Kim, J.K. Synergistic influence of keratin and TPU: An approach towards bioinspired artificial skin. Mater. Chem. Phys. 2019, 223, 196–201. [Google Scholar] [CrossRef]
- Aranberri, I.; Montes, S.; Azcune, I.; Rekondo, A.; Grande, H.-J. Flexible Biocomposites with Enhanced Interfacial Compatibility Based on Keratin Fibers and Sulfur-Containing Poly (urea-urethane) s. Polymers 2018, 10, 1056. [Google Scholar] [CrossRef] [Green Version]
- Adeniyi, A.; Agboola, O.; Sadiku, E.R.; Durowoju, M.O.; Olubambi, P.A.; Babul Reddy, A.; Ibrahim, I.D.; Kupolati, W.K. Thermoplastic-Thermoset Nanostructured Polymer Blends. In Design and Applications of Nanostructured Polymer Blends and Nanocomposite Systems; Thomas, S., Shanks, R., Chandrasekharakurup, S., Eds.; Micro and Nano Technologies; Elsevier: Boston, MA, USA, 2016; pp. 15–38. ISBN 978-0-323-39408-6. [Google Scholar]
- Guo, Q.; Zheng, H. Miscibility and crystallization of thermosetting polymer blends of unsaturated polyester resin and poly(ϵ-caprolactone). Polymer 1999, 40, 637–646. [Google Scholar] [CrossRef]
- Brandt, H.-D.; Nentwig, W.; Rooney, N.; LaFlair, R.T.; Wolf, U.U.; Duffy, J.; Puskas, J.E.; Kaszas, G.; Drewitt, M.; Glander, S. Rubber, 5. Solution Rubbers. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; ISBN 9783527306732. [Google Scholar]
- Kim, J.; Oh, T.; Lee, D. Morphology and rheological properties of nanocomposites based on nitrile rubber and organophilic layered silicates. Polym. Int. 2003, 52, 1203–1208. [Google Scholar] [CrossRef]
- Tadiello, L.; D’Arienzo, M.; Di Credico, B.; Hanel, T.; Matejka, L.; Mauri, M.; Morazzoni, F.; Simonutti, R.; Spirkova, M.; Scotti, R. The filler–rubber interface in styrene butadiene nanocomposites with anisotropic silica particles: Morphology and dynamic properties. Soft Matter 2015, 11, 4022–4033. [Google Scholar] [CrossRef] [PubMed]
- Han, D.-H.; Choi, M.-C.; Jeong, J.-H.; Choi, K.-M.; Kim, H.-S. Properties of acrylonitrile butadiene rubber (NBR)/poly (lactic acid) (PLA) blends and their foams. Compos. Interfaces 2016, 23, 771–780. [Google Scholar] [CrossRef]
- Prochoń, M.; Przepiórkowska, A.; Zaborski, M. Keratin as a filler for carboxylated acrylonitrile-butadiene rubber XNBR. J. Appl. Polym. Sci. 2007, 106, 3674–3687. [Google Scholar] [CrossRef]
- Prochon, M.; Janowska, G.; Przepiorkowska, A.; Kucharska-Jastrzabek, A. Thermal properties and combustibility of elastomer–protein composites. Part I. Composites SBR–keratin. J. Therm. Anal. Calorim. 2012, 109, 1563–1570. [Google Scholar] [CrossRef]
- Janowska, G.; Kucharska-Jastrzabek, A.; Prochon, M.; Przepiorkowska, A. Thermal properties and combustibility of elastomer–protein composites. Part II. Composites NBR–keratin. J. Therm. Anal. Calorim. 2013, 113, 933–938. [Google Scholar] [CrossRef] [Green Version]
- Tshela Ntumba, Y.-H.; Prochoń, M. The effect of modified keratin on the thermal properties of a cellulosic–elastomeric material. J. Therm. Anal. Calorim. 2016, 125, 1151–1160. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Castillo, C.; Salazar-Cruz, B.A.; Rivera-Armenta, J.L.; Chávez-Cinco, M.Y.; Méndez-Hernández, M.L.; Estrada-Moreno, I.A.; Lara Ceniceros, T.E. Evaluation of Elastomeric Composites Reinforced with Chicken Feathers. In Futuristic Composites: Behavior, Characterization, and Manufacturing; Sidhu, S.S., Bains, P.S., Zitoune, R., Yazdani, M., Eds.; Springer: Singapore, 2018; pp. 297–318. ISBN 978-981-13-2417-8. [Google Scholar]
- Méndez-Hernández, M.L.; Rivera-Armenta, J.L.; Sandoval-Arellano, Z.; Salazar-Cruz, B.A.; Chavez-Cinco, M.Y. Evaluation of Styrene Content over Physical and Chemical Properties of Elastomer/TPS-EVOH/Chicken Feather Composites. In Applications of Modified Starches; Huicochea, E.F., Villalobos, R.R., Eds.; InTech: Rijeka, Croatia, 2018. [Google Scholar]
- Matějka, L.; Lövy, J.; Pokorný, S.; Bouchal, K.; Dušek, K. Curing epoxy resins with anhydrides. Model reactions and reaction mechanism. J. Polym. Sci. Polym. Chem. Ed. 1983, 21, 2873–2885. [Google Scholar] [CrossRef]
- Matějka, L.; Dušek, K.; Dobáš, I. Curing of epoxy resins with amines - Gelation of polyepoxides derived from diglycidylaniline. Polym. Bull. 1985, 14, 309–315. [Google Scholar]
- Matějka, L.; Pokorný, S.; Dušek, K. Acid curing of epoxy resins. A comparison between the polymerization of diepoxide-diacid and monoepoxide-cyclic anhydride systems. Makromol. Chem. 1985, 186, 2025–2036. [Google Scholar] [CrossRef]
- May, C.A. Epoxy Resins. Chemistry and Technology, 2nd ed.; Dekker, M., Ed.; CRC Press: New York, NY, USA, 1988; ISBN 9780824776909. [Google Scholar]
- Jin, F.-L.; Li, X.; Park, S.-J. Synthesis and application of epoxy resins: A review. J. Ind. Eng. Chem. 2015, 29, 1–11. [Google Scholar] [CrossRef]
- Kumar, S.; Samal, S.K.; Mohanty, S.; Nayak, S.K. Recent Development of Biobased Epoxy Resins: A Review. Polym. Plast. Technol. Eng. 2018, 57, 133–155. [Google Scholar] [CrossRef]
- Baroncini, E.A.; Kumar Yadav, S.; Palmese, G.R.; Stanzione, J.F. Recent advances in bio-based epoxy resins and bio-based epoxy curing agents. J. Appl. Polym. Sci. 2016, 133, 44103. [Google Scholar] [CrossRef] [Green Version]
- Zhan, M.; Wool, R.P.; Xiao, J.Q. Electrical properties of chicken feather fiber reinforced epoxy composites. Compos. Part A Appl. Sci. Manuf. 2011, 42, 229–233. [Google Scholar] [CrossRef]
- Zhan, M.; Wool, R.P. Thermal expansivity of chicken feather fiber reinforced epoxy composites. J. Appl. Polym. Sci. 2013, 128, 997–1003. [Google Scholar] [CrossRef]
- Zhan, M.; Wool, R.P. Mechanical properties of composites with chicken feather and glass fibers. J. Appl. Polym. Sci. 2016, 133, 44013. [Google Scholar] [CrossRef]
- Bessa, J.; Souza, J.; Lopes, J.B.; Sampaio, J.; Mota, C.; Cunha, F.; Fangueiro, R. Characterization of thermal and acoustic insulation of chicken feather reinforced composites. Procedia Eng. 2017, 200, 472–479. [Google Scholar] [CrossRef]
- Verma, A.; Negi, P.; Singh, V.K. Experimental Analysis on Carbon Residuum Transformed Epoxy Resin: Chicken Feather Fiber Hybrid Composite. Polym. Compos. 2019, 40, 2690–2699. [Google Scholar] [CrossRef]
- Hong, C.K.; Wool, R.P. Development of a bio-based composite material from soybean oil and keratin fibers. J. Appl. Polym. Sci. 2005, 95, 1524–1538. [Google Scholar] [CrossRef]
- Zhan, M.; Wool, R.P. Design and evaluation of bio-based composites for printed circuit board application. Compos. Part A Appl. Sci. Manuf. 2013, 47, 22–30. [Google Scholar] [CrossRef]
- Senoz, E.; Wool, R.P. Microporous carbon-nitrogen fibers from keratin fibers by pyrolysis. J. Appl. Polym. Sci. 2010, 118, 1752–1765. [Google Scholar] [CrossRef]
- Senoz, E.; Wool, R.P.; McChalicher, C.W.J.; Hong, C.K. Physical and chemical changes in feather keratin during pyrolysis. Polym. Degrad. Stab. 2012, 97, 297–307. [Google Scholar] [CrossRef]
- Senoz, E.; Wool, R.P. Hydrogen storage on pyrolyzed chicken feather fibers. Int. J. Hydrogen Energy 2011, 36, 7122–7127. [Google Scholar] [CrossRef]
- Senoz, E.; Stanzione, J.F.; Reno, K.H.; Wool, R.P.; Miller, M.E.N. Pyrolyzed chicken feather fibers for biobased composite reinforcement. J. Appl. Polym. Sci. 2013, 128, 983–989. [Google Scholar] [CrossRef]
- Tollens, B.I. Ueber einige Derivate des Formaldehyds. Berichte der Deutschen Chemischen Gesellschaft 1884, 17, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Diem, H.; Matthias, G.; Wagner, R.A. Amino Resins. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; ISBN 9783527306732. [Google Scholar]
- Dunky, M.; Pizzl, A. Wood adhesives. In Adhesion Science and Engineering; Dillard, D.A., Pocius, A.V., Chaudhury, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2002; pp. 1039–1103. ISBN 978-0-444-51140-9. [Google Scholar]
- Nuryawan, A.; Risnasari, I.; Sucipto, T.; Heri Iswanto, A.; Rosmala Dewi, R. Urea-formaldehyde resins: Production, application, and testing. IOP Conf. Ser. Mater. Sci. Eng. 2017, 223, 012053. [Google Scholar] [CrossRef]
- Pang, J.Y.; Sun, C.; Zhang, S.C.; Cui, H.X. Study on Modification of Urea Formaldehyde Resin with Keratin. Adv. Mater. Res. 2010, 113, 1787–1791. [Google Scholar] [CrossRef]
- Dim, P.E. Application of Keratin-Modified Urea-Formaldehyde Resin for Bonding Particleboard. Aust. J. Basic Appl. Sci. 2011, 5, 196–200. [Google Scholar]
- Imperial Chemical Industries, ltd. Plastics Division. Landmarks of the Plastics Industry, 1st ed.; Imperial Chemical Industries (Plastics Division): Welwyn Garden City, UK, 1962. [Google Scholar]
- Brydson, J.A. Phenolic Resins. In Plastics Materials; Brydson, J.A., Ed.; Elsevier: Oxford, UK, 1999; pp. 635–667. ISBN 978-0-7506-4132-6. [Google Scholar]
- Cygan, M.; Szemień, M.; Krompiec, S. Statistical screening analysis of the chemical composition and kinetic study of phenol-formaldehyde resins synthesized in the presence of polyamines as co-catalysts. PLoS ONE 2018, 13, e0195069. [Google Scholar] [CrossRef]
- Gusse, A.C.; Miller, P.D.; Volk, T.J. White-Rot Fungi Demonstrate First Biodegradation of Phenolic Resin. Environ. Sci. Technol. 2006, 40, 4196–4199. [Google Scholar] [CrossRef] [PubMed]
- El Mansouri, N.E.; Yuan, Q.; Huang, F. Preparation and Characterization of Phenol-Formaldehyde Resins Modified with Alkaline Rice Straw Lignin. BioResources 2018, 13, 8061–8075. [Google Scholar] [CrossRef]
- Siddiqui, H.; Mahmood, N.; Yuan, Z.; Crapulli, F.; Dessbesell, L.; Rizkalla, A.; Ray, A.; Xu, C. (Charles) Sustainable Bio-Based Phenol-Formaldehyde Resoles Using Hydrolytically Depolymerized Kraft Lignin. Molecules 2017, 22, 1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winandy, J.E.; Muehl, J.H.; Glaeser, J.A.; Schmidt, W. Chicken Feather Fiber as an Additive in MDF Composites. J. Nat. Fibers 2007, 4, 35–48. [Google Scholar] [CrossRef]
- Jiang, Z.; Qin, D.; Hse, C.-Y.; Kuo, M.; Luo, Z.; Wang, G.; Yu, Y. Preliminary Study on Chicken Feather Protein–Based Wood Adhesives. J. Wood Chem. Technol. 2008, 28, 240–246. [Google Scholar] [CrossRef]
- Kawahara, Y.; Ishibashi, N.; Yamamoto, K.; Wakizaka, H.; Iwashita, N.; Kenjo, S.; Nishikawa, G. Activated carbon production by co-carbonization of feathers using water-soluble phenolic resin under controlled graphitization. Sustain. Mater. Technol. 2015, 4, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y. Electrospinning of Direct Carbonizable Phenolic Resin-based Nanofibers. J. Text. Sci. Eng. 2016, 6, 257. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, H.P.; Tharanathan, R.N. Carbohydrates—The Renewable Raw Materials of High Biotechnological Value. Crit. Rev. Biotechnol. 2003, 23, 149–173. [Google Scholar] [CrossRef]
- Thomas, B.; Raj, M.C.; Joy, J.; Moores, A.; Drisko, G.L.; Sanchez, C. Nanocellulose, a Versatile Green Platform: From Biosources to Materials and Their Applications. Chem. Rev. 2018, 118, 11575–11625. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Barone, J.R. Lignocellulosic Fiber-Reinforced Keratin Polymer Composites. J. Polym. Environ. 2009, 17, 143–151. [Google Scholar] [CrossRef]
- Song, K.; Xu, H.; Xie, K.; Yang, Y. Keratin-Based Biocomposites Reinforced and Cross-Linked with Dual-Functional Cellulose Nanocrystals. ACS Sustain. Chem. Eng. 2017, 5, 5669–5678. [Google Scholar] [CrossRef]
- Kaur, M.; Arshad, M.; Ullah, A. In-Situ Nanoreinforced Green Bionanomaterials from Natural Keratin and Montmorillonite (MMT)/Cellulose Nanocrystals (CNC). ACS Sustain. Chem. Eng. 2018, 6, 1977–1987. [Google Scholar] [CrossRef]
- Liebeck, B.; Hidalgo, N.; Roth, G.; Popescu, C.; Böker, A. Synthesis and Characterization of Methyl Cellulose/Keratin Hydrolysate Composite Membranes. Polymers 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Chen, X.; Zhou, H.; Zhou, X.; Xu, H.; Chen, H. Elaboration of a feather keratin/carboxymethyl cellulose complex exhibiting pH sensitivity for sustained pesticide release. J. Appl. Polym. Sci. 2019, 136, 47160. [Google Scholar] [CrossRef]
- Wang, X.; Lu, C.; Chen, C. Effect of chicken-feather protein-based flame retardant on flame retarding performance of cotton fabric. J. Appl. Polym. Sci. 2014, 131, 40584. [Google Scholar] [CrossRef]
- De Silva, R.; Wang, X.; Byrne, N. Tri-component bio-composite materials prepared using an eco-friendly processing route. Cellulose 2013, 20, 2461–2468. [Google Scholar] [CrossRef]
- De Silva, R.; Vongsanga, K.; Wang, X.; Byrne, N. Development of a novel regenerated cellulose composite material. Carbohydr. Polym. 2015, 121, 382–387. [Google Scholar] [CrossRef]
- De Silva, R.; Wang, X.; Byrne, N. Development of a novel cellulose/duck feather composite fibre regenerated in ionic liquid. Carbohydr. Polym. 2016, 153, 115–123. [Google Scholar] [CrossRef]
- Kammiovirta, K.; Jääskeläinen, A.-S.; Kuutti, L.; Holopainen-Mantila, U.; Paananen, A.; Suurnäkki, A.; Orelma, H. Keratin-reinforced cellulose filaments from ionic liquid solutions. RSC Adv. 2016, 6, 88797–88806. [Google Scholar] [CrossRef]
- Tran, C.D.; Mututuvari, T.M. Cellulose, Chitosan and Keratin Composite Materials: Facile and Recyclable Synthesis, Conformation and Properties. ACS Sustain. Chem. Eng. 2016, 4, 1850–1861. [Google Scholar] [CrossRef]
- Tran, C.D.; Mututuvari, T.M. Cellulose, Chitosan, and Keratin Composite Materials. Controlled Drug Release. Langmuir 2015, 31, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Sahariah, P.; Másson, M. Antimicrobial Chitosan and Chitosan Derivatives: A Review of the Structure–Activity Relationship. Biomacromolecules 2017, 18, 3846–3868. [Google Scholar] [CrossRef] [PubMed]
- Zargar, V.; Asghari, M.; Dashti, A. A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Gassner, G.; Schmidt, W.; Line, M.J.; Thomas, C.; Waters, R. Fiber and Fiber Products Produced From Feathers. U.S. Patent No. 5,705,030, 6 January 1998. [Google Scholar]
- Flores-Hernández, C.; Colín-Cruz, A.; Velasco-Santos, C.; Castaño, V.; Rivera-Armenta, J.; Almendarez-Camarillo, A.; García-Casillas, P.; Martínez-Hernández, A. All Green Composites from Fully Renewable Biopolymers: Chitosan-Starch Reinforced with Keratin from Feathers. Polymers 2014, 6, 686–705. [Google Scholar] [CrossRef] [Green Version]
- Flores-Hernandez, C.G.; Martinez-Hernandez, A.L.; Colin-Cruz, A.; Martinez-Bustos, F.; Castaño, V.M.; Olivas-Armendariz, I.; Almendarez-Camarillo, A.; Velasco-Santos, C. Starch Modified With Chitosan and Reinforced With Feather Keratin Materials Produced by Extrusion Process: An Alternative to Starch Polymers. Starch Stärke 2018, 70, 1700295. [Google Scholar] [CrossRef]
- Hsieh, S.-H.; Huang, Z.K.; Huang, Z.Z.; Tseng, Z.S. Antimicrobial and physical properties of woolen fabrics cured with citric acid and chitosan. J. Appl. Polym. Sci. 2004, 94, 1999–2007. [Google Scholar] [CrossRef]
- Ghosh, A.; Grosvenor, A.J.; Dyer, J.M. Improving the properties of chemically damaged wool fabrics with carbohydrate polymers. J. Appl. Polym. Sci. 2013, 130, 3105–3111. [Google Scholar] [CrossRef]
- Ranjbar-Mohammadi, M.; Hajir Bahrami, S.; Arami, M. Eco-friendly grafting of natural biopolymer chitosan onto acylated wool fabrics using ultrasonic and study its properties. J. Appl. Polym. Sci. 2013, 129, 707–713. [Google Scholar] [CrossRef]
- Shanmugasundaram, O.L.; Ahmed, K.S.Z.; Sujatha, K.; Ponnmurugan, P.; Srivastava, A.; Ramesh, R.; Sukumar, R.; Elanithi, K. Fabrication and characterization of chicken feather keratin/polysaccharides blended polymer coated nonwoven dressing materials for wound healing applications. Mater. Sci. Eng. C 2018, 92, 26–33. [Google Scholar] [CrossRef]
- Saravanan, S.; Sameera, D.K.; Moorthi, A.; Selvamurugan, N. Chitosan scaffolds containing chicken feather keratin nanoparticles for bone tissue engineering. Int. J. Biol. Macromol. 2013, 62, 481–486. [Google Scholar] [CrossRef]
- Ma, B.; Chen, W.; Qiao, X.; Pan, G.; Jakpa, W.; Hou, X.; Yang, Y. Tunable wettability and tensile strength of chitosan membranes using keratin microparticles as reinforcement. J. Appl. Polym. Sci. 2017, 134, 44667. [Google Scholar] [CrossRef]
- Eslahi, N.; Simchi, A.; Mehrjoo, M.; Shokrgozar, M.A.; Bonakdar, S. Hybrid cross-linked hydrogels based on fibrous protein/block copolymers and layered silicate nanoparticles: Tunable thermosensitivity, biodegradability and mechanical durability. RSC Adv. 2016, 6, 62944–62957. [Google Scholar] [CrossRef]
- Zahedi, E.; Esmaeili, A.; Eslahi, N.; Shokrgozar, M.A.; Simchi, A. Fabrication and Characterization of Core-Shell Electrospun Fibrous Mats Containing Medicinal Herbs for Wound Healing and Skin Tissue Engineering. Mar. Drugs 2019, 17, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-W.; Chen, Y.-K.; Lu, M.; Lou, K.-L.; Yu, J. Photo-Crosslinked Keratin/Chitosan Membranes as Potential Wound Dressing Materials. Polymers 2018, 10, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakkar, P.; Verma, S.; Manjubala, I.; Madhan, B. Development of keratin–chitosan–gelatin composite scaffold for soft tissue engineering. Mater. Sci. Eng. C 2014, 45, 343–347. [Google Scholar] [CrossRef]
- Balaji, S.; Kumar, R.; Sripriya, R.; Kakkar, P.; Ramesh, D.V.; Reddy, P.N.K.; Sehgal, P.K. Preparation and comparative characterization of keratin–chitosan and keratin–gelatin composite scaffolds for tissue engineering applications. Mater. Sci. Eng. C 2012, 32, 975–982. [Google Scholar] [CrossRef]
- Singaravelu, S.; Ramanathan, G.; Raja, M.D.; Barge, S.; Sivagnanam, U.T. Preparation and characterization of keratin-based biosheet from bovine horn waste as wound dressing material. Mater. Lett. 2015, 152, 90–93. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [Green Version]
- Wrześniewska-Tosik, K.; Adamiec, J. Biocomposites with a content of keratin from chicken feathers. Fibres Text. East. Eur. 2007, 15, 106–112. [Google Scholar]
- Hamasaki, S.; Tachibana, A.; Tada, D.; Yamauchi, K.; Tanabe, T. Fabrication of highly porous keratin sponges by freeze-drying in the presence of calcium alginate beads. Mater. Sci. Eng. C 2008, 28, 1250–1254. [Google Scholar] [CrossRef]
- Gupta, P.; Nayak, K.K. Compatibility study of alginate/keratin blend for biopolymer development. J. Appl. Biomater. Funct. Mater. 2015, 13, 332–339. [Google Scholar] [CrossRef]
- He, M.; Zhang, B.; Dou, Y.; Yin, G.; Cui, Y. Blend modification of feather keratin-based films using sodium alginate. J. Appl. Polym. Sci. 2017, 134, 44680. [Google Scholar] [CrossRef]
- Srisuwan, Y.; Srihanam, P. Preparation and Characterization of Keratin/Alginate Blend Microparticles. Adv. Mater. Sci. Eng. 2018, 2018, 8129218. [Google Scholar] [CrossRef] [Green Version]
- Bertoft, E. Understanding Starch Structure: Recent Progress. Agronomy 2017, 7, 56. [Google Scholar] [CrossRef]
- Wojtowicz, A.; Janssen, L.P.B.M.; Moscicki, L. Blends of Natural and Synthetic Polymers. In Thermoplastic Starch; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 35–53. ISBN 9783527628216. [Google Scholar]
- Rabe, S.; Sanchez-Olivares, G.; Pérez-Chávez, R.; Schartel, B. Natural Keratin and Coconut Fibres from Industrial Wastes in Flame Retarded Thermoplastic Starch Biocomposites. Materials 2019, 12, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertweck, C. Biosynthesis and Charging of Pyrrolysine, the 22nd Genetically Encoded Amino Acid. Angew. Chemie Int. Ed. 2011, 50, 9540–9541. [Google Scholar] [CrossRef]
- Hu, X.; Cebe, P.; Weiss, A.S.; Omenetto, F.; Kaplan, D.L. Protein-based composite materials. Mater. Today 2012, 15, 208–215. [Google Scholar] [CrossRef]
- Cui, L.; Wang, Q.; Wang, P.; Huan, Q.; Fan, X. Transglutaminase-mediated crosslinking of gelatin onto wool surfaces to improve the fabric properties. J. Appl. Polym. Sci. 2009, 113, 2598–2604. [Google Scholar] [CrossRef]
- Prasong, S.; Wasan, T. Preparation and Characterization of Hair Keratin/Gelatin Blend Films. Pakistan J. Biol. Sci. 2011, 14, 351–356. [Google Scholar] [CrossRef]
- Thonpho, A.; Srihanam, P. Preparation and Characterization of Keratin Blended Films using Biopolymers for Drug Controlled Release Application. Orient. J. Chem. 2016, 32, 1739–1748. [Google Scholar] [CrossRef] [Green Version]
- Ramadoss, P.; Thanigai Arul, K.; Ramana Ramya, J.; Rigana Begam, M.; Sarath Chandra, V.; Manikandan, E. Enhanced mechanical strength and sustained drug release of gelatin/keratin scaffolds. Mater. Lett. 2017, 186, 109–112. [Google Scholar] [CrossRef]
- Maruthi, Y.; Sudhakar, H.; Rao, U.S.; Babu, P.K.; Rao, K.C.; Subha, M.C.S. Blend Membranes of Sodium alginate and Soya protein for Pervaporation Dehydration of Isopropanol. Adv. Polym. Sci. Technol. 2014, 4, 12–21. [Google Scholar]
- Li, S.; Donner, E.; Xiao, H.; Thompson, M.; Zhang, Y.; Rempel, C.; Liu, Q. Preparation and characterization of soy protein films with a durable water resistance-adjustable and antimicrobial surface. Mater. Sci. Eng. C 2016, 69, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Tansaz, S.; Liverani, L.; Vester, L.; Boccaccini, A.R. Soy protein meets bioactive glass: Electrospun composite fibers for tissue engineering applications. Mater. Lett. 2017, 199, 143–146. [Google Scholar] [CrossRef]
- Hammann, F.; Schmid, M. Determination and Quantification of Molecular Interactions in Protein Films: A Review. Materials 2014, 7, 7975–7996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santis, M.A.; Giuliani, M.M.; Giuzio, L.; De Vita, P.; Lovegrove, A.; Shewry, P.R.; Flagella, Z. Differences in gluten protein composition between old and modern durum wheat genotypes in relation to 20th century breeding in Italy. Eur. J. Agron. 2017, 87, 19–29. [Google Scholar] [CrossRef]
- Lucas, I.; Becker, T.; Jekle, M. Gluten Polymer Networks—A Microstructural Classification in Complex Systems. Polymers 2018, 10, 617. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meng, D.; Wang, S.; Zhang, Z.; Yang, R.; Zhao, W. Modification of wheat gluten for improvement of binding capacity with keratin in hair. R. Soc. Open Sci. 2018, 5, 171216. [Google Scholar] [CrossRef] [Green Version]
- Garrido, T.; Leceta, I.; de la Caba, K.; Guerrero, P. Chicken feathers as a natural source of sulphur to develop sustainable protein films with enhanced properties. Int. J. Biol. Macromol. 2018, 106, 523–531. [Google Scholar] [CrossRef]
- Lefèvre, T.; Rousseau, M.-E.; Pézolet, M. Protein Secondary Structure and Orientation in Silk as Revealed by Raman Spectromicroscopy. Biophys. J. 2007, 92, 2885–2895. [Google Scholar] [CrossRef] [Green Version]
- Jao, D.; Mou, X.; Hu, X. Tissue Regeneration: A Silk Road. J. Funct. Biomater. 2016, 7, 22. [Google Scholar] [CrossRef]
- Vu, T.; Xue, Y.; Vuong, T.; Erbe, M.; Bennet, C.; Palazzo, B.; Popielski, L.; Rodriguez, N.; Hu, X. Comparative Study of Ultrasonication-Induced and Naturally Self-Assembled Silk Fibroin-Wool Keratin Hydrogel Biomaterials. Int. J. Mol. Sci. 2016, 17, 1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Youbo, D.; Zhou, Z.; Xing, W.; Chunli, Q.; Libin, G. Preparation and characterization of protein/viscose fiber and its action in self-heating. J. Appl. Polym. Sci. 2019, 136, 47146. [Google Scholar] [CrossRef]
- Baden, H.P. Structure of epidermal keratin and variations in its polypeptide composition. Curr. Probl. Dermatol. 1980, 10, 345–363. [Google Scholar] [PubMed]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; Nič, M., Jirát, J., Košata, B., Jenkins, A., McNaught, A., Eds.; IUPAC: Carolina Research Triagle Park, NC, USA, 2006; ISBN 0-9678550-9-8. [Google Scholar]
- Wang, X.; Peng, Y. Comparative study of the structure and properties of wool treated by a chicken-feather keratin agent, plasma, and their combination. J. Appl. Polym. Sci. 2011, 119, 1627–1634. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Associated Polymer | Source | Amount (wt.-%) | Additives * | Mechanical Properties (MPa) ** | Thermal Properties (°C) *** | Preparation Method | Obs. **** | Ref. |
|---|---|---|---|---|---|---|---|---|
| PP | feather | 30 | MPR/APS | TS ~ 35; E ~ 2500; σ ~ 50; Ebend ~ 3000; IS ~ 55(J/m) | - | melt mixing | - | [108] |
| 30 | MAPP | E = 3590; TS = 40.71; Ebend = 3530; σ = 75.20 | - | compression moulding | composite panels | [104] | ||
| 5–15 | - | E = 1167–1451; TS = 40.71; Ebend = 3530; σ = 75.20 | Tg(α) = 109–137; Tm ~ 156–169; Td(max) ~ 300–400 | melt mixing/compression moulding | - | [110] | ||
| wool | 30 | MAPP | - | Td(max) ~ 450 | melt mixing | fire retardant | [107] | |
| LDPE | feather | 20 | - | E ~ 80; YS ~ 17 | - | melt mixing | [106] | |
| PEO | wool | 20–60 | - | - | Tm = 64.4–53.5; Tc = 41.6–9.7 | solvent casting | - | [95] |
| 10–70 | - | E = 12–7; σbreak = 4.7–1.6 | Tm ~ 200–220 | Electrospinning | fibres | [116] | ||
| hair | 90 | EGDE | Td(max) ~ 290–420 | Electrospinning | fibres | [119] | ||
| feather | 90 | graphene | E ~ 1200; Er ~ 12000; Hi ~ 570 | - | solvent casting | films | [120] | |
| PEG | hair | 43 | - | E ~ 0.030; Ecomp = 0.045 | - | photo-crosslink (thiol-norbornene “click”) | hydrogel | [121] |
| PMMA | wool | - | - | TS ~ 152 | Tm ~ 230; Td(50%) ~ 390 | graft-copolymerization | fibres | [139,140] |
| HEMA | wool | - | - | Lmax ~ 0.20(kN), εmax = 35.71(%) | - | graft-copolymerization | fibres | [141] |
| PBzMA | wool | - | - | TS = 377(g), εbreak = 40.0(%) | Tm ~ 232–278, Td(10%) = 300, Tg ~90 | graft-copolymerization | fibres | [143] |
| PMMA PEMA;PBMA PHMA | feather | - | - | σbreak ~ 7.0; εbreak = 45.5 | Td = 380–520 | graft-copolymerization/compression moulding | films | [146] |
| PAM | wool | - | - | TS = 247(g); εbreak = 1.7(%) | Td(10%) ~ 235; Td(50%) ~ 665 | graft-copolymerization | fibres | [148] |
| PVOH | wool | 10–30 | - | Tn = 1.49–1.57(cN/dtex); εbreak = 22.1–41.1(%) | Td(max) ~ 260 | wet-spinning | fibres | [159] |
| 5–25 | - | TS = 15.8(cN), εbreak = 77.6(%) | Td(5%) ~ 230 | wet-spinning | fibres | [160] | ||
| Feather | 20 | Citric acid/glyoxal | TS ~ 12; εbreak ~ 150(%) | Tm ~ 60, 200 and 270; Td(5%) ~ 250; Td(50%) ~ 360 | Electrospinning | fibres | [162] | |
| 80 | DAS | TS ~ 15–21; εbreak ~ 2.5–9.0(%) | Tg = 100–125; Tm ~ 220; Td ~ 250 | solution casting | films | [165] | ||
| 50–90 | Tris | E ~ 16–2007; TS ~ 4–23(%); εbreak = 2–262(%) | - | solution casting | films | [167] | ||
| hair | 92 | β-mercapto ethanol/IL/thioglycolic acid | TS ~ 6 | Tm ~ 290 | Electrospinning | fibres | [163] | |
| PA6 | feather | 20–80 | - | E = 825–1444; Sn = 1.42–2.86(N m−1) | - | spin-coating | films | [173] |
| PCL | hair | 10–40 | - | E = 4.5–8; σbreak = 1–2 | - | Electrospinning | membranes | [179] |
| 10 | MgO | E = 5.5; TS = 3.0; | - | Electrospinning | membranes | [180] | ||
| 30 | Glutaraldehyde/Ca3(PO4)2 | E = 25.92; TS = 16.53; εbreak = 153(%) | - | Electrospinning | scaffolds | [181] | ||
| PLA | Wool/fleece | 1–5 | - | σbreak = 13–28; εbreak = 9–197(%); E = 420–1200 | Tg = 61–64; Tc = 100–109; Tm ~ 180; Td(max) ~ 300–360 | solvent casting | films | [185] |
| feather | 30 | MPR/APS | TS ~ 50; E ~ 4500; σ ~ 90; Ebend ~ 9000; IS ~ 25(J/m) | Tg ~ 55; Tc ~ 87; Tm ~ 170; Td(50%) ~ 400 | melt mixing | - | [108] | |
| 2–4 | chitosan | E ~ 2300–3300; TS ~ 35–65; εbreak = 1.3–3.8(%); IS ~ 7–11(KJ/m2) | Td(50%) ~ 330–365 | melt mixing | - | [188] | ||
| 50–60 | copolymer blend/PEG400 | E ~ 1500–2100; TS ~ 5–22; εbreak = 1.4–1.9(%) | Tg ~ 47–66; Tm ~ 145–179; Td(50%) ~ 321–325 | melt mixing/compression moulding | films | [189] | ||
| 5–25 (vol.-%) | - | E ~ 2800–3100; TS ~ 21–36; εbreak = 1.6–1.9(%) | - | melt mixing/compression moulding | films | [190] | ||
| TPU | feather | 11–21 | PPG | - | Tg ~ 50; Tm ~ 133; Td(max) ~ 418–424 | graft-copolymerization | membranes | [204] |
| 10–80 | - | Ec = 60–200; TS ~ 5–29; εbreak = 183–276(%); Ecomp = 40–145(scaffold) | Tg ~ −42 | solvent casting | scaffolds | [206] | ||
| 10–70 | - | E ~ 100–1190; εmax = 0.6–2.5(%) | Tg ~ −45; Td(5%) ~ 249–309 | solvent casting/compression moulding | films | [207] | ||
| hair | 0.5–10 | - | E = 31.4; TS ~ 18.5; εbreak = 570(%) | - | melt mixing/compression moulding | artificial skin | [209] | |
| TPUU | feather | 40–75 | bis(4-aminophenyl) disulphide/bis(4-aminophenyl) methane | E ~ 67–408; TS ~ 2.8–11.2; εbreak = 1.9–30.7(%) | Td(5%) ~ 204–237; Td(50%) ~ 340–361 | melt mixing/compression moulding | films | [210] |
| PHBV | feather | 0.5–50 | - | E ~ 540–865; εbreak = 2.8–4.7(%) | Tc ~ 102; Tm ~ 147–157 | melt mixing/compression moulding | films | [197] |
| 0.5–5 | - | E ~ 770–1840;TS ~ 16–30; εbreak = 0.7–5.3(%) | Tc ~ 101–115; Tm ~ 136–184 | electrospinning/solvent casting/melt mixing/compression moulding | films | [198] | ||
| PBAT | Feather | 50–60 | PEG400 | E ~ 300–600; TS ~ 3–5; εbreak = 1.8–2.8(%) | Tg ~ −30; Tm ~ 117; Td(50%) ~ 369–377 | melt mixing/compression moulding | films | [189] |
| Associated Polymer | Source | Amount (wt.-%) | Additives* | Mechanical Properties (MPa) ** | Thermal Properties (°C) *** | Preparation Method | Obs. **** | Ref. |
|---|---|---|---|---|---|---|---|---|
| XNBR | hair | 5 | ZnO\EG | E = 2200–3000; TS ~ 11.4–14.4; εbreak = 371–406(%); Hs = 54–58(N) | Tg = 3.0–6.6 | roll-milling/vulcanization | elastomer film | [217] |
| SBR | hair | 5–10 | ZnO | TS ~ 1.4–2.6; εbreak = 134–156(%) | Td(5%) = 235–250; Td(50%) = 395–405 | roll-milling/vulcanization | elastomer film | [218] |
| feather | 1–5 | TPS/EVOH | E = 3000–5000 | Tg = (−95)–(−82) and (−4)–131; Td(5%) ~ 250–300; Td(50%) ~ 430 | melt-mixing/compression moulding | elastomer film | [222] | |
| NBR | hair | 5–30 | ZnO/MMC | TS ~ 1.9–5.6; εbreak = 350–895(%) | Tg ~ −50; Td(5%) = 280–335; Td(50%) = 410–420 | roll-milling/vulcanization | elastomer film | [219] |
| XSBL | hair | - | cotton fabric | film [TS ~ 11–13; εbreak = 310–345(%); Hs = 53–55(°)]. coating [TS ~ 24–26; εbreak ~17(%); Hs = 52–55(°) | film [Tg ~ −8.5; Td(5%) ~ 340; Td(50%) ~ 428]. coating [Tg ~ 85; Td(5%) ~ 306; Td(50%) = 333–362] | coating/solvent casting | fibre coating | [220] |
| Epoxy (DGEBF) | feather | 11–69 (vol.-%) | diamine/E-glass | E ~ 3500–10500; σ = 50–310; Ebend = 2350–13400; εbreak = 2.5–6.0(%) | Tg ~ 120–130 | moulding | films | [232] |
| Epoxy (DGEBA) | feather | 1–7 | TETA/CR | E ~ 521–1126; TS ~ 16–38; σcomp ~ 55–81; Ebend = 3010–4420; σ = 57–79; IS = 2.06–3.43(kJ/m2) | - | hand lay-up | coating | [234] |
| Epoxy (AESO) | feather | 5–45 | glass fibre | E = 1291–2085; Lmax = 97–131(N); Ef = 1.61–1.98(kJ/m2); Ebend = 971–1938; σ ~ 36–58 | Tg ~ 70 | moulding | mats | [235] |
| 5–32 | MLAU | Es = 20–300(at 25°C); E = 10–150(at 25°C); Ef = 2.2–7.5(kJ/m2) | Tg = 23–29 | moulding | films | [240] | ||
| Epoxy (PAESO) | feather | ~30 (vol.-%) | E-glass | Ebend = 8860–10570; σ ~ 84–100; εbreak ~ 1.23(%) | Tg ~ 107–112; Td(5%) = 308–324 306; Td(50%) = 391–414 | compression moulding | circuit board | [236] |
| Urea-formaldehyde | feather | 25–75 | sawmill hardwood residue | TS = 0.65–1.85; σ = 2.4–3.2; σcomp = 0.86–1.42; τ = 0.80–0.98; | - | compression moulding | particle board | [246] |
| Phenol-formaldehyde | feather | 2.5–95 | wood fibre | Ebend = 1470–3170; σ ~ 11–25 | - | compression moulding | MDF | [253] |
| ~33 | wood fibreboard | Ebend = 2339–3179; σ ~ 32–43; IB = 0.29–0.76 | - | compression moulding | MDF | [254] |
| Polymer Source | Keratin Source | Keratin (wt.-%) | Additives * | Mechanical Properties (MPa) ** | Thermal Properties (°C) *** | Preparation Method | Obs. **** | Ref. |
|---|---|---|---|---|---|---|---|---|
| lignocellulose | feather | 30–35 | glycerol | E = 36–60; σbreak ~ 1.8–4.3; εbreak = 0.076–0.171(cm/cm) | Td(max) ~ 300-330; | compression moulding | films | [260] |
| CNC | feather | 90–99 | - | E ~ 451; σbreak ~ 5–23; εbreak ~ 8–28(%) | - | solution casting | films | [261] |
| chitosan/glycerol | TS ~ 4.6–5.3; εbreak ~ 21.1–27.6(%) | Tg(α) ~ 35–65; Tm ~ 229–254; Td(max) ~ 310 | compression moulding | films | [262] | |||
| wood cellulose | feather | 22–58 | glycerol | E ~ 149–544; YS ~ 11–28; εbreak ~ 45-94(%) | Td(max) ~ 300–360 | solution casting | films | [263] |
| 10–70 | ILs | E ~ 6900–7500; TS = 88.4–142.4; εbreak ~ 9.0–19.3(%); Sn = 5917–7166(N/m); Tn = 20–46(cN/tex) | - | wet-spinning | Fibres/filaments | [269] | ||
| wool | 25–75 | ILs | TS ~ 9–38 | Td ~ 270–305 | solution casting | films | [270] | |
| cotton cellulose | wool | 5–25 | ILs | E ~ 1610–1790; σbreak ~ 25–48; εbreak ~ 1.3-3.3(%) | Td ~ 227–278 | co-solvent coagulation | films | [266] |
| feather | 2.5–20 | ILs | E ~ 1640–1730; σbreak ~ 26–53; εbreak ~ 4.5–11.6(%) | Td ~ 278–289 | co-solvent coagulation | films | [267] | |
| 5–20 | ILs | E ~ 5540–17220; σbreak ~ 75–132; εbreak ~ 1.4–7.1(%) | - | wet-spinning | fibres | [268] | ||
| chitosan | Wool | - | - | σbreak ~ 13–20; εbreak ~ 26–45(%) | Tm ~ 230 | Crosslinking | fibre coating | [277] |
| - | cellulose | [recovered properties] σbreak ~ 17; εbreak ~ 42(%) | - | solution coating | fibre coating | [278] | ||
| 77–90 | glycerol | E ~ 14–735; TS ~ 3–37; εbreak ~ 4–31(%) | - | solution casting | films | [93] | ||
| ~10 | Pluronic F127/genepin/laponite | G’ = 2.5–71.3 | - | Crosslinking | Injectable hydrogel | [283] | ||
| 5–15 | PCL/PEO | TS ~ 3.2–5.3; εbreak ~ 10–63(%); Ebreak = 0.34–2.34(J/m3) | - | Electrospinning | fibres | [284] | ||
| Feather | 5–20 | sorbitol/starch | E ~ 241–1142 | Tg(α) ~ 159–196; Tm ~ 200; Td(50%) ~ 300–340 | solution casting | films | [275] | |
| 5–10 | sorbitol/starch | Es ~ 1158–2972; E ~ 29–826; σmax ~ 2.0–15.6 | Tg(α) ~ 70–104; Td(50%) ~ 290–300 | solution casting/melt-mixing | films | [276] | ||
| 1–15 | - | TS ~ 40–65; εbreak ~ 10–15(%) | Td(max) ~ 277–331 | solution casting/coagulation | membranes | [282] | ||
| Hair | 25–50 | - | TS ~ 22–28 | - | Crosslinking | membranes | [285] | |
| Hoof | 25 | gelatine | Ecomp = 0.005–0.009; E ~ 0.010–0.096 | Td(50%) ~ 361 | freeze-drying | scaffold | [286] | |
| Horn | 67 | - | TS ~ 1.6; εbreak ~ 21.6(%); Lmax = 6.3(N); εmax = 5.12(mm) | Tm ~ 216; Tc ~ 180 | freeze-drying | scaffold | [287] | |
| 25–50 | ethylene glycol | E ~ 0.5–3.2; TS ~ 7.4–21.1; εbreak ~ 6.2–16.0(%) | Td ~ 300; Tm ~ 145–175 | solution casting | films | [288] | ||
| alginate | Feather | 10 | glycerol | E ~ 0.08–0.38; εbreak ~ 31–60(%) | - | solution casting | films | [292] |
| 50–90 | sorbitol | E ~ 6.0–16.3; εbreak ~ 25–29(%) | Tc = 211–218 | solution casting | films | [293] | ||
| starch | Hair | 15–30 | APP | E ~ 165–247; TS ~ 10–13; Tn = 2–19; εbreak ~ 26–190(%); IS = 81–397(J/m) | Td(5%) ~ 211–250; Td(max) ~ 361–397; | melt-mixing | flame retardant | [297] |
| Polymer Source | Keratin Source | Keratin (wt.-%) | Additives * | Mechanical Properties (MPa) ** | Thermal Properties (°C) *** | Preparation Method | Obs. **** | Ref. |
|---|---|---|---|---|---|---|---|---|
| gelatine | wool | - | TGas | TS ~ 26.8 (270–360N) | - | crosslinking | fibre coating | [300] |
| hair | 33–67 | - | - | Tm ~ 250; Td(max) ~ 310–340; | solution casting | films | [301,302] | |
| n.d. | 10 | glutaraldehyde | E ~ 0.23 | - | freeze-drying | scaffolds | [303] | |
| soy gluten | feather | 3–9 | glycerol | E ~ 97–109; TS ~ 4.8–9.5; εbreak = 94–110(%) | Tg(α) ~ −30/55; Td(max) ~ 310 | solution casting/compression moulding | films | [311] |
| Silk fibroin | wool | 20–50 | - | E = 1409–2724; TS = 9153–29755; εbreak = 0.36–0.68(mm) | Tg ~ 178; Tm ~ 224–278; Tc ~ 226; Td ~ 274–296 | solution casting | films | [94] |
| 10–90 | - | - | Tg ~ 154–171; Td ~ 274–300; | crosslinking | hydrogels | [314] | ||
| 0.7–2.36 | PVF/cotton | Tn ~ 2.1–2.6(cN/dtex); εbreak ~ 23.4–30.2(%) | - | crosslinking | fibre | [315] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, R.K.; Mija, A. Keratin Associations with Synthetic, Biosynthetic and Natural Polymers: An Extensive Review. Polymers 2020, 12, 32. https://doi.org/10.3390/polym12010032
Donato RK, Mija A. Keratin Associations with Synthetic, Biosynthetic and Natural Polymers: An Extensive Review. Polymers. 2020; 12(1):32. https://doi.org/10.3390/polym12010032
Chicago/Turabian StyleDonato, Ricardo K., and Alice Mija. 2020. "Keratin Associations with Synthetic, Biosynthetic and Natural Polymers: An Extensive Review" Polymers 12, no. 1: 32. https://doi.org/10.3390/polym12010032
APA StyleDonato, R. K., & Mija, A. (2020). Keratin Associations with Synthetic, Biosynthetic and Natural Polymers: An Extensive Review. Polymers, 12(1), 32. https://doi.org/10.3390/polym12010032