Proteasome Biology: Chemistry and Bioengineering Insights

Abstract
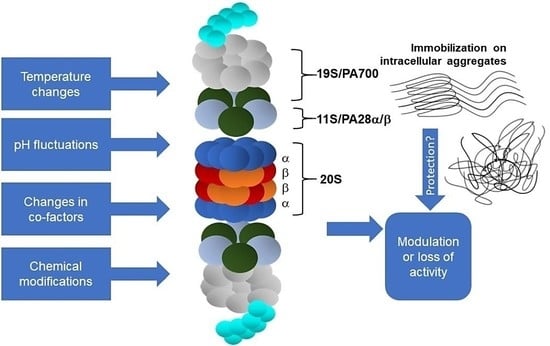
:
1. Introduction
Kinetics of Proteasomal Degradation
2. Physico-Chemical Parameters of the Cellular Environment
2.1. Effect of pH Changes
2.2. Effect of Temperature Variation
2.3. Effect of Changes in ATP and Mg2+ Cofactor Levels
3. Intracellular Chemical Modifications of the Proteasome
3.1. Regulatory Covalent Modifications
3.1.1. S-Glutathionylation
3.1.2. S-Nitrosylation and Other S-Modifications
3.1.3. Poly-ADP Ribosylation
3.1.4. Phosphorylation
3.1.5. O-GlcNAcylation
3.1.6. Nα- and Nε-Acetylation
3.1.7. Ubiquitination
3.1.8. Other N-Modifications (SUMOylation, N-Myristoylation, N-Methylation)
3.2. Covalent Modifications of Amino Acid Side Chains by Strong Oxidants and Electrophiles
3.2.1. Protein Carbonylation
3.2.2. HNE Modification
3.2.3. Glycation and Glycoxidation
3.2.4. Tyrosine Nitration
4. Proteasome and Protein Aggregates
4.1. Inhibition vs. Immobilization
4.2. Proteasome Immobilisation In Vivo: Detriments vs. Benefits?
5. Summary and Remaining Questions
Author Contributions
Funding
Conflicts of Interest
References
- Morozov, A.V.; Karpov, V.L. Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Sherman, D.J.; Li, J. Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [Green Version]
- Enenkel, C. Proteasome dynamics. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Kish-Trier, E.; Hill, C.P. Structural Biology of the Proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, M.; Wilk, S. Catalytic Activities of the 20 S Proteasome, a Multicatalytic Proteinase Complex. Arch. Biochem. Biophys. 2000, 383, 1–16. [Google Scholar] [CrossRef]
- Stadtmueller, B.M.; Hill, C.P. Proteasome Activators. Mol. Cell 2011, 41, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Lasker, K.; Förster, F.; Bohn, S.; Walzthoeni, T.; Villa, E.; Unverdorben, P.; Beck, F.; Aebersold, R.; Sali, A.; Baumeister, W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. USA 2012, 109, 1380–1387. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Kulkarni, K.A.; Da Fonseca, P.C.; Krutauz, D.; Glickman, M.H.; Barford, D.; Morris, E.P. The Structure of the 26S Proteasome Subunit Rpn2 Reveals Its PC Repeat Domain as a Closed Toroid of Two Concentric α-Helical Rings. Structure 2012, 20, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, B.-H.; Zhang, N.; De Poot, S.A.H.; Tuebing, F.; Sun, S.; et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 2016, 351, aad9421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nat. Cell Biol. 2008, 453, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nocker, S.; Sadis, S.; Rubin, D.M.; Glickman, M.; Fu, H.; Coux, O.; Wefes, I.; Finley, D.; Vierstra, R.D. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol. Cell. Biol. 1996, 16, 6020–6028. [Google Scholar] [CrossRef] [Green Version]
- Unverdorben, P.; Beck, F.; Śledź, P.; Schweitzer, A.; Pfeifer, G.; Plitzko, J.M.; Baumeister, W.; Förster, F. Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. USA 2014, 111, 5544–5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehmer, M.; Rudack, T.; Beck, F.; Aufderheide, A.; Pfeifer, G.; Plitzko, J.M.; Förster, F.; Schulten, K.; Baumeister, W.; Sakata, E. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. USA 2017, 114, 1305–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wu, J.; Lu, Y.; Ma, Y.-B.; Lee, B.-H.; Yu, Z.; Ouyang, Q.; Finley, D.J.; Kirschner, M.W.; Mao, Y. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, 12991–12996. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, W.L.; Yu, D.; Ouyang, Q.; Lu, Y.; Mao, Y. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef] [Green Version]
- Reis, J.; Guan, X.Q.; Kisselev, A.F.; Papasian, C.J.; Qureshi, A.A.; Morrison, D.C.; Van Way, C.W.; Vogel, S.N.; Qureshi, N. LPS-Induced Formation of Immunoproteasomes: TNF-α and Nitric Oxide Production are Regulated by Altered Composition of Proteasome-Active Sites. Cell Biophys. 2011, 60, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Freudenburg, W.; Gautam, M.; Chakraborty, P.; James, J.; Richards, J.; Salvatori, A.S.; Baldwin, A.; Schriewer, J.; Buller, R.M.L.; Corbett, J.A.; et al. Reduction in ATP Levels Triggers Immunoproteasome Activation by the 11S (PA28) Regulator during Early Antiviral Response Mediated by IFNβ in Mouse Pancreatic β-Cells. PLoS ONE 2013, 8, e52408. [Google Scholar] [CrossRef] [Green Version]
- Kotamraju, S.; Matalon, S.; Matsunaga, T.; Shang, T.; Hickman-Davis, J.; Kalyanaraman, B. Upregulation of immunoproteasomes by nitric oxide: Potential antioxidative mechanism in endothelial cells. Free. Radic. Biol. Med. 2006, 40, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moebius, J.; Broek, M.V.D.; Groettrup, M.; Basler, M. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur. J. Immunol. 2010, 40, 3439–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussong, S.A.; Roehrich, H.; Kapphahn, R.J.; Maldonado, M.; Pardue, M.T.; Ferrington, D.A. A novel role for the immunoproteasome in retinal function. Investig. Opthalmology Vis. Sci. 2011, 52, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Muchamuel, T.; Basler, M.; Aujay, M.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, R. Proteasome activator PA28γ regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 2008, 27, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Drews, O.; Wildgruber, R.; Zong, C.; Sukop, U.; Nissum, M.; Weber, G.; Gomes, A.V.; Ping, P. Mammalian Proteasome Subpopulations with Distinct Molecular Compositions and Proteolytic Activities. Mol. Cell. Proteom. 2007, 6, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Dahlmann, B.; Ruppert, T.; Kuehn, L.; Merforth, S.; Kloetzel, P.-M. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J. Mol. Biol. 2000, 303, 643–653. [Google Scholar] [CrossRef]
- De, M.; Jayarapu, K.; Elenich, L.; Monaco, J.J.; Colbert, R.A.; Griffin, T.A. β2 Subunit Propeptides Influence Cooperative Proteasome Assembly. J. Biol. Chem. 2002, 278, 6153–6159. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Eynde, B.J.V.D. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, K.; Takada, K.; Ohte, Y.; Kondo, H.; Sorimachi, H.; Tanaka, K.; Takahama, Y.; Murata, S. Thymoproteasomes produce unique peptide motifs for positive selection of CD8+ T cells. Nat. Commun. 2015, 6, 7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Haratake, K.; Miyahara, H.; Chiba, T. Proteasome activators, PA28γ and PA200, play indispensable roles in male fertility. Sci. Rep. 2016, 6, 23171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blickwedehl, J.; Agarwal, M.; Seong, C.; Pandita, R.K.; Melendy, T.; Sung, P.; Bangia, N.; Pandita, T.K. Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc. Natl. Acad. Sci. USA 2008, 105, 16165–16170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Grune, T. The proteasome and the degradation of oxidized proteins: Part I—structure of proteasomes. Redox Biol. 2013, 1, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part II – protein oxidation and proteasomal degradation. Redox Biol. 2014, 2, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Raynes, R.; Pomatto, L.C.D.; Davies, K.J.A. Degradation of oxidized proteins by the proteasome: Distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol. Asp. Med. 2016, 50, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Pickering, A.M.; Davies, K.J.A. Differential roles of proteasome and immunoproteasome regulators Pa28αβ, Pa28γ and Pa200 in the degradation of oxidized proteins. Arch. Biochem. Biophys. 2012, 523, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Luciani, F.; Kesmir, C.; Mishto, M.; Or-Guil, M.; De Boer, R.J. A Mathematical Model of Protein Degradation by the Proteasome. Biophys. J. 2005, 88, 2422–2432. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.L.; Melandri, A.F.; Dick, L. Kinetic Characterization of the Chymotryptic Activity of the 20S Proteasome. Biochemistry 1996, 35, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Akopian, T.N.; Castillo, V.; Goldberg, A.L. Proteasome Active Sites Allosterically Regulate Each Other, Suggesting a Cyclical Bite-Chew Mechanism for Protein Breakdown. Mol. Cell 1999, 4, 395–402. [Google Scholar] [CrossRef]
- Stohwasser, R.; Salzmann, U.; Giesebrecht, J.; Kloetzel, P.-M.; Holzhütter, H.-G. Kinetic evidences for facilitation of peptide channelling by the proteasome activator PA28. JBIC J. Biol. Inorg. Chem. 2000, 267, 6221–6230. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Emch, S.; Groettrup, M.; Holzhütter, H.-G. Evidence for the Existence of a Non-catalytic Modifier Site of Peptide Hydrolysis by the 20 S Proteasome. J. Biol. Chem. 2000, 275, 22056–22063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzhütter, H.-G.; Kloetzel, P.-M. A Kinetic Model of Vertebrate 20S Proteasome Accounting for the Generation of Major Proteolytic Fragments from Oligomeric Peptide Substrates. Biophys. J. 2000, 79, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Köhler, A.; Cascio, P.; Leggett, D.S.; Woo, K.M.; Goldberg, A.L.; Finley, D. The Axial Channel of the Proteasome Core Particle Is Gated by the Rpt2 ATPase and Controls Both Substrate Entry and Product Release. Mol. Cell 2001, 7, 1143–1152. [Google Scholar] [CrossRef]
- Mishto, M.; Luciani, F.; Holzhütter, H.-G.; Bellavista, E.; Santoro, A.; Textoris-Taube, K.; Franceschi, C.; Kloetzel, P.M.; Zaikin, A. Modeling the in Vitro 20S Proteasome Activity: The Effect of PA28–αβ and of the Sequence and Length of Polypeptides on the Degradation Kinetics. J. Mol. Biol. 2008, 377, 1607–1617. [Google Scholar] [CrossRef]
- Xu, L.; Qu, Z. Roles of Protein Ubiquitination and Degradation Kinetics in Biological Oscillations. PloS ONE 2012, 7, e34616. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Lee, B.-H.; King, R.W.; Finley, D.; Kirschner, M.W. Substrate degradation by the proteasome: A single-molecule kinetic analysis. Science 2015, 348, 1250834. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Xu, C.; Sahu, I.; Wang, Y.; Fu, Z.; Huang, M.; Wong, C.C.; Glickman, M.H.; Cong, Y. Structural Snapshots of 26S Proteasome Reveal Tetraubiquitin-Induced Conformations. Mol. Cell 2019, 73, 1150–1161.e6. [Google Scholar] [CrossRef] [Green Version]
- Reichard, E.L.; Chirico, G.G.; Dewey, W.J.; Nassif, N.D.; Bard, K.E.; Millas, N.E.; Kraut, D.A. Substrate Ubiquitination Controls the Unfolding Ability of the Proteasome. J. Biol. Chem. 2016, 291, 18547–18561. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.A.; Bashore, C.; Dong, K.C.; Martin, A. The 26S Proteasome Utilizes a Kinetic Gateway to Prioritize Substrate Degradation. Cell 2019, 177, 286–298.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, P. PA28αβ: The Enigmatic Magic Ring of the Proteasome? Biomolecules 2014, 4, 566–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, A.; Honarvar, A.; Zetterberg, M. Changes in Activity and Kinetic Properties of the Proteasome in Different Rat Organs during Development and Maturation. Curr. Gerontol. Geriatr. Res. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Höhn, T.J.A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part III—Redox regulation of the proteasomal system. Redox Biol. 2014, 2, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear Erythroid Factor 2-mediated Proteasome Activation Delays Senescence in Human Fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Motosugi, R.; Murata, S. Dynamic Regulation of Proteasome Expression. Front. Mol. Biosci. 2019, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Tsujita, T.; Kobayashi, E.H.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yamamoto, M. A Homeostatic Shift Facilitates Endoplasmic Reticulum Proteostasis through Transcriptional Integration of Proteostatic Stress Response Pathways. Mol. Cell. Biol. 2017, 37, 00439-16. [Google Scholar] [CrossRef] [Green Version]
- Moritz, K.E.; McCormack, N.M.; Abera, M.B.; Viollet, C.; Yauger, Y.J.; Sukumar, G.; Dalgard, C.L.; Burnett, B.G. The role of the immunoproteasome in interferon-γ-mediated microglial activation. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Johnston-Carey, H.K.; Pomatto, L.C.D.; Davies, K.J.A. The Immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Luan, Y.; Xiang, D.; Tan, X.; Chen, H.; Deng, Q.; Zhang, J.; Chen, M.; Huang, H.; Wang, W.; et al. The 11S Proteasome Subunit PSME3 Is a Positive Feedforward Regulator of NF-κB and Important for Host Defense against Bacterial Pathogens. Cell Rep. 2016, 14, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, E.; Münzel, T. Intracellular pH: A fundamental modulator of vascular function. Circulation 2011, 124, 1806–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rêgo, A.T.; Da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147.e5. [Google Scholar] [CrossRef] [Green Version]
- Images 6RGQ, 5A5B, 5MX5, 6E5B, 6KWY, 2RAM, 2LZ1, 6XMJ, 1G0U Were Created Using Mol* (Sehnal, D.; Rose, A.S.; Kovca, J.; Burley, S.K.; Velankar, S. Mol*: Towards a Common Library and Tools for Web molecular graphics MolVA/EuroVis Proceedings. 2018, doi:10.2312/molva.20181103.) and RCSB PDB (rcsb.org) H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne. Nucleic Acids Research. 2020, Volume 28, pp. 235–242. Available online: https://academic.oup.com/nar/article/28/1/235/2384399 (accessed on 15 November 2020).
- Aufderheide, A.; Beck, F.; Stengel, F.; Hartwig, M.; Schweitzer, A.; Pfeifer, G.; Goldberg, A.L.; Sakata, E.; Baumeister, W.; Förster, F. Structural characterization of the interaction of Ubp6 with the 26S proteasome. Proc. Natl. Acad. Sci. USA 2015, 112, 8626–8631. [Google Scholar] [CrossRef] [Green Version]
- Huber, E.M.; Groll, M. The Mammalian Proteasome Activator PA28 Forms an Asymmetric α4β3 Complex. Structure 2017, 25, 1473–1480.e3. [Google Scholar] [CrossRef] [Green Version]
- Ladi, E.; Everett, C.; Stivala, C.E.; Daniels, B.E.; Durk, M.R.; Harris, S.F.; Huestis, M.P.; Purkey, H.E.; Staben, S.T.; Augustin, M.; et al. Design and Evaluation of Highly Selective Human Immunoproteasome Inhibitors Reveal a Compensatory Process That Preserves Immune Cell Viability. J. Med. Chem. 2019, 62, 7032–7041. [Google Scholar] [CrossRef]
- Guan, H.; Wang, Y.; Yu, T.; Huang, Y.; Li, M.; Saeed, A.F.U.H.; Perčulija, V.; Li, D.; Xiao, J.; Wang, D.; et al. Cryo-EM structures of the human PA200 and PA200-20S complex reveal regulation of proteasome gate opening and two PA200 apertures. PLoS Biol. 2020, 18, e3000654. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-Q.; Ghosh, S.; Ghosh, G. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Mol. Biol. 1998, 5, 67–73. [Google Scholar] [CrossRef]
- Eletsky, A.; Pulavarti, S.V.S.R.K.; Lee, D.; Kohan, E.; Janjua, H.; Xiao, R.; Acton, T.B.; Everett, J.K.; Montelione, G.T. Solution NMR Structure of the DNA-Binding Domain of Human NF-E2-Related Factor 2 (2012) Northeast Structural Genomics Consortium (NESG) Target HR3520O. 2020, to Be Published, doi:10.2210/pdb2lz1/pdb. Image from the RCSB PDB (Rcsb.Org) of PDB ID: 2LZ1. Available online: https://www.rcsb.org/structure/2lz1 (accessed on 15 November 2020).
- Bailey, J.L.; Wang, X.; England, B.K.; Price, S.R.; Ding, X.; Mitch, W. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J. Clin. Investig. 1996, 97, 1447–1453. [Google Scholar] [CrossRef]
- Peters, L.Z.; Hazan, R.; Breker, M.; Schuldiner, M.; Ben-Aroya, S. Formation and dissociation of proteasome storage granules are regulated by cytosolic pH. J. Cell Biol. 2013, 201, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. To save or degrade: Balancing proteasome homeostasis to maximize cell survival. Autophagy 2018, 14, 2029–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Zheng, D.; Wang, X.; Zhang, Y.; Chen, S.; Cai, X.; Mo, L.; Hu, Z.; Li, H.; Zhou, Z.; et al. Cancer Cell enters reversible quiescence through Intracellular Acidification to resist Paclitaxel Cytotoxicity. Int. J. Med. Sci. 2020, 17, 1652–1664. [Google Scholar] [CrossRef]
- Wojcik, C.; Di Napoli, M. Ubiquitin-Proteasome System and Proteasome Inhibition: New Strategies in Stroke Therapy. Stroke 2004, 35, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.C.; Midtvedt, D.; Franzmann, T.; Nuske, E.; Otto, O.; Herbig, M.; Ulbricht, E.; Müller, P.; Taubenberger, A.; Maharana, S.; et al. A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. eLife 2016, 5, 09347. [Google Scholar] [CrossRef] [PubMed]
- Ugai, S.-I.; Tamura, T.-A.; Tanahashi, N.; Takai, S.; Komi, N.; Chung, C.H.; Tanaka, K.; Ichihara, A. Purification and Characterization of the 26S Proteasome Complex Catalyzing ATP-Dependent Breakdown of Ubiquitin-Ligated Prot from Rat Liver. J. Biochem. 1993, 113, 754–768. [Google Scholar] [CrossRef]
- Klinkradt, S.; Naudé, R.J.; Muramoto, K.; Oelofsen, W. Purification and characterization of proteasome from ostrich liver. Int. J. Biochem. Cell Biol. 1997, 29, 611–622. [Google Scholar] [CrossRef]
- Ostrowska, H.; Ostrowska, J.K.; Worowski, K.; Radziwon, P. Human platelet 20S proteasome: Inhibition of its chymotrypsin-like activity and identification of the proteasome activator PA28. A preliminary report. Platelets 2003, 14, 151–157. [Google Scholar] [CrossRef]
- Lam, T.I.; Brennan-Minnella, A.M.; Won, S.J.; Shen, Y.; Hefner, C.; Shi, Y.; Sun, D.; Swanson, R.A. Intracellular pH reduction prevents excitotoxic and ischemic neuronal death by inhibiting NADPH oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, E4362–E4368. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.-L.; Wang, S.-M.; Chen, C.-C.; Fong, T.-H.; Wu, M.-L. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J. Physiol. 1997, 502, 161–174. [Google Scholar] [CrossRef]
- Reinheckel, T.; Sitte, N.; Ullrich, O.; Kuckelkorn, U.; Davies, K.J.A.; Grune, T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 1998, 335, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, A.M.; Cuccioloni, M.; Bellesi, J.; Lupidi, G.; Fioretti, E.; Angeletti, M. Interaction of Hsp90 with 20S proteasome: Thermodynamic and kinetic characterization. Proteins Struct. Funct. Bioinform. 2002, 48, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.; Barford, D.; Harper, J.W. Purification and Assay of the Budding Yeast Anaphase-Promoting Complex. Methods Enzymol. 2005, 398, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Zünd, G.; Uezono, S.; Stahl, G.L.; Dzus, A.L.; McGowan, F.X.; Hickey, P.R.; Colgan, S.P. Hypoxia enhances induction of endothelial ICAM-1: Role for metabolic acidosis and proteasomes. Am. J. Physiol. Physiol. 1997, 273, C1571–C1580. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Mathur, S.K.; Morimoto, R.I. Heat Shock Response and Protein Degradation: Regulation of HSF2 by the Ubiquitin-Proteasome Pathway. Mol. Cell. Biol. 1998, 18, 5091–5098. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.-J.; Sun, X.; Hasselgren, P.-O. Hyperthermia stimulates energy-proteasome-dependent protein degradation in cultured myotubes. Am. J. Physiol. Integr. Comp. Physiol. 2000, 278, R749–R756. [Google Scholar] [CrossRef] [Green Version]
- Beedholm, R.; Clark, B.F.; Rattan, S.I. Mild heat stress stimulates 20S proteasome and its 11S activator in human fibroblasts undergoing aging in vitro. Cell Stress Chaperon 2004, 9, 49–57. [Google Scholar] [CrossRef]
- Pispa, J.; Matilainen, O.; Holmberg, C.I. Tissue-specific effects of temperature on proteasome function. Cell Stress Chaperon 2020, 25, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Bozaykut, P.; Özer, N.K.; Karademir, B. Nrf2 silencing to inhibit proteolytic defense induced by hyperthermia in HT22 cells. Redox Biol. 2016, 8, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Pajonk, F.; Van Ophoven, A.; McBride, W.H. Hyperthermia-Induced Proteasome Inhibition and Loss of Androgen Receptor Expression in Human Prostate Cancer Cells. Cancer Res. 2005, 65, 4836–4843. [Google Scholar] [CrossRef] [Green Version]
- Kuckelkorn, U.; Knuehl, C.; Boes-Fabian, B.; Drung, I.; Kloetzel, P.-M. The Effect of Heat Shock on 20S/26S Proteasomes. Biol. Chem. 2000, 381, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Romero, J.; Saini, V.; Baker, T.A.; Picken, M.M.; Gamelli, R.L.; Majetschak, M. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem. Biophys. Res. Commun. 2009, 390, 1136–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.M.; Fraga, H.; Reis, C.; Kafri, G.; Goldberg, A.L. ATP Binds to Proteasomal ATPases in Pairs with Distinct Functional Effects, Implying an Ordered Reaction Cycle. Cell 2011, 144, 526–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peth, A.; Nathan, J.A.; Goldberg, A.L. The ATP Costs and Time Required to Degrade Ubiquitinated Proteins by the 26 S Proteasome. J. Biol. Chem. 2013, 288, 29215–29222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höglinger, G.U.; Carrard, G.; Michel, P.P.; Medja, F.; Lombès, A.; Ruberg, M.; Friguet, B.; Hirsch, E.C. Dysfunction of mitochondrial complex I and the proteasome: Interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J. Neurochem. 2003, 86, 1297–1307. [Google Scholar] [CrossRef]
- Vernace, V.A.; Arnaud, L.; Schmidt-Glenewinkel, T.; Figueiredo-Pereira, M.E. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J. 2007, 21, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Meul, T.; Berschneider, K.; Schmitt, S.; Mayr, C.H.; Mattner, L.F.; Schiller, H.B.; Yazgili, A.S.; Wang, X.; Lukas, C.; Schlesser, C.; et al. Mitochondrial Regulation of the 26S Proteasome. Cell Rep. 2020, 32, 108059. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, X.; Li, S.; Liu, N.; Lian, W.; McDowell, E.; Zhou, P.; Zhao, C.; Guo, H.; Zhang, C.; et al. Physiological levels of ATP negatively regulate proteasome function. Cell Res. 2010, 20, 1372–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, S.R.; Davies, K.J.; Divald, A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J. Mol. Cell. Cardiol. 2007, 42, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottenheijm, C.A.C.; Heunks, L.M.A.; Li, Y.-P.; Jin, B.; Minnaard, R.; Van Hees, H.W.H.; Dekhuijzen, P.N.R. Activation of the Ubiquitin–Proteasome Pathway in the Diaphragm in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2006, 174, 997–1002. [Google Scholar] [CrossRef]
- Kors, S.; Geijtenbeek, K.; Reits, E.; Schipper-Krom, S. Regulation of Proteasome Activity by (Post-)transcriptional Mechanisms. Front. Mol. Biosci. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Scruggs, S.B.; Gilda, J.E.; Ping, P.; Gomes, A.V. Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J. Mol. Cell. Cardiol. 2014, 71, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, H.; Kimura, Y.; Kimura, A. Biological significance of co- and post-translational modifications of the yeast 26S proteasome. J. Proteom. 2016, 134, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Sanchez, R.; Riddle, M.; Woo, J.; Momand, J. Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 2008, 17, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Demasi, M.; Netto, L.E.S.; Silva, G.M.; Hand, A.; De Oliveira, C.L.P.; Bicev, R.N.; Gozzo, F.; De Barros, M.H.; Leme, J.D.M.M.; Ohara, E. Redox regulation of the proteasome via S-glutathionylation. Redox Biol. 2014, 2, 44–51. [Google Scholar] [CrossRef]
- Silva, G.M.; Netto, L.; Simões, V.; Santos, L.F.A.; Gozzo, F.C.; Demasi, M.A.A.; De Oliveira, C.L.P.; Bicev, R.N.; Klitzke, C.F.; Sogayar, M.C.; et al. Redox Control of 20S Proteasome Gating. Antioxid. Redox Signal. 2012, 16, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Leme, J.M.; Ohara, E.; Santiago, V.F.; Barros, M.H.; Netto, L.E.; Pimenta, D.C.; Mariano, D.O.; Oliveira, C.L.; Bicev, R.N.; Barreto-Chaves, M.L.; et al. Mutations of Cys and Ser residues in the α5-subunit of the 20S proteasome from Saccharomyces cerevisiae affects gating and chronological lifespan. Arch. Biochem. Biophys. 2019, 666, 63–72. [Google Scholar] [CrossRef]
- Demasi, M.; Shringarpure, R.; Davies, K.J. Glutathiolation of the Proteasome Is Enhanced by Proteolytic Inhibitors. Arch. Biochem. Biophys. 2001, 389, 254–263. [Google Scholar] [CrossRef]
- Silva, G.M.; Netto, L.E.S.; Discola, K.F.; Piassa-Filho, G.M.; Pimenta, D.C.; Bárcena, J.A.; Demasi, M. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J. 2008, 275, 2942–2955. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Kevei, É.; Kleifeld, O.; Krutauz, D.; Segref, A.; Rinaldi, T.; Erpapazoglou, Z.; Cohen, M.; Reis, N.; Hoppe, T.; et al. Reversible 26S Proteasome Disassembly upon Mitochondrial Stress. Cell Rep. 2014, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Banerjee, S.; Abraham, E. S-Glutathionylation of the Rpn2 Regulatory Subunit Inhibits 26 S Proteasomal Function. J. Biol. Chem. 2009, 284, 22213–22221. [Google Scholar] [CrossRef] [Green Version]
- Grune, T.; Catalgol, B.; Licht, A.; Ermak, G.; Pickering, A.M.; Ngo, J.K.; Davies, K.J.A. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free. Radic. Biol. Med. 2011, 51, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Reinheckel, T.; Ullrich, O.; Sitte, N.; Grune, T. Differential Impairment of 20S and 26S Proteasome Activities in Human Hematopoietic K562 Cells during Oxidative Stress. Arch. Biochem. Biophys. 2000, 377, 65–68. [Google Scholar] [CrossRef]
- Obin, M.; Shang, F.; Gong, X.; Handelman, G.; Blumberg, J.; Taylor, A. Redox regulation of ubiquitin-conjugating enzymes: Mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998, 12, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Gould, N.; Doulias, P.-T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of Protein Function and Signaling by Reversible Cysteine S-Nitrosylation. J. Biol. Chem. 2013, 288, 26473–26479. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.I.; Uhrigshardt, H.; O’Meally, R.N.; Cole, R.N.; Van Eyk, J.E. Identification and Quantification of S-Nitrosylation by Cysteine Reactive Tandem Mass Tag Switch Assay. Mol. Cell. Proteom. 2012, 11, 013441. [Google Scholar] [CrossRef] [Green Version]
- Kapadia, M.R.; Eng, J.W.; Jiang, Q.; Stoyanovsky, D.A.; Kibbe, M.R. Nitric oxide regulates the 26S proteasome in vascular smooth muscle cells. Nitric Oxide 2009, 20, 279–288. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Xu, Z.; Yue, W.; Zhuang, Y.; Chen, Y.; Chen, Y. S-nitrosylation of PDE5 increases its ubiquitin–proteasomal degradation. Free. Radic. Biol. Med. 2015, 86, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-Nitrosylation of Bcl-2 Inhibits Its Ubiquitin-Proteasomal Degradation: A Novel Antiapoptotic Mechanism That Suppresses Apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcone, S.; Evans, P.; Fitzgerald, D.J. 15-Deoxy-Δ12,14-Prostaglandin J2 Modifies Components of the Proteasome and Inhibits Inflammatory Responses in Human Endothelial Cells. Front. Immunol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free. Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 1–24. [Google Scholar] [CrossRef]
- Zong, C.; Young, G.W.; Wang, Y.; Lu, H.; Deng, N.; Drews, O.; Ping, P. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 2008, 8, 5025–5037. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, V.; Dantzer, F.; Ame, J.-C.; De Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Hu, K.; Wu, W.; Li, Y.; Lin, L.; Chen, D.; Yan, H.; Xiao, X.; Chen, H.; Chen, Z.; Zhang, Y.; et al. Poly(ADP -ribosyl)ation of BRD 7 by PARP 1 confers resistance to DNA -damaging chemotherapeutic agents. EMBO Rep. 2019, 20, 46166. [Google Scholar] [CrossRef]
- Ullrich, O.; Reinheckel, T.; Sitte, N.; Hass, R.; Grune, T.; Davies, K.J. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc. Natl. Acad. Sci. USA 1999, 96, 6223–6228. [Google Scholar] [CrossRef] [Green Version]
- Catalgol, B.; Wendt, B.; Grimm, S.; Breusing, N.; Özer, N.K.; Grune, T. Chromatin repair after oxidative stress: Role of PARP-mediated proteasome activation. Free. Radic. Biol. Med. 2010, 48, 673–680. [Google Scholar] [CrossRef]
- Ullrich, O.; Diestel, A.; Bechmann, I.; Homberg, M.; Grune, T.; Hass, R.; Nitsch, R. Turnover of oxidatively damaged nuclear proteins in BV-2 microglial cells is linked to their activation state by poly(ADP-ribose)polymerase. FASEB J. 2001, 15, 1460–1462. [Google Scholar] [CrossRef]
- Sha, Z.; Peth, A.; Goldberg, A.L. Keeping proteasomes under control—a role for phosphorylation in the nucleus. Proc. Natl. Acad. Sci. USA 2011, 108, 18573–18574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VerPlank, J.J.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Huang, X.; Chen, M.J. Reversible phosphorylation of the 26S proteasome. Protein Cell 2017, 8, 255–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Engel, J.L.; Xiao, J.; Tagliabracci, V.S.; Wang, X.; Huang, L.; E Dixon, J. UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc. Natl. Acad. Sci. USA 2011, 108, 18649–18654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, G.G.F.; Hendil, K.B.; Rivett, A.J. Phosphorylation of Proteasomes in Mammalian Cells. Identification of Two Phosphorylated Subunits and the Effect of Phosphorylation on Activity. JBIC J. Biol. Inorg. Chem. 1996, 238, 453–462. [Google Scholar] [CrossRef]
- Mason, G.G.; Murray, R.Z.; Pappin, D.; Rivett, A.J. Phosphorylation of ATPase subunits of the 26S proteasome. FEBS Lett. 1998, 430, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Stratford, F.L.L.; Broadfoot, K.I.; Mason, G.G.F.; Rivett, A.J. Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma-interferon. Biochem. J. 2004, 378, 177–184. [Google Scholar] [CrossRef]
- Iwafune, Y.; Kawasaki, H.; Hirano, H. Electrophoretic analysis of phosphorylation of the yeast 20S proteasome. Electrophoresis 2002, 23, 329–338. [Google Scholar] [CrossRef]
- Castaño, J.G.; Mahillo, E.; Arizti, P.; Arribas, J. Phosphorylation of C8 and C9 Subunits of the Multicatalytic Proteinase by Casein Kinase II and Identification of the C8 Phosphorylation Sites by Direct Mutagenesis. Biochemistry 1996, 35, 3782–3789. [Google Scholar] [CrossRef]
- Li, N.; Lerea, K.M.; Etlinger, J.D. Phosphorylation of the Proteasome Activator PA28 Is Required for Proteasome Activation. Biochem. Biophys. Res. Commun. 1996, 225, 855–860. [Google Scholar] [CrossRef]
- Feng, Y.; Longo, D.L.; Ferris, D.K. Polo-like kinase interacts with proteasomes and regulates their activity. Cell Growth Differ. 2001, 12, 29–37. [Google Scholar] [PubMed]
- Lu, H.; Zong, C.; Wang, Y.; Young, G.W.; Deng, N.; Souda, P.; Li, X.; Whitelegge, J.P.; Drews, O.; Yang, P.-Y.; et al. Revealing the Dynamics of the 20 S Proteasome Phosphoproteome: A combined CID and electron transfer dissociation approach. Mol. Cell. Proteom. 2008, 7, 2073–2089. [Google Scholar] [CrossRef] [Green Version]
- Zong, C.; Gomes, A.V.; Drews, O.; Li, X.; Young, G.W.; Berhane, B.; Qiao, X.; French, S.; Bardag-Gorce, F.; Ping, P. Regulation of Murine Cardiac 20S Proteasomes. Circ. Res. 2006, 99, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marambaud, P.; Wilk, S.; Checler, F. Protein Kinase A Phosphorylation of the Proteasome: A Contribution to the α-Secretase Pathway in Human Cells. J. Neurochem. 2002, 67, 2616–2619. [Google Scholar] [CrossRef]
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.-C.; Goldberg, A.L.; Cheng, Y. Mechanism of Gate Opening in the 20S Proteasome by the Proteasomal ATPases. Mol. Cell 2008, 30, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Hu, Y.; Huang, P.; Toleman, C.A.; Paterson, A.J.; Kudlow, J.E. Proteasome Function Is Regulated by Cyclic AMP-dependent Protein Kinase through Phosphorylation of Rpt. J. Biol. Chem. 2007, 282, 22460–22471. [Google Scholar] [CrossRef] [Green Version]
- Djakovic, S.N.; Schwarz, L.A.; Barylko, B.; DeMartino, G.N.; Patrick, G.N. Regulation of the Proteasome by Neuronal Activity and Calcium/Calmodulin-dependent Protein Kinase II. J. Biol. Chem. 2009, 284, 26655–26665. [Google Scholar] [CrossRef] [Green Version]
- Satoh, K.; Sasajima, H.; Nyoumura, K.-I.; Yokosawa, H.; Sawada, H. Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 2001, 40, 314–319. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.; Wang, Z.; Banerjee, S.; Yang, J.; Huang, L.; Dixon, J.E. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 2016, 18, 202–212. [Google Scholar] [CrossRef]
- Fan, K.; Wang, F.; Li, Y.; Chen, L.; Gao, Z.; Zhang, Y.; Duan, J.-Y.; Huang, T.; Zhong, J.; Liu, R.-B.; et al. CRL4DCAF2 is required for mature T-cell expansion via Aurora B-regulated proteasome activity. J. Autoimmun. 2019, 96, 74–85. [Google Scholar] [CrossRef]
- Pathare, G.R.; Nagy, I.; Bohn, S.; Unverdorben, P.; Hubert, A.; Körner, R.; Nickell, S.; Lasker, K.; Sali, A.; Tamura, T.; et al. The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl. Acad. Sci. USA 2012, 109, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VerPlank, J.J.S.; Lokireddy, S.; Zhao, J.; Goldberg, A.L. 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc. Natl. Acad. Sci. USA 2019, 116, 4228–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokireddy, S.; Kukushkin, N.V.; Goldberg, A.L. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E7176–E7185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; E Duff, K. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Tomita, T.; Hirayama, S.; Sakurai, Y.; Ohte, Y.; Yoshihara, H.; Saeki, Y.; Hamazaki, J.; Murata, S. Specific Modification of Aged Proteasomes Revealed by Tag-Exchangeable Knock-In Mice. Mol. Cell. Biol. 2018, 39, 00426-18. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, W.; Li, C.; Li, P.; Yuan, J.; Li, X.; Qiu, X.-B.; Ma, Q.; Cao, C. Interaction between c-Abl and Arg Tyrosine Kinases and Proteasome Subunit PSMA7 Regulates Proteasome Degradation. Mol. Cell 2006, 22, 317–327. [Google Scholar] [CrossRef]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative Stress-Mediated Regulation of Proteasome Complexes. Mol. Cell. Proteom. 2011, 10, R110.006924. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W.; Im, E.; Park, J.; Oh, Y.; Min, B.; Lee, H.J.; Yoon, J.-B.; Chung, K.C. ASK1 Negatively Regulates the 26 S Proteasome. J. Biol. Chem. 2010, 285, 36434–36446. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S Proteasome Complex during Oxidative Stress. Sci. Signal. 2010, 3, ra88. [Google Scholar] [CrossRef] [Green Version]
- Wani, P.S.; Suppahia, A.; Capalla, X.; Ondracek, A.; Roelofs, J. Phosphorylation of the C-terminal tail of proteasome subunit α7 is required for binding of the proteasome quality control factor Ecm29. Sci. Rep. 2016, 6, 27873. [Google Scholar] [CrossRef] [Green Version]
- Iwafune, Y.; Kawasaki, H.; Hirano, H. Identification of three phosphorylation sites in the α7 subunit of the yeast 20S proteasome in vivo using mass spectrometry. Arch. Biochem. Biophys. 2004, 431, 9–15. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, Y.; Yoon, S.K.; Yoon, J.-B. Osmotic Stress Inhibits Proteasome by p38 MAPK-dependent Phosphorylation. J. Biol. Chem. 2010, 285, 41280–41289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Su, K.; Yang, X.; Bowe, D.B.; Paterson, A.J.; Kudlow, J.E. O-GlcNAc Modification Is an Endogenous Inhibitor of the Proteasome. Cell 2003, 115, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Overath, T.; Kuckelkorn, U.; Henklein, P.; Strehl, B.; Bonar, D.; Kloss, A.; Siele, D.; Kloetzel, P.-M.; Janek, K. Mapping of O-GlcNAc Sites of 20 S Proteasome Subunits and Hsp90 by a Novel Biotin-Cystamine Tag. Mol. Cell. Proteom. 2012, 11, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Ngoh, G.A.; Watson, L.J.; Facundo, H.T.; Jones, S.P. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids 2011, 40, 895–911. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-Specific GlcNAcylation of Human Erythrocyte Proteins: Potential Biomarker(s) for Diabetes. Diabetes 2008, 58, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Butkinaree, C.; Park, K.; Hart, G.W. O-linked β-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2010, 1800, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Sümegi, M.; Hunyadi-Gulyas, E.; Medzihradszky, K.F.; Udvardy, A. 26S proteasome subunits are O-linked N-acetylglucosamine-modified in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2003, 312, 1284–1289. [Google Scholar] [CrossRef]
- Ruan, H.-B.; Nie, Y.; Yang, X. Regulation of Protein Degradation by O-GlcNAcylation: Crosstalk with Ubiquitination. Mol. Cell. Proteom. 2013, 12, 3489–3497. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Fang, C.; Zong, N.C.; Liem, D.A.; Cadeiras, M.; Scruggs, S.B.; Yu, H.; Kim, A.K.; Yang, P.; Deng, M.; et al. Regulation of acetylation restores proteolytic function of diseased myocardium in mouse and human. Mol. Cell. Proteom. 2013, 12, 3793–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, Y.; Li, L.; Zhou, L.; Wei, H.; Zhou, Q.; Wang, W.; Ji, L.; Shan, P.; Wang, Y.; et al. Site-specific Acetylation of the Proteasome Activator REGγ Directs Its Heptameric Structure and Functions. J. Biol. Chem. 2013, 288, 16567–16578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, L.; Xu, H.; Wang, J.; Qu, L.; Jiang, B.; Zeng, Y.; Meng, L.; Jin, H.; Shou, C. N-α-acetyltransferase 10 protein is a negative regulator of 28S proteasome through interaction with PA28β. FEBS Lett. 2013, 587, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Arendt, C.S.; Hochstrasser, M. Eukaryotic 20S proteasome catalytic subunit propeptides prevent active site inactivation by N-terminal acetylation and promote particle assembly. EMBO J. 1999, 18, 3575–3585. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Hochstrasser, M. Autocatalytic Subunit Processing Couples Active Site Formation in the 20S Proteasome to Completion of Assembly. Cell 1996, 86, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Boyault, C.; Khochbin, S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. BioEssays 2005, 27, 408–415. [Google Scholar] [CrossRef]
- Wang, X.; Taplick, J.; Geva, N.; Oren, M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett. 2004, 561, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Gregory, R.C.; Taniguchi, T.; D’Andrea, A.D. Regulation of the Fanconi anemia pathway by monoubiquitination. Semin. Cancer Biol. 2003, 13, 77–82. [Google Scholar] [CrossRef]
- Schmitt, S.M.; Neslund-Dudas, C.; Shen, M.; Cui, C.; Mitra, B.; Dou, Q.P. Involvement of ALAD-20S Proteasome Complexes in Ubiquitination and Acetylation of Proteasomal α2 Subunits. J. Cell. Biochem. 2015, 117, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isasa, M.; Katz, E.J.; Kim, W.; Yugo, V.; González, S.; Kirkpatrick, D.S.; Thomson, T.M.; Finley, D.; Gygi, S.P.; Crosas, B. Monoubiquitination of RPN10 Regulates Substrate Recruitment to the Proteasome. Mol. Cell 2010, 38, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuin, A.; Bichmann, A.; Isasa, M.; Puig-Sàrries, P.; Díaz, L.M.; Crosas, B. Rpn10 monoubiquitination orchestrates the association of the ubiquilin-type DSK2 receptor with the proteasome. Biochem. J. 2015, 472, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Kaplan, T.; Peters, L.Z.; Levin-Kravets, O.; Attali, I.; Kleifeld, O.; Shohat, N.; Artzi, S.; Zucker, O.; Pilzer, I.; Reis, N.; et al. Structure of ubiquitylated-Rpn10 provides insight into its autoregulation mechanism. Nat. Commun. 2016, 7, 12960. [Google Scholar] [CrossRef] [Green Version]
- Uchiki, T.; Kim, H.T.; Zhai, B.; Gygi, S.P.; A Johnston, J.; O’Bryan, J.P.; Goldberg, A.L. The Ubiquitin-interacting Motif Protein, S5a, Is Ubiquitinated by All Types of Ubiquitin Ligases by a Mechanism Different from Typical Substrate Recognition. J. Biol. Chem. 2009, 284, 12622–12632. [Google Scholar] [CrossRef] [Green Version]
- Besche, H.C.; Sha, Z.; Kukushkin, N.V.; Peth, A.; Hock, E.; Kim, W.; Gygi, S.; A Gutierrez, J.; Liao, H.; Dick, L.; et al. Autoubiquitination of the 26S Proteasome on Rpn13 Regulates Breakdown of Ubiquitin Conjugates. EMBO J. 2014, 33, 1159–1176. [Google Scholar] [CrossRef]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-Wide Changes to SUMO Modifications in Response to Heat Shock. Sci. Signal. 2009, 2, ra24. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Gygi, S.P.; Azuma, Y.; Arnaoutov, A.; Dasso, M. SUMOylation of Psmd1 Controls Adrm1 Interaction with the Proteasome. Cell Rep. 2014, 7, 1842–1848. [Google Scholar] [CrossRef] [Green Version]
- Lamoliatte, F.; McManus, F.P.; Maarifi, G.; Chelbi-Alix, M.K.; Thibault, P. Uncovering the SUMOylation and ubiquitylation crosstalk in human cells using sequential peptide immunopurification. Nat. Commun. 2017, 8, 14109. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Kato, Y.; Hirano, H. N-myristoylation of the Rpt2 subunit regulates intracellular localization of the yeast 26S proteasome. Biochemistry 2012, 51, 8856–8866. [Google Scholar] [CrossRef]
- Kimura, A.; Kurata, Y.; Nakabayashi, J.; Kagawa, H.; Hirano, H. N-Myristoylation of the Rpt2 subunit of the yeast 26S proteasome is implicated in the subcellular compartment-specific protein quality control system. J. Proteom. 2016, 130, 33–41. [Google Scholar] [CrossRef]
- Erce, M.A.; Pang, C.N.I.; Hart-Smith, G.; Wilkins, M.R. The methylproteome and the intracellular methylation network. Proteomics 2012, 12, 564–586. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kurata, Y.; Ishikawa, A.; Okayama, A.; Kamita, M.; Hirano, H. N-Terminal methylation of proteasome subunit Rpt1 in yeast. Proteomics 2013, 13, 3167–3174. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.V.; Young, G.W.; Wang, Y.; Zong, C.; Eghbali, M.; Drews, O.; Lu, H.; Stefani, E.; Ping, P. Contrasting Proteome Biology and Functional Heterogeneity of the 20S Proteasome Complexes in Mammalian Tissues. Mol. Cell. Proteom. 2008, 8, 302–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osna, N.A.; White, R.L.; Donohue, T.M.; Beard, M.R.; Tuma, D.J.; Kharbanda, K.K. Impaired methylation as a novel mechanism for proteasome suppression in liver cells. Biochem. Biophys. Res. Commun. 2010, 391, 1291–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osna, N.A.; Bardag-Gorce, F.; White, R.L.; Weinman, S.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Ethanol and Hepatitis C Virus Suppress Peptide-MHC Class I Presentation in Hepatocytes by Altering Proteasome Function. Alcohol. Clin. Exp. Res. 2012, 36, 2028–2035. [Google Scholar] [CrossRef] [Green Version]
- Knowles, J.R. Chemical modification and the reactivity of amino acids in proteins. MTP Intl. Rev. Sci. Biochem. 1974, 1, 149. [Google Scholar]
- Baslé, E.; Joubert, N.; Pucheault, M. Protein Chemical Modification on Endogenous Amino Acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef]
- Davies, J.M.; Horwitz, D.A.; Davies, K.J. Potential roles of hypochlorous acid and N-chloroamines in collagen breakdown by phagocytic cells in synovitis. Free. Radic. Biol. Med. 1993, 15, 637–643. [Google Scholar] [CrossRef]
- Ishii, T.; Sakurai, T.; Usami, H.; Uchida, K. Oxidative Modification of Proteasome: Identification of an Oxidation-Sensitive Subunit in 26S Proteasome. Biochemistry 2005, 44, 13893–13901. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Møller, I.M.; Rogowska-Wrzesinska, A.; Rao, S.P. Protein carbonylation and metal-catalyzed protein oxidation in a cellular perspective. J. Proteom. 2011, 74, 2228–2242. [Google Scholar] [CrossRef] [PubMed]
- Divald, A.; Kivity, S.; Wang, P.; Hochhauser, E.; Roberts, B.; Teichberg, S.; Gomes, A.V.; Powell, S.R. Myocardial Ischemic Preconditioning Preserves Postischemic Function of the 26S Proteasome Through Diminished Oxidative Damage to 19S Regulatory Particle Subunits. Circ. Res. 2010, 106, 1829–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.N.; Westfall, M.V.; Dyke, D.B.; Pagani, F.D.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillette, T.G.; Kumar, B.; Thompson, D.; Slaughter, C.A.; DeMartino, G.N. Differential Roles of the COOH Termini of AAA Subunits of PA700 (19 S Regulator) in Asymmetric Assembly and Activation of the 26 S Proteasome. J. Biol. Chem. 2008, 283, 31813–31822. [Google Scholar] [CrossRef] [Green Version]
- Aslebagh, R.; Pfeffer, B.A.; Fliesler, S.J.; Darie, C.C. Mass spectrometry-based proteomics of oxidative stress: Identification of 4-hydroxy-2-nonenal (HNE) adducts of amino acids using lysozyme and bovine serum albumin as model proteins. Electrophoresis 2016, 37, 2615–2623. [Google Scholar] [CrossRef] [Green Version]
- Doorn, J.A.; Petersen, D.R. Covalent Modification of Amino Acid Nucleophiles by the Lipid Peroxidation Products 4-Hydroxy-2-nonenal and 4-Oxo-2-nonenal. Chem. Res. Toxicol. 2002, 15, 1445–1450. [Google Scholar] [CrossRef]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Just, J.; Jung, T.; Friis, N.A.; Lykkemark, S.; Drasbek, K.R.; Siboska, G.; Grune, T.; Kristensen, P.L. Identification of an unstable 4-hydroxynoneal modification on the 20S proteasome subunit α7 by recombinant antibody technology. Free. Radic. Biol. Med. 2015, 89, 786–792. [Google Scholar] [CrossRef] [Green Version]
- Musatov, A.; Carroll, C.A.; Liu, Y.-C.; Henderson, G.I.; Weintraub, S.T.; Robinson, N.C. Identification of Bovine Heart Cytochrome c Oxidase Subunits Modified by the Lipid Peroxidation Product 4-Hydroxy-2-nonenal. Biochemistry 2002, 41, 8212–8220. [Google Scholar] [CrossRef]
- Humphries, K.M.; Szweda, L.I. Selective Inactivation of α-Ketoglutarate Dehydrogenase and Pyruvate Dehydrogenase: Reaction of Lipoic Acid with 4-Hydroxy-2-nonenal. Biochemistry 1998, 37, 15835–15841. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free. Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.-L.; Lundberg, K.C.; Humphries, K.M.; Sadek, H.A.; Szweda, P.A.; Friguet, B.; Szweda, L.I. Oxidative Modification and Inactivation of the Proteasome during Coronary Occlusion/Reperfusion. J. Biol. Chem. 2001, 276, 30057–30063. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Huang, F.F.; Zhu, H.; Yu, J.; Ho, Y.-S.; Kindy, M.S. Oxidative Stress-Associated Impairment of Proteasome Activity during Ischemia–Reperfusion Injury. Br. J. Pharmacol. 2000, 20, 1467–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, K.; Wangpoengtrakul, C.; Osawa, T.; Toyokuni, S.; Tanaka, K.; Uchida, K. 4-Hydroxy-2-nonenal-mediated Impairment of Intracellular Proteolysis during Oxidative Stress. J. Biol. Chem. 1999, 274, 23787–23793. [Google Scholar] [CrossRef] [Green Version]
- Carrard, G.; Dieu, M.; Raes, M.; Toussaint, O.; Friguet, B. Impact of ageing on proteasome structure and function in human lymphocytes. Int. J. Biochem. Cell Biol. 2003, 35, 728–739. [Google Scholar] [CrossRef]
- Keller, J.; Huang, F.; Markesbery, W. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 2000, 98, 149–156. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Kapphahn, R.J. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004, 578, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Amici, M.; Lupidi, G.; Angeletti, M.; Fioretti, E.; Eleuteri, A.M. Peroxynitrite-induced oxidation and its effects on isolated proteasomal systems. Free. Radic. Biol. Med. 2003, 34, 987–996. [Google Scholar] [CrossRef]
- Gavilán, M.P.; Castaño, A.; Torres, M.; Portavella, M.; Caballero, C.; Jiménez, S.; García-Martínez, A.; Parrado, J.; Vitorica, J.; Ruano, D. Age-related increase in the immunoproteasome content in rat hippocampus: Molecular and functional aspects. J. Neurochem. 2009, 108, 260–272. [Google Scholar] [CrossRef]
- Thorpe, S.R.; Baynes, J.W. Role of the Maillard Reaction in Diabetes Mellitus and Diseases of Aging. Drugs Aging 1996, 9, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ames, J.M.; Smith, R.D.; Baynes, J.W.; Metz, T.O. A Perspective on the Maillard Reaction and the Analysis of Protein Glycation by Mass Spectrometry: Probing the Pathogenesis of Chronic Disease. J. Proteome Res. 2009, 8, 754–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.-P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia Impairs Proteasome Function by Methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Račková, L.; Šnirc, V.; Jung, T.; Stefek, M.; Karasu, Ç.; Grune, T. Metabolism-induced oxidative stress is a mediator of glucose toxicity in HT22 neuronal cells. Free. Radic. Res. 2009, 43, 876–886. [Google Scholar] [CrossRef]
- Moheimani, F.; Morgan, P.E.; Van Reyk, D.; Davies, M.J. Deleterious effects of reactive aldehydes and glycated proteins on macrophage proteasomal function: Possible links between diabetes and atherosclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, A.F.; Van Der Linden, W.A.; Overkleeft, H.S. Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Gräwert, M.A.; Gallastegui, N.; Stein, M.; Schmidt, B.; Kloetzel, P.-M.; Huber, R.; Groll, M. Elucidation of the α-Keto-Aldehyde Binding Mechanism: A Lead Structure Motif for Proteasome Inhibition. Angew. Chem. Int. Ed. 2010, 50, 542–544. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Radi, R. Protein Tyrosine Nitration: Biochemical Mechanisms and Structural Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, S.; Zhang, M.; Wang, Q.; Asfa, S.; Zou, M.-H. Tyrosine Nitration of PA700 Links Proteasome Activation to Endothelial Dysfunction in Mouse Models with Cardiovascular Risk Factors. PloS ONE 2012, 7, e29649. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, S.; Wu, Y.; Song, P.; Zou, M.-H. Tyrosine Nitration of PA700 Activates the 26S Proteasome to Induce Endothelial Dysfunction in Mice with Angiotensin II–Induced Hypertension. Hypertension 2009, 54, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Blasig, I.E.; Sitte, N.; Roloff, B.; Haseloff, R.; Davies, K.J. Peroxynitrite Increases the Degradation of Aconitase and Other Cellular Proteins by Proteasome. J. Biol. Chem. 1998, 273, 10857–10862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osna, N.A.; Haorah, J.; Krutik, V.M.; Donohue, T.M. Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology 2004, 40, 574–582. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nat. Cell Biol. 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular Chaperones in the Cytosol: From Nascent Chain to Folded Protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [Green Version]
- Routledge, K.E.; Tartaglia, G.G.; Platt, G.W.; Vendruscolo, M.; Radford, S.E. Competition between Intramolecular and Intermolecular Interactions in an Amyloid-Forming Protein. J. Mol. Biol. 2009, 389, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Markossian, K.A.; Kurganov, B.I. Protein Folding, Misfolding, and Aggregation. Formation of Inclusion Bodies and Aggresomes. Biochemistry 2004, 69, 971–984. [Google Scholar] [CrossRef]
- Ross, C.A.; A Poirier, M. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Esteras-Chopo, A.; Pastor, M.T.; Serrano, L. Protein Misfolding and β-Amyloid Formation. In Protein Folding, Misfolding and Aggregation; Muñoz, V., Ed.; Royal Society of Chemistry (RSC): Milton Road, Cambridge, UK, 2008; pp. 214–240. [Google Scholar] [CrossRef]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PloS Pathog. 2013, 9, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Lansbury, P.T. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Mirzaei, H.; Regnier, F.E. Protein:protein aggregation induced by protein oxidation. J. Chromatogr. B 2008, 873, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Weids, A.J.; Ibstedt, S.; Tamás, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, D.C. Aging and the aggregating proteome. Front. Genet. 2012, 3, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitraki, A. Protein Aggregation: From inclusion bodies to amyloid and biomaterials. Adv. Protein. Chem. Struct. Biol. 2010, 79, 89–125. [Google Scholar] [CrossRef]
- Bolognesi, B.; Kumita, J.R.; Barros, T.P.; Esbjorner, E.K.; Luheshi, L.M.; Crowther, D.C.; Wilson, M.R.; Dobson, C.M.; Favrin, G.; Yerbury, J.J. ANS Binding Reveals Common Features of Cytotoxic Amyloid Species. ACS Chem. Biol. 2010, 5, 735–740. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell. Free. Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Boellaard, J.W.; Schlote, W.; Höfer, W. Species-Specific Ultrastructure of Neuronal Lipofuscin in Hippocampus and Neocortex of Subhuman Mammals and Humans. Ultrastruct. Pathol. 2004, 28, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [CrossRef]
- Höhn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: Role in senescent cells. Free. Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: Formation, Distribution, and Metabolic Consequences. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kikugawa, K.; Kato, T.; Beppu, M.; Hayasaka, A. Fluorescent and Cross-Linked Proteins Formed by Free Radical and Aldehyde Species Generated during Lipid Oxidation. Biol. Mammary Gland 1990, 266, 345–357. [Google Scholar] [CrossRef]
- Leeuwenburgh, C.; Rasmussen, J.E.; Hsu, F.F.; Mueller, D.M.; Pennathur, S.; Heinecke, J.W. Mass Spectrometric Quantification of Markers for Protein Oxidation by Tyrosyl Radical, Copper, and Hydroxyl Radical in Low Density Lipoprotein Isolated from Human Atherosclerotic Plaques. J. Biol. Chem. 1997, 272, 3520–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höhn, A.; Sittig, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free. Radic. Biol. Med. 2012, 53, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A Cellular Response to Misfolded Proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- García-Mata, R.; Bebök, Z.; Sorscher, E.J.; Sztul, E. Characterization and Dynamics of Aggresome Formation by a Cytosolic Gfp-Chimera. J. Cell Biol. 1999, 146, 1239–1254. [Google Scholar] [CrossRef] [Green Version]
- Mayer, R.J.; Lowe, J.; Lennox, G.; Landon, M.; MacLennan, K.; Doherty, F.J. Intermediate filament-ubiquitin diseases: Implications for cell sanitization. Biochem. Soc. Symp. 1989, 55, 193–201. [Google Scholar]
- Wigley, W.C.; Fabunmi, R.P.; Lee, M.G.; Marino, C.R.; Muallem, S.; DeMartino, G.N.; Thomas, P.J. Dynamic Association of Proteasomal Machinery with the Centrosome. J. Cell Biol. 1999, 145, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.-P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.-S.; Olzmann, J.A.; Li, L. Aggresome Formation and Neurodegenerative Diseases: Therapeutic Implications. Curr. Med. Chem. 2008, 15, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.; Shashidharan, P.; Perl, D.P.; Jenner, P.; Olanow, C.W. Aggresome-related biogenesis of Lewy bodies. Eur. J. Neurosci. 2002, 16, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P.; Engel, W.K.; McFerrin, J.; Davies, K.J.; Lin, S.W.; Askanas, V. Proteasome Inhibition and Aggresome Formation in Sporadic Inclusion-Body Myositis and in Amyloid-β Precursor Protein-Overexpressing Cultured Human Muscle Fibers. Am. J. Pathol. 2005, 167, 517–526. [Google Scholar] [CrossRef]
- Mah, A.L.; Perry, G.; Smith, M.A.; Monteiro, M.J. Identification of Ubiquilin, a Novel Presenilin Interactor That Increases Presenilin Protein Accumulation. J. Cell Biol. 2000, 151, 847–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardley, H.C.; Scott, G.B.; Rose, S.A.; Tan, N.G.S.; Markham, A.F.; Robinson, P.A. Inhibition of Proteasomal Activity Causes Inclusion Formation in Neuronal and Non-Neuronal Cells Overexpressing Parkin. Mol. Biol. Cell 2003, 14, 4541–4556. [Google Scholar] [CrossRef]
- Lee, H.-J.; Shin, S.Y.; Choi, C.; Lee, Y.H.; Lee, S.J. Formation and Removal of α-Synuclein Aggregates in Cells Exposed to Mitochondrial Inhibitors. J. Biol. Chem. 2001, 277, 5411–5417. [Google Scholar] [CrossRef] [Green Version]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of Mutant Huntingtin Fragments in Aggresome-like Inclusion Bodies as a Result of Insufficient Protein Degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef] [Green Version]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S.P. Lewy-body formation is an aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef]
- Lowe, J.; Blanchard, A.; Morrell, K.; Lennox, G.; Reynolds, L.; Billett, M.; Landon, M.; Mayer, R.J. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J. Pathol. 1988, 155, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Matilla, A.; Gorbea, C.; Einum, D.D.; Townsend, J.; Michalik, A.; Van Broeckhoven, C.; Jensen, C.C.; Murphy, K.J.; Ptácek, L.J.; Fu, Y.-H. Association of ataxin-7 with the proteasome subunit S4 of the 19S regulatory complex. Hum. Mol. Genet. 2001, 10, 2821–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deriziotis, P.; André, R.; Smith, D.M.; Goold, R.; Kinghorn, K.J.; Kristiansen, M.; A Nathan, J.; Rosenzweig, R.; Krutauz, D.; Glickman, M.H.; et al. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J. 2011, 30, 3065–3077. [Google Scholar] [CrossRef] [PubMed]
- Elnashar, M.M.M. The Art of Immobilization Using Biopolymers, Biomaterials and Nanobiotechnology. In Biotechnology of Biopolymers; IntechOpen: Rijeka, Croatia, 2011; p. 3. [Google Scholar]
- Castillo, J.; Sasso, L.; Svendsen, W.E. Self-Assembled Peptide Nanostructures: Advances and Applications in Nanobiotechnology; CRC Press: Boca Raton, FL, USA, 2012; p. 56. [Google Scholar]
- Raynes, J.K.; Pearce, F.; Meade, S.J.; Gerrard, J.A. Immobilization of organophosphate hydrolase on an amyloid fibril nanoscaffold: Towards bioremediation and chemical detoxification. Biotechnol. Prog. 2011, 27, 360–367. [Google Scholar] [CrossRef]
- Bhak, G.; Lee, S.; Park, J.W.; Cho, S.; Paik, S.R. Amyloid hydrogel derived from curly protein fibrils of α-synuclein. Biomaterials 2010, 31, 5986–5995. [Google Scholar] [CrossRef]
- Rinas, U.; Garcia-Fruitós, E.; Corchero, J.L.; Vázquez, E.; Seras-Franzoso, J.; Villaverde, A. Bacterial Inclusion Bodies: Discovering Their Better Half. Trends Biochem. Sci. 2017, 42, 726–737. [Google Scholar] [CrossRef]
- Corchero, J.L. Eukaryotic aggresomes: From a model of conformational diseases to an emerging type of immobilized biocatalyzers. Appl. Microbiol. Biotechnol. 2015, 100, 559–569. [Google Scholar] [CrossRef]
- Engasser, J.-M.; Horvath, C. Diffusion and Kinetics with Immobilized Enzymes. In Applied Biochemistry and Bioengineering: Immobilized Enzyme Principles; Wingard, L.B., Katchalski-Katzir, E., Goldstein, L., Eds.; Academic Press: New York, NY, USA, 1976; Volume 1, pp. 127–220. [Google Scholar]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, C.; E Staniszewski, K.; Mensah, K.N.; Matouschek, A.; Morimoto, R.I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004, 23, 4307–4318. [Google Scholar] [CrossRef] [Green Version]
- Stenoien, D.L.; Mielke, M.; Mancini, M.A. Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat. Cell Biol. 2002, 4, 806–810. [Google Scholar] [CrossRef]
- Höhn, A.; Jung, T.; Grimm, S.; Catalgol, B.; Weber, D.; Grune, T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free. Radic. Biol. Med. 2011, 50, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Xia, X.; Wei, A.; Wang, X.; Cai, W.; Huo, D.; Wei, A. Preparation of Cu(II)-chelated poly(vinyl alcohol) nanofibrous membranes for catalase immobilization. J. Appl. Polym. Sci. 2011, 120, 3291–3296. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.W.; Pithadia, A.S.; DeToma, A.S.; Korshavn, K.J.; Lim, M.H. Ligand Design to Target and Modulate Metal-Protein Interactions in Neurodegenerative Diseases. In Ligand Design in Medicinal Inorganic Chemistry; Storr, T., Ed.; John Wiley & Sons: Chichester, Germany, 2014; pp. 257–286. [Google Scholar]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Secundo, F. Conformational changes of enzymes upon immobilisation. Chem. Soc. Rev. 2013, 42, 6250–6261. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell. 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Gregori, L.; Hainfeld, J.F.; Simon, M.N.; Goldgaber, D. Binding of Amyloid β Protein to the 20 S Proteasome. J. Biol. Chem. 1997, 272, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Andre, R.; Tabrizi, S.J. Misfolded PrP and a novel mechanism of proteasome inhibition. Prion 2012, 6, 32–36. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and Monomeric α-Synuclein Bind to the S6′ Proteasomal Protein and Inhibit Proteasomal Function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [Green Version]
- De la Pena, A.H.; Opoku-Nsiah, K.A.; Williams, S.K.; Chopra, N.; Sali, A.; Gestwicki, J.E.; Lander, G.C. The Yphi Motif Defines the Structure-Activity Relationships of Human 20S Proteasome Activators. 2020, to Be Published, doi:10.2210/pdb6xmj/pdb. Image from the RCSB PDB (Rcsb.Org) of PDB ID: 6XMJ. Available online: https://www.rcsb.org/structure/6xmj (accessed on 15 November 2020).
- Groll, M.; Bajorek, M.; Kohler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Hernandez, M.; Valera, A.G.; Moran, M.A.; Gomez-Ramos, P.; Alvarez-Castelao, B.; Castano, J.G.; Hernandez, F.; Lucas, J.J. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 2006, 98, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Bence, N.F.; Jayakumar, R.; Kopito, R.R. Global Impairment of the Ubiquitin-Proteasome System by Nuclear or Cytoplasmic Protein Aggregates Precedes Inclusion Body Formation. Mol. Cell 2005, 17, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Hernández, K.; Barbosa, O.; Rueda, N.; Garcia-Galan, C.; Dos Santos, J.C.S.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Immobilization of Proteins in Poly-Styrene-Divinylbenzene Matrices: Functional Properties and Applications. Curr. Org. Chem. 2015, 19, 1707–1718. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.B.; Yde, P.; Giehm, L.; Sundbye, S.; Christiansen, G.; Mathiesen, J.; Jensen, M.H.; Jensen, P.H.; Otzen, D. Multiple Roles of Heparin in the Aggregation of p25α. J. Mol. Biol. 2012, 421, 601–615. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, S.; Meng, Y.; Xiongwei, D.; Zhang, D.; Yang, Y.; Wang, L.; Liu, C. Polyanion binding accelerates the formation of stable and low-toxic aggregates of ALS-linked SOD1 mutant A4V. Proteins: Struct. Funct. Bioinform. 2014, 82, 3356–3372. [Google Scholar] [CrossRef]
- Carr, P.W.; Bowers, L.D. Immobilized Enzymes in Analytical and Clinical Chemistry: Fundamentals and Applications (Chemical Analysis: A Series of Monographs on Analytical Chemistry and Its Applications); John Wiley & Sons: New York, NY, USA, 1980; Volume 56, pp. 148–196. [Google Scholar]
- Kennedy, J.F.; Cabral, J.M.S. Enzyme immobilization. In Biotechnology Enzyme Technology; Rehm, H.-I., Reed, G., Eds.; VCH: Weinheim, Germany, 1987; Volume 7a, pp. 347–404. [Google Scholar]
- Bar-Eli, A.; Katchalski, E. Preparation and properties of water-insoluble derivatives of trypsin. J. Biol. Chem. 1963, 238, 1690–1698. [Google Scholar]
- Seemüller, E.; Lupas, A.; Baumeister, W. Autocatalytic processing of the 20S proteasome. Nat. Cell Biol. 1996, 382, 468–470. [Google Scholar] [CrossRef]
- Tanaka, K.; Ichihara, A. Autodegradation of rat liver proteasomes (large multicatalytic proteinase complexes). Biochem. Biophys. Res. Commun. 1989, 158, 548–554. [Google Scholar] [CrossRef]
- Palomo, J.M.; Fernández-Lorente, G.; Mateo, C.; Segura, R.L.; Ortiz, C.; Fernandez-Lafuente, R.; Guisan, J.M. Purification, Immobilization, Hyperactivation, and Stabilization of Lipases by Selective Adsorption on Hydrophobic Supports. In Immobilization of Enzymes and Cells; Guisan, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 143–152. [Google Scholar]
- Jana, N.R.; Zemskov, E.A.; Wang, G.-H.; Nukina, N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 2001, 10, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, K.; Fernandez-Lafuente, R. Lipase B from Candida antarctica immobilized on octadecyl Sepabeads: A very stable biocatalyst in the presence of hydrogen peroxide. Process. Biochem. 2011, 46, 873–878. [Google Scholar] [CrossRef]
- Mateo, C.; Fernandes, B.; Van Rantwijk, F.; Stolz, A.; A Sheldon, R. Stabilisation of oxygen-labile nitrilases via co-aggregation with poly(ethyleneimine). J. Mol. Catal. B Enzym. 2006, 38, 154–157. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; A Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nat. Cell Biol. 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Cummings, C.J.; Reinstein, E.; Sun, Y.; Antalffy, B.; Jiang, Y.-H.; Ciechanover, A.; Orr, H.T.; Beaudet, A.L.; Zoghbi, H.Y. Mutation of the E6-AP Ubiquitin Ligase Reduces Nuclear Inclusion Frequency While Accelerating Polyglutamine-Induced Pathology in SCA1 Mice. Neuron 1999, 24, 879–892. [Google Scholar] [CrossRef] [Green Version]
- Schipper-Krom, S.; Juenemann, K.; Jansen, A.H.; Wiemhoefer, A.; Nieuwendijk, R.V.D.; Smith, D.L.; Hink, M.A.; Bates, G.P.; Overkleeft, H.; Ovaa, H.; et al. Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. 2013, 588, 151–159. [Google Scholar] [CrossRef]
- Kim, S.; Nollen, E.A.A.; Kitagawa, K.; Bindokas, V.P.; Morimoto, R.I. Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 2002, 4, 826–831. [Google Scholar] [CrossRef]
- Hrabárová, E.; Achbergerová, L.; Nahalka, J. Insoluble Protein Applications: The Use of Bacterial Inclusion Bodies as Biocatalysts. Methods Mol. Biol. 2014, 1258, 411–422. [Google Scholar] [CrossRef]
- Jäger, V.D.; Kloss, R.; Grünberger, A.; Seide, S.; Hahn, D.; Karmainski, T.; Piqueray, M.; Embruch, J.; Longerich, S.; Mackfeld, U.; et al. Tailoring the properties of (catalytically)-active inclusion bodies. Microb. Cell Factories 2019, 18, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmann, B.; Christmann, A.; Heiseler, T.; Fritz, J.; Kolmar, H. In Vivo Enzyme Immobilization by Inclusion Body Display. Appl. Environ. Microbiol. 2010, 76, 5563–5569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsten, K.; De Vrij, F.M.; Verhoef, L.G.; Fischer, D.F.; Van Leeuwen, F.W.; Hol, E.M.; Masucci, M.G.; Dantuma, N.P. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 2002, 157, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Han, D.H.; Na, H.-K.; Choi, W.H.; Lee, J.H.; Kim, Y.-K.; Won, C.; Lee, S.-H.; Kim, K.P.; Kuret, J.; Min, D.-H.; et al. Direct cellular delivery of human proteasomes to delay tau aggregation. Nat. Commun. 2014, 5, 5633. [Google Scholar] [CrossRef] [Green Version]

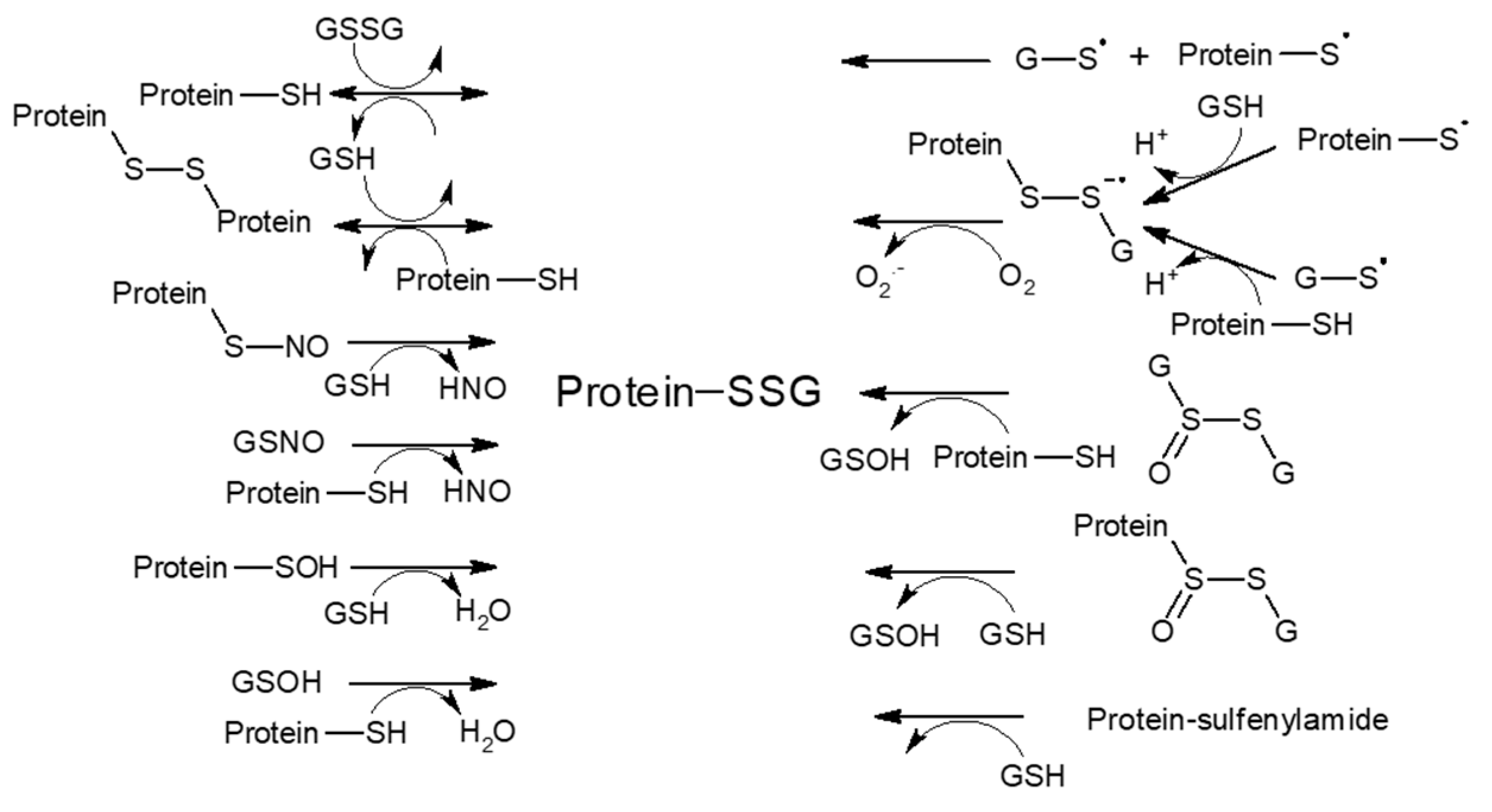
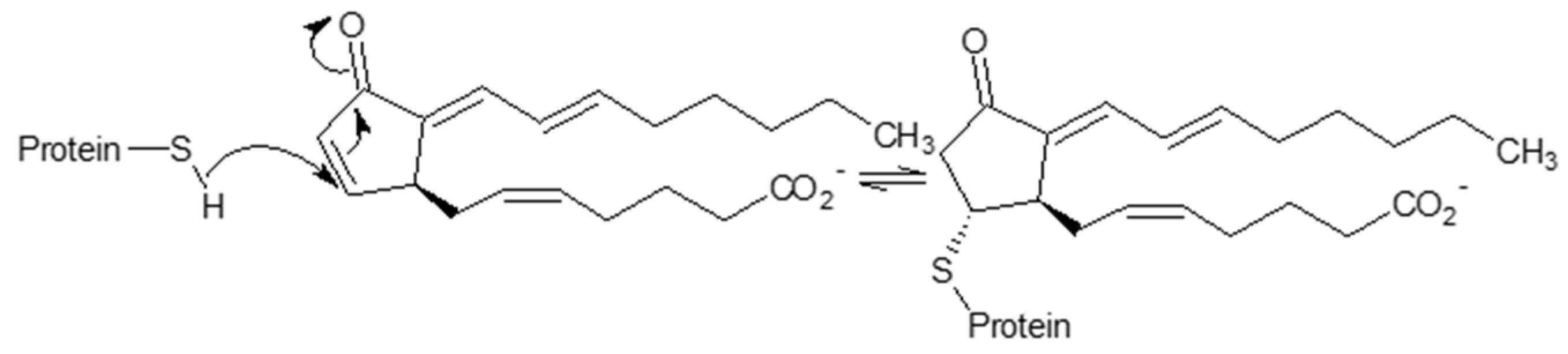
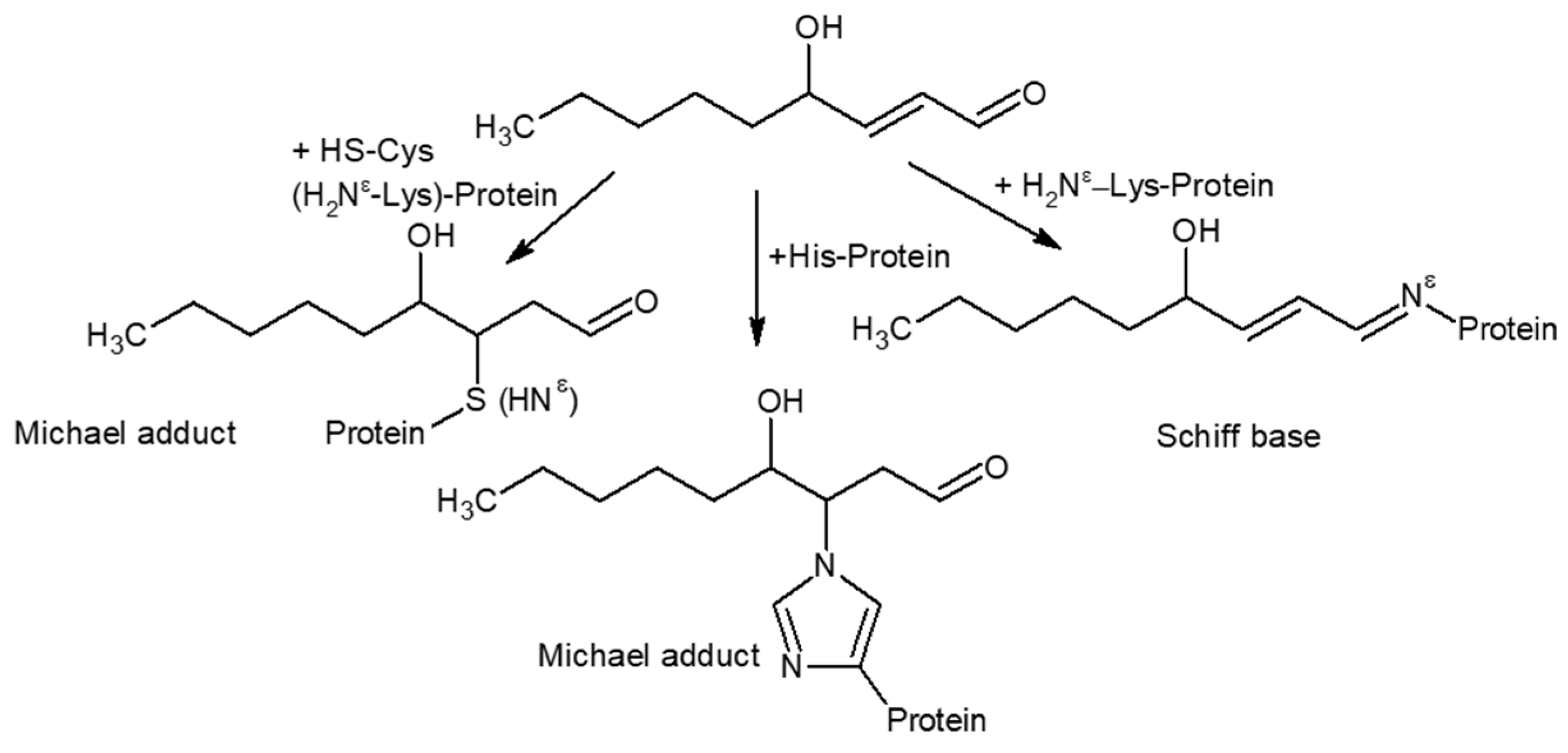



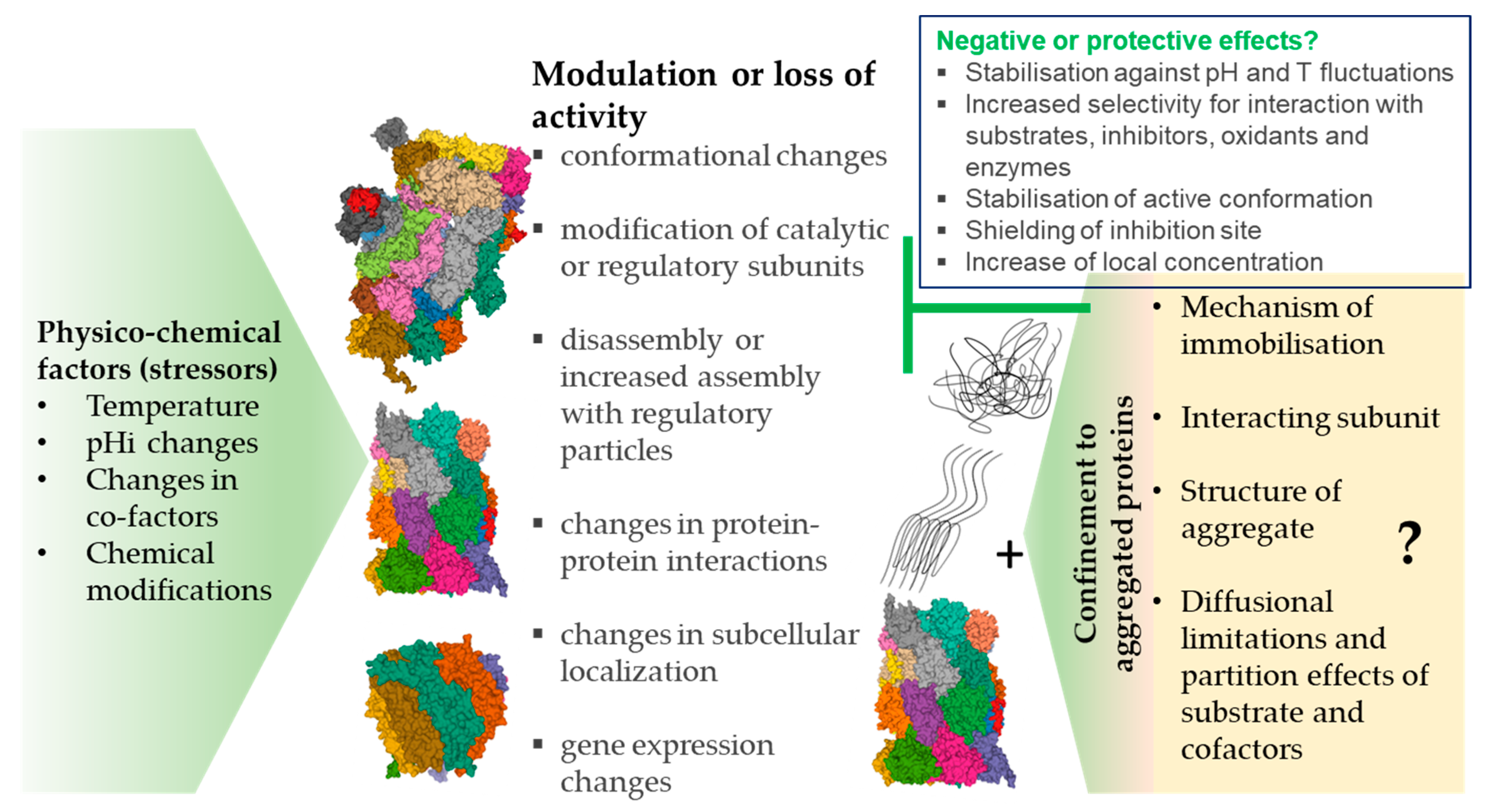

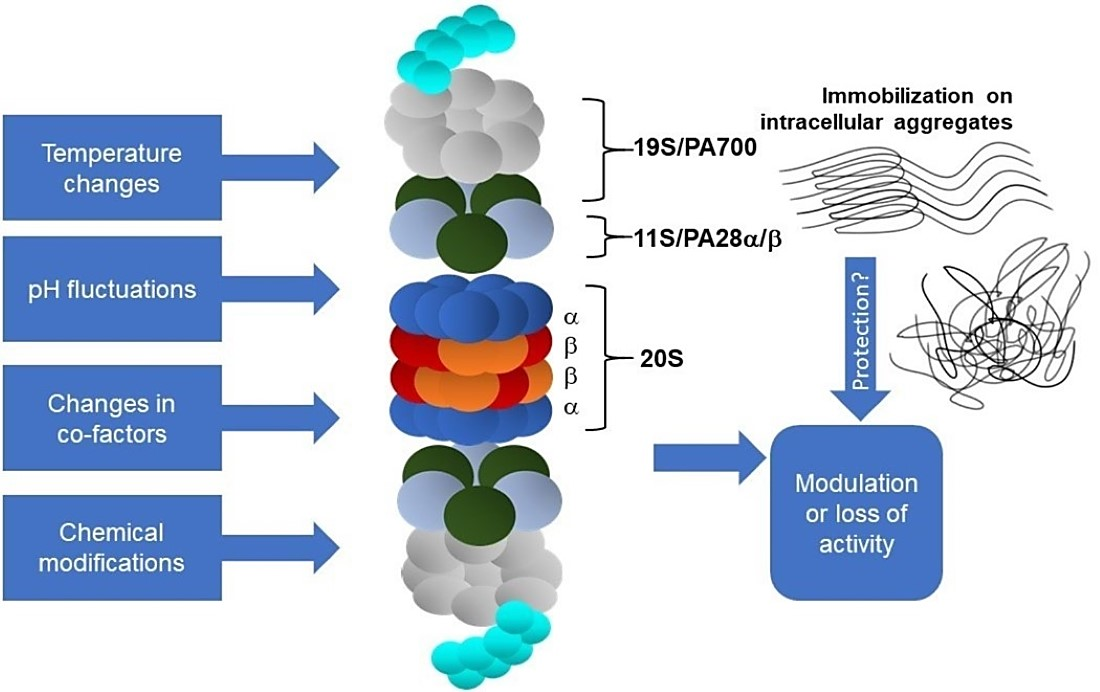{kind=link}
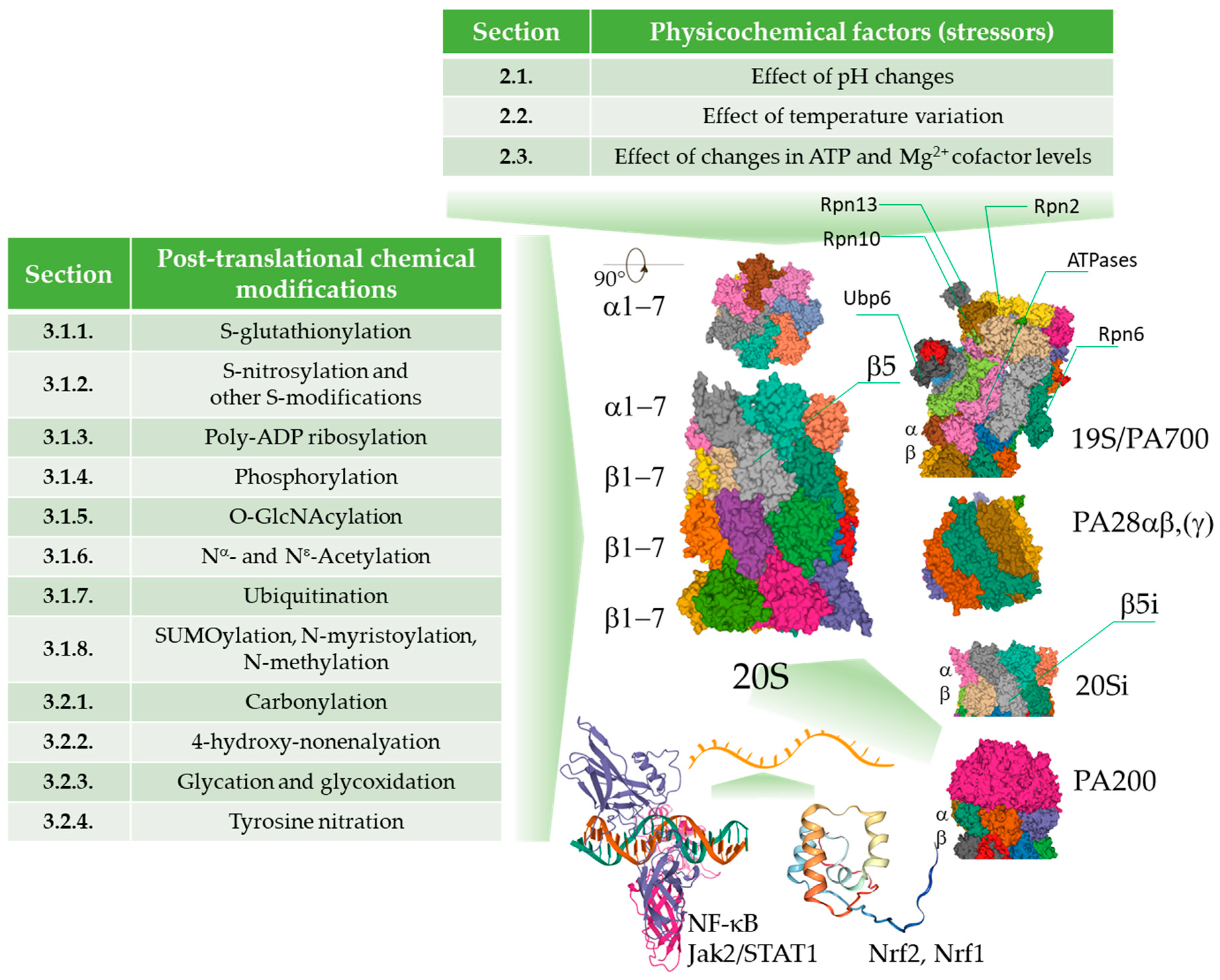{kind=link}
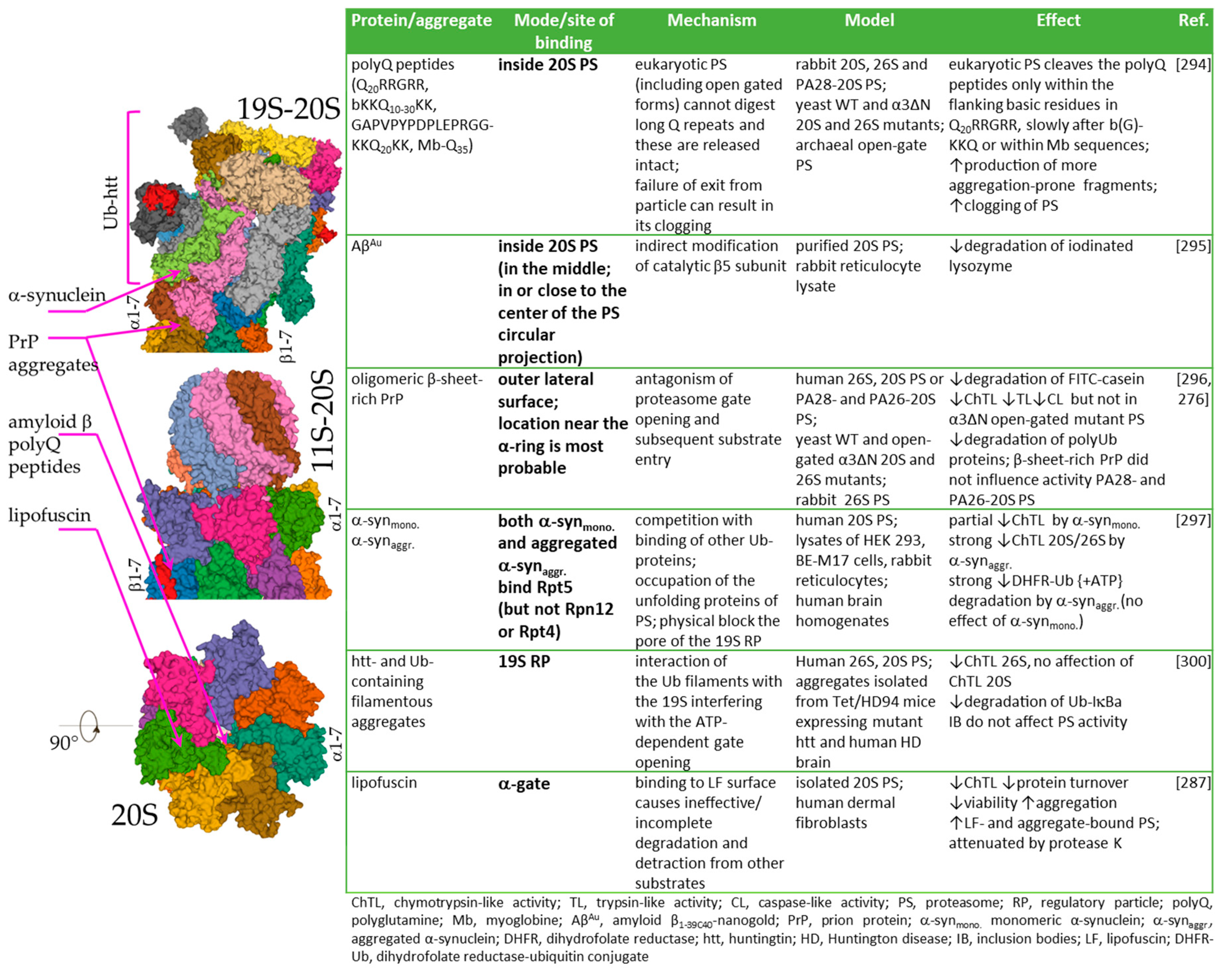{kind=link}
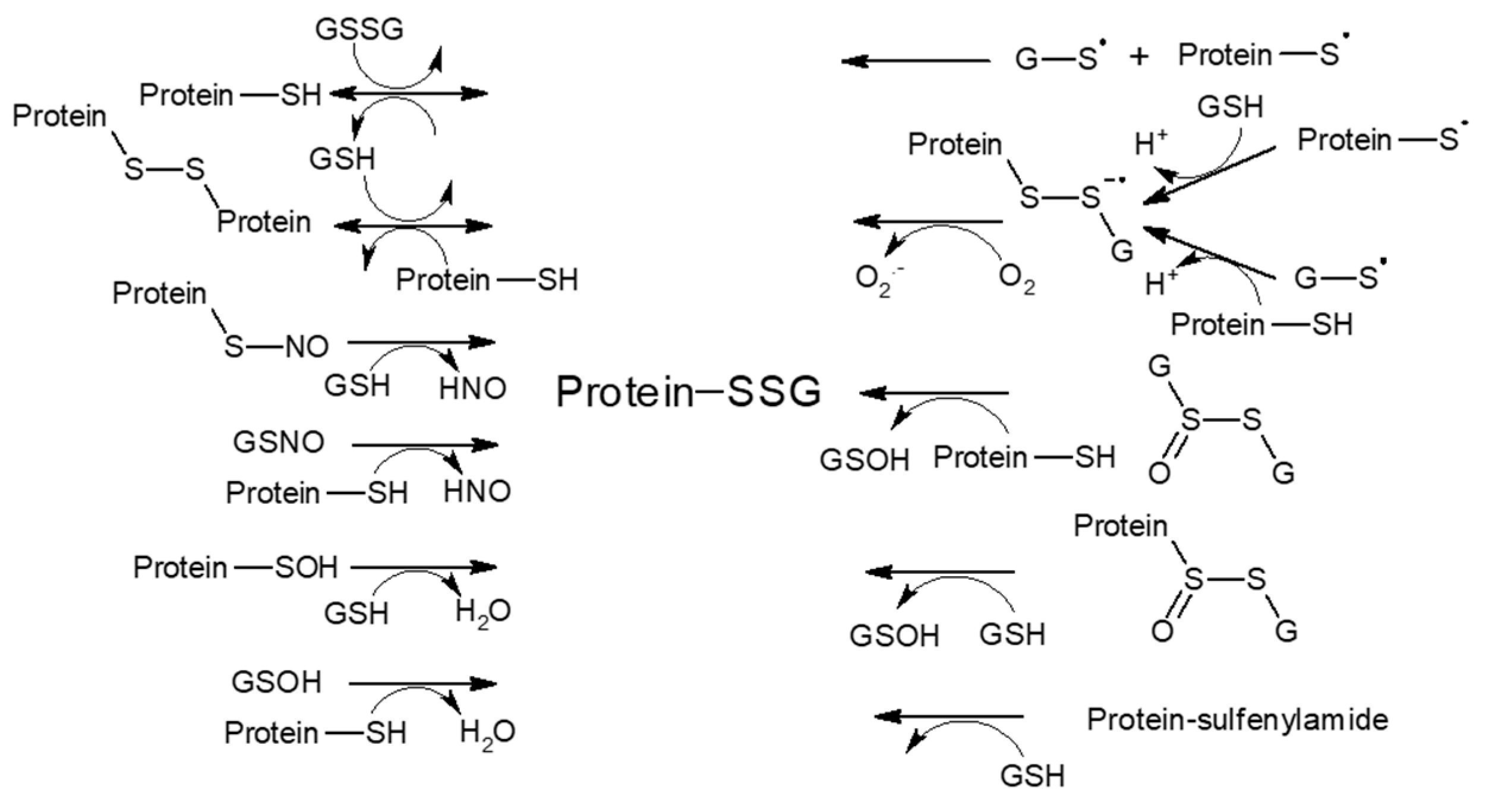{kind=link}
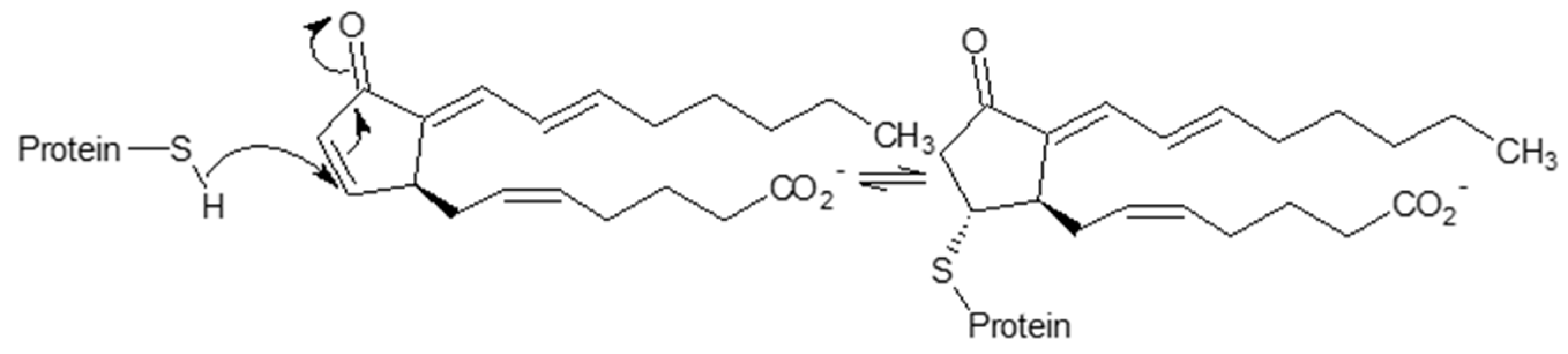{kind=link}
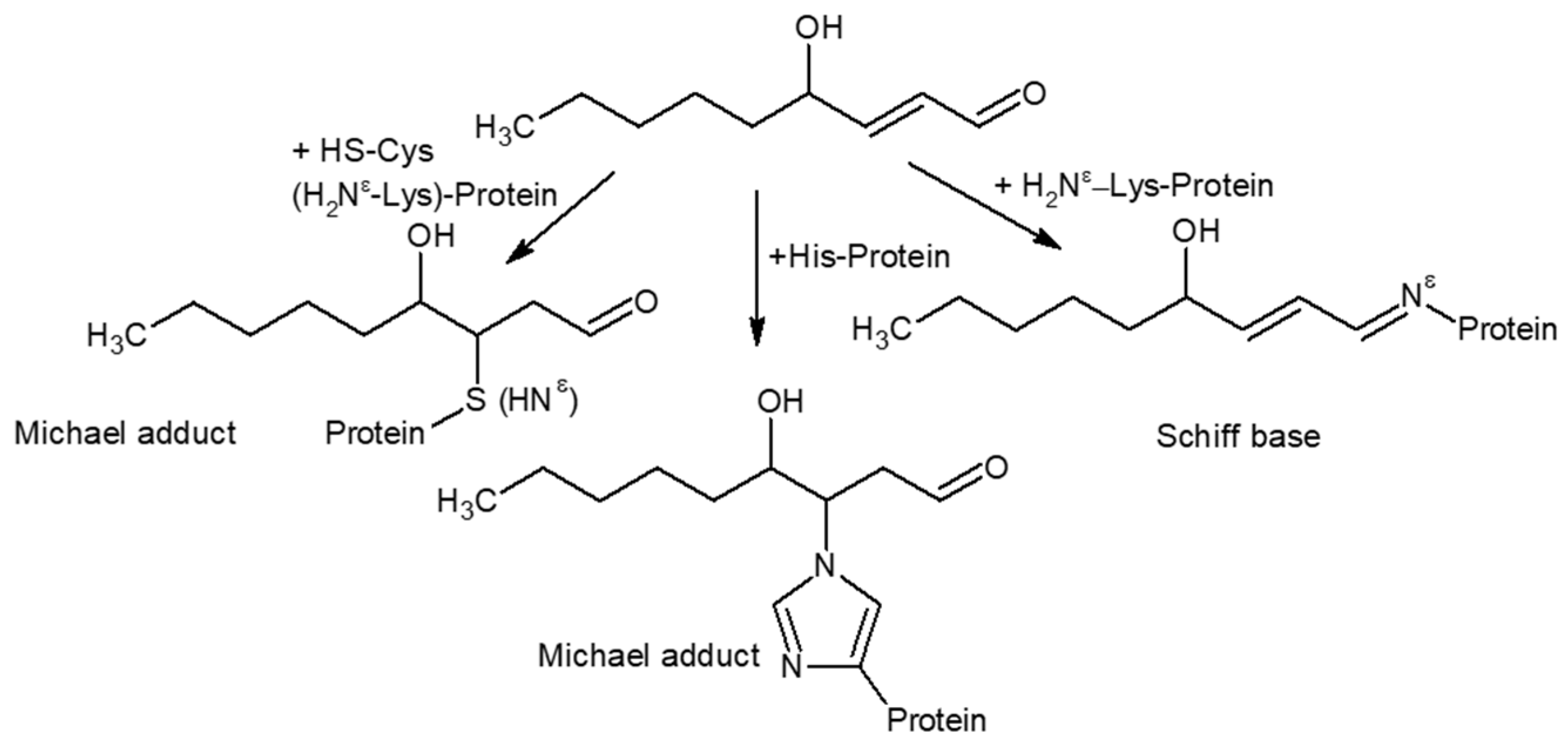{kind=link}
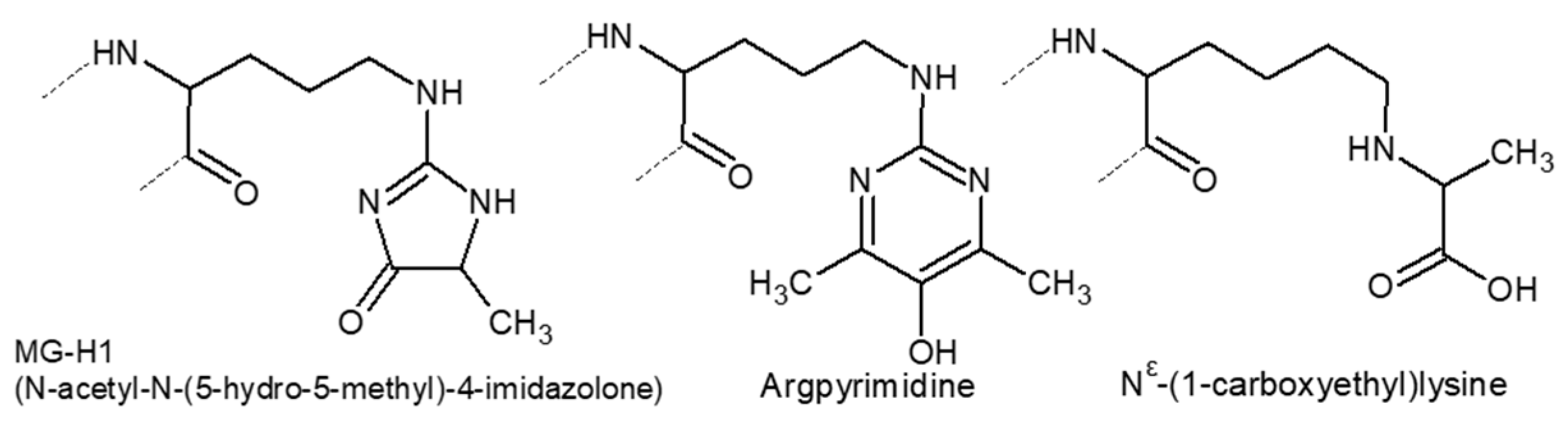{kind=link}
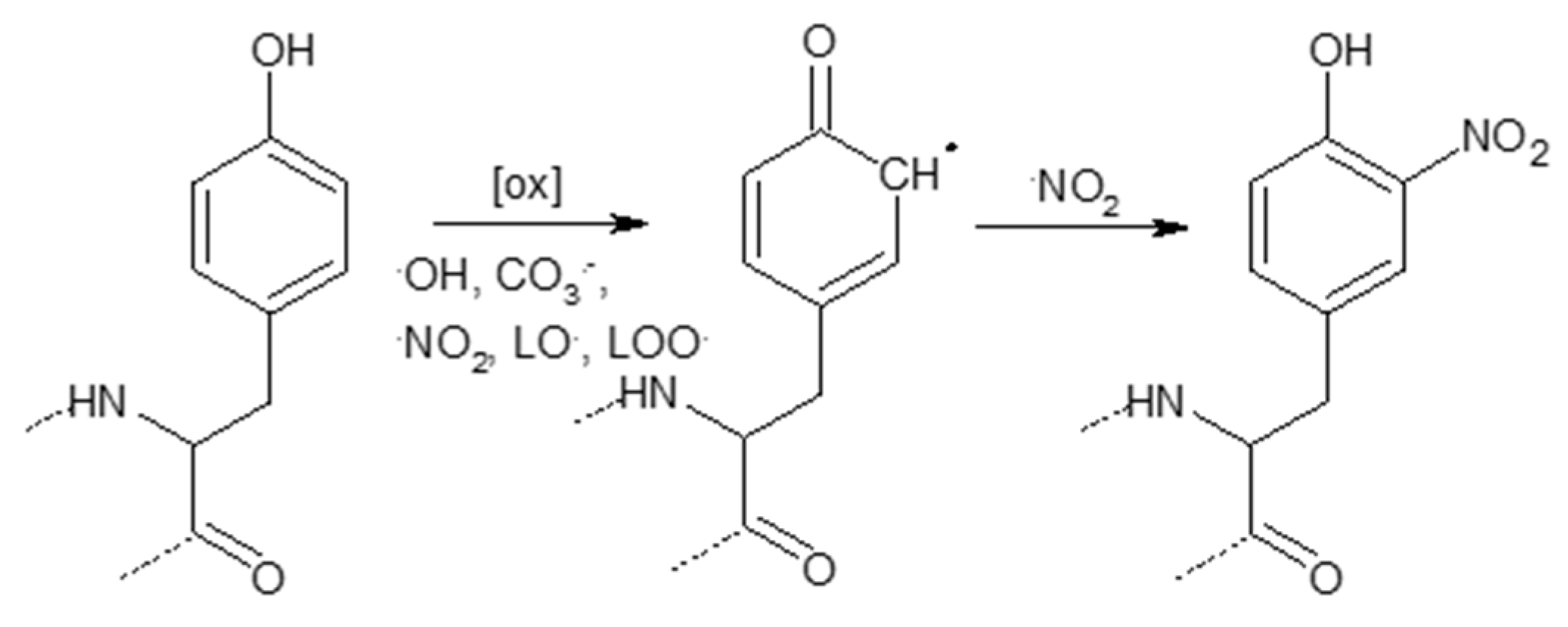{kind=link}
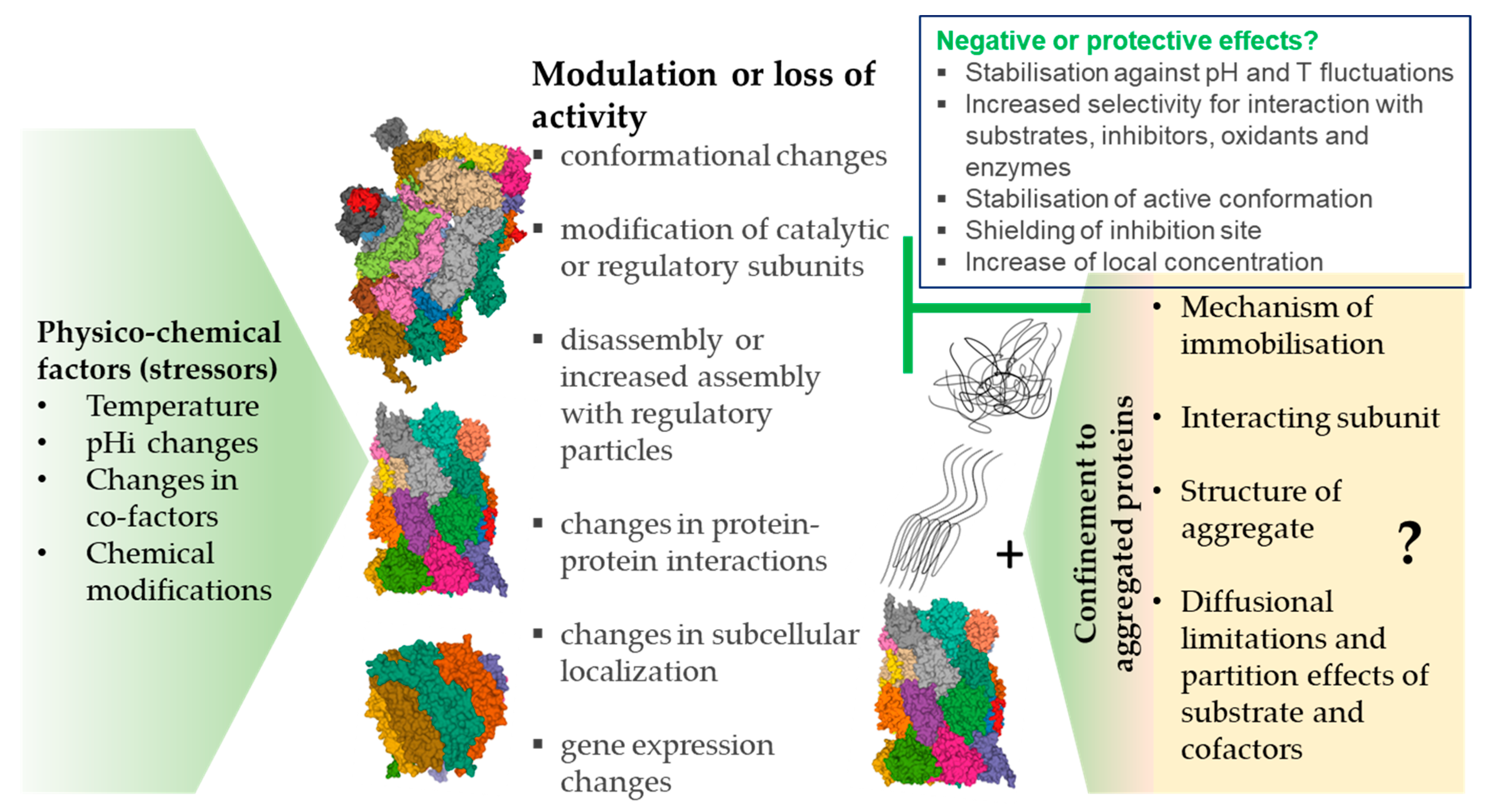{kind=link}
| Factor | Substrate | Assay Conditions | Model | Mechanism | Effect | Ref. |
|---|---|---|---|---|---|---|
| 2.1 Effect of pH changes | Total proteins | Incubation in calcium-free KRB (+/− inhibitors) for 2 h, 37 °C | Renal failure-associated acidosis in Sprague-Dawley rats, rat muscle tissue; NaHCO3 diet, MG132, E-6 and other lysosomal inhibitors | ↑mRNA encoding Ub and PS subunits a2 and a4 | ↑ATP-dependent muscle proteolysis {eliminated by MG132, ATP depletion or NaHCO3 diet} | [72] |
| - | Dextrose buffers with pH 7.5 and 4; Loss of pHi control promoted by CCCP or by reducing Pma1 levels | Yeast cells (vacuolar-ATPase mutants and WT) | Signalling mechanism engaging pH in response to carbon source exhaustion | Protection of PS from autophagy during starvation ↑accumulation of PSG ↑relocalization of PS, actin and Hos-2 | [73] | |
| Total proteins | pHi 7.3–7.5 (proliferative cancer cells) and 6.7–6.8 (quiescent cancer cells), 37 °C | Human cancer HepG2 and UMUC-3 cells Paclitaxel, NHE1 inhibitors | NHE1 downregulation Upregulation of UPS {promotion of ubiquitination} | Entry to quiescence and ↑resistance to chemotherapy reversed by MG132 | [75] | |
| FPS | pH range from neutral to alkaline, 37 °C | Purified 20S and 26S PS from rat liver | pH effect on enzyme activity | 26S, 20S PS: Max. ChTL, TL, CL at ~pH 8; 26S PS: ↓ChTL ↓TL ↓CL at pH 7.5–7.0 | [78] | |
| FPS | pH range from 3.5 to 10.5 {50 mM Na citrate (pH 3.5–6.0), 50 mM TrisHCl (pH 7.0–8.0) and 50 mM glycine-NaOH (pH 8.5–10.5)}, 37 °C | Purified PS from ostrich liver | pH effect on enzyme activity | pH optima for ChTL, TL, CL: neutral or slightly alkaline Max. ChTL, CL at pH 7.0 Max. TL at pH 8.0 | [79] | |
| FPS | pH 3.5–6.0 (25 mmol/l Hepes–acetic acid/acetate buffer) pH 6.5–9.0 (25 mmol/l Hepes–NaOH buffer), 30 or 60 min, 37 °C | Purified 20S PS from human platelets | pH effect on enzyme activity | pH optimum for ChTL: pH 7.5–8.0 and 5.0–5.5 | [80] | |
| FPS | pH 8, 30 min, 37°C {peptidase activity} Kinetic association/dissociation of Hsp90-20S proteasome complex measured in pH range 6.0–8.0 | 20S PS and HSP90 purified from bovine thymus | Kinetic properties of the HSP90-20S proteasome complex controlled by protons | ↑TL HSP90 and 20S do not associate at pH < 6.5 and at pH > 7.5, high affinity binding at physiological pH (kas 107 M−1.s−1). | [84] | |
| - | Lysates +/− PS inhibitors ALLN, ALLM {in medium} | Human endothelial cells (HUVECs and HPMVECs); hypoxia | - | ↑PS activity promoted by ↓extracellular pH and pHi ↑LPS-stimulated expression of ICAM-1 and lymphocyte adhesion ↓cytoplasmic IκB | [86] |
| Factor | Substrate | Assay Conditions | Model | Mechanism | Effect | Ref. |
|---|---|---|---|---|---|---|
| FPS | 20S and 26S PS incubated with FPS at 20–75 °C | Purified 20S and 26S PS from rat liver | High temperature effect on enzyme activity | 20S PS: Max. ChTL, TL, CL at ~40 °C; 26S PS: ChTL unstable at T > ~35 °C, Comparable effect of T on TL and CL of 20S and 26S; impairment of ChTL of 26S PS reversed by SDS | [78] | |
| 2.2 Effect of temperature | [3,5-3H]Tyr-labelled long-lived and short-lived proteins | Protein degradation measured at 37 and 41 °C (+/− energy-depleting medium, E64, Ca-free medium, lysosomal and PS inhibitors) | L6 rat skeletal muscle (myotubes) | Not dependent on changes in mRNA levels for Ub and subunit β2 | ↑degradation of short- and long-lived proteins (max. effect at 41 °C, with greater effect in long-lived proteins) suppressed by PS inhibitors or ATP depletion | [88] |
| FPS | Cell lysates, 37 °C | Human skin fibroblast undergoing ageing in vitro exposed to RMHS at 41 °C for 60 min twice a week | ↑ levels of 11S and 11S bound to 20S PS in early- and mid-passage cells only | ↑ChTL ↑TL ↑CL in early- and mid-passage cultures; loss of upregulation in late passage | [89] | |
| - | Shift from 20 to 25 °C for 1 day (in vivo) | C. elegans (whole animal lysates and in tissue-specific transgenic reporter strains) hsp-4p and hsp-16.2p::GFP reporters; UbG76V- Dendra2; UIM2::GFP::MODC | PS subunit translation or posttranscriptional regulation, regulatory interactors, substrate ubiquitination, or protein folding | ↑UPS activity in the intestine, but not in the body wall muscle tissue; ↑polyUb proteins in body wall muscle tissue | [90] | |
| FPS | Cell lysates, 37 °C | HT22 cells (whole and nuclear lysates); Nrf2-siRNA transfected cells; hyperthermia at 42 °C for 1 h with or without recovery | ↑Nrf2 expression | ↑nuclear ChTL and ↑β5 subunit and ↑HSP70 expression, ↑HO-1 and GSTα expression | [91] | |
| FPS | Cell lysates, 37 °C (+/− ATP or SDS) | PC-3, LnCaP, DU-145 human prostate cancer cells; hyperthermia at 44 °C for 1 h | Thermosensitivity of 19S | ↓ChTL of 26S, no effect on 20S; ↑apoptosis and radiosensitization; ↑stabilization IkB ↓androgen receptor | [92] | |
| FPS | 20S and 26S PS fractions from density gradient or purified proteins (+/− ATP or SDS) | Murine RMA cells, 3T3 and B8 fibroblasts Heat shock for 25 min at 42 °C with or without recovery | ↑locked inactive latent conformation of 20S PS; ↓mRNA PS levels and de novo PS maturation | ↓ChTL activity of 26S and 20S PS; ↑cellular redistribution {nucleus and membrane}; positive effects during heat shock response | [93] | |
| FPS | Rat heart extracts, 4 °C or 37 °C, physiological or actual ATP | Cold myocardial ischemia, 4 °C purified 20S/26S PS preparations from human erythrocytes | Hypothermia effect on enzyme activity; reduced competing effect of natural protein substrates {activation effect} | 4-5-fold ↑activation ChTL at 4 °C ChTL {non-ischemic hearts} 8-fold↓ at 4°C vs 37 °C | [94] |
| Factor | Substrate | Assay Conditions | Model | Mechanism | Effect | Ref. |
|---|---|---|---|---|---|---|
| 2.3. Effect of changes in ATP and Mg2+ cofactor levels | FPS, 32P-Ub5- DHFR Ubn-Sic1 Poly-Ub-proteins | Purified PS (+/− ATP or ATP𝛾S) 32P-Ub5-DHFR (+folate} | Purified 26S PS yeast—WT and mutants preventing ATP binding to Rpt3, Rpt5, or Rpt6; Purified 26S PS from mouse fibroblasts | Polypeptide structure determines energy expenditure; cooperative function of ATPase subunits | ↓ similar basal and stimulated ATP hydrolysis and ↓degradation of Ub-substrates in mutants ↑ time of degradation and ATP expenditure in tightly folded substrates by WT PS | [96] |
| FPS | Cell lysates (endogenous ATP) | Primary mesencephalic rat cell cultures Rotenone, MPP+ epoxomicin, MG132, Gluc, NAC | ATP depletion and highly energy-dependent PS | ↓ChTL ↓TL ↓CL ↑ROS production ↑complex I-inhibition-induced neurotoxicity enhanced by PS inhibition | [97] | |
| FPS | Whole fly extracts (endogenous ATP) Colorimetric and in gel assay | Drosophila melanogaster flies 1–2 days of age (young) flies 43–47 days of age (old) | Ageing-promoted decline (~50%) in ATP affecting assembly and activity of 26S PS | ↓ChTL ↓TL ↓CL of 26S PS in old flies; ↓assembly of 26S ↓ATP level ↑Ub-proteins ↓locomotor function | [98] | |
| FPS | Cell lysates (endogenous ATP) Luminometric and in gel assay | Mouse embryonic fibroblasts from mtDNA mutator mice | Impaired activity of respiratory complex I; TCA reprogramming with shortage of aspartate and e− acceptors | ↓ ChTL and assembly of 26S PS in mutator cells despite of stable ATP production via glycolysis | [99] | |
| FPS GFP-CL1 GFP-dgn/RFP | Cell lysates (endogenous ATP) Purified PS (0-4mM ATP) Fluorometric assay {WB for degrons} | Purified 26S PS; K562, P388, NCI-H460, and SH-SY5Y cells; Oligomycin, 2DG | Negative regulatory effect of basal level of ATP on PS serving likely as a layer of safeguard for the PS reserve | <50 μM ATP ↑ChTL >100 μM ATP ↓ChTL ↑substrates degradation at low ATP; ↑resistance of leukemic cells to PS inhibition at low ATP levels | [100] | |
| FPS | Tissue cytosolic extracts (0–2 mmol/L ATP) Optimized assay conditions | Sprague Dawley rats; cardiac cytosolic tissue isolates | - | 6 μmol/L ATP/10 μg protein max ↑ChTL; 2 mmol/L ATP/30–90 μg protein↓ChTL; Optimal c(ATP) = 6–100 μmol/L for ChTL, TL | [101] | |
| FPS | Purified PS (endogenous ATP) | Isolated 20S PS from diaphragm biopsies from patients with and without COPD | - | ↑ChTL and ↑CL 20S PS ↑MAFbx mRNA ↓myosin levels | [102] | |
| FPS | Rat heart extracts, Purified PS, 4 °C or 37 °C, physiological or actual ATP | Cold myocardial ischemia, 4 °C; Purified 20S/26S PS preparations from human erythrocytes | ~30% subset of 26S PS are stable and activated at very low levels of ATP; ATP hydrolysis is required for 26S activation | ↑ChTL ↑TL ↑CL at low μmol/L range ATP ↓ChTL ↓TL ↓CL at ATP physiol. range; activating effect of low ATP on 26S PS activity suppressed by Mg2+ chelation | [94] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| 3.1.1. Glutathionylation | α5 (Cys76, Cys221) | BSAox–DNPH, casein-FITC, Grx, FPS | respiratory conditions, Grx, cytosolic Trxs,proteolytic inhibitors, GSH/GSSG, Cys | Yeast, rat liver epithelium cells, purified 20S PS from yeast, rabbit reticulocytes and human erythrocytes, yeast cells with mutated (ΔNα3α7, α5-C76S, 5-C221S) 20S PS | Positive and negative regulation of α-gate opening ↑degradation of protein substrate, ↓ChTL ↑ChTL {mmolar GSH, GSSG, Cys} | [109,110,111,112,113] |
| Rpn1, Rpn2 | FPS | GSH/H2O2 | HEK 293 cells, neutrophils; lung extracts from mice with inactivated catalase; purified human 20S and 26S PS | ↓ChTL ↓TL | [115] | |
| 3.1.2. S-Nitrosylation | Rpt1 (Cys389), Rpt4 (Cys170, Cys347), Rpt5 (Cys387, Cys396) Rpn2 (Cys806) Rpn6 (Cys222) Rpn9 (Cys114), α1 (Cys154, Cys161), α7 (Cys42), β3 (Cys19), S15 (Cys81) | - | NO donors (CysS-NO, GS-NO) | Human pulmonary arterial endothelial cells and lysates | - | [120] |
| 20S PS (10 cysteines) | FPS | NO-donors (e.g., SNAP, GS-NO) DTT cGMP/cAMP analogues | Rat vascular smooth muscle cells | ↓ChTL ↓TL ↓CL ↑ β1, β1i, α5, α6 | [121] | |
| 3.1.2. S-15d-PGJ2 | Rpn1-3, Rpn5-9, Rpn11, Rpt3-6 (confirmed by immunoprecipitation for Rpn1, Rpn2, Rpn3, Rpn6) | FPS | 15d-PGJ2 IκB-α and p105 | Human aortic endothelial cells Purified 19S RP | ↓ChTL ↑Accumulation of Ub-proteins ↓Proteolysis of IκB-α and p105 ↓Migration and adhesion of monocytes | [124] |
| 3.1.2. Other S-modifications (oxidations) | β1, β2, β5 | CaMox, FPS | Ageing | Aged F344BN rat fast-twitch skeletal muscle – homogenates and purified PS | ↓ChTL ↓TL ↓CL ↓CaMox degradation restored by DTT | [126] |
| α2, β1, β3, β5i | FPS | Paraquat | Purified murine cardiac 20 PS | ↓ChTL | [127] | |
| 3.1.3. Poly-ADP ribosylation | 20S PS | FPS [3H]Leu- and [3H]-histones Fluorescamine-proteins [35S]-Met-proteins | PARP1 H2O2, TNFα Lactacystin, MG-132 3-ABA, PJ-34 | Primary human fibroblasts, PC12, RAW264.7, K562, BV2, PARP expressing cells; Isolated histones, nuclei and 20S PS | ↑ChTL ↑degradation of radio-labelled and fluorescamine-proteins ↑20S PS and PARP association | [130,131,132] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| 3.1.4. Phosphorylation | α2, α3 (Ser or Thr), α7 (Tyr) | FPS | CKII, cGMP PP, PP inhibitors | L-132 human embryonic lung cells, Rat-1, COS cells, yeast, purified 20S and 26S PS | ↑26S PS assembly ↑ChTL (↑Km), ↑TL ↑CL | [137,138,139,140] |
| α7 (Ser258, Ser263, Ser264) | - | CKII | Purified yeast 20S PS α7, Rpt5 and Ecm truncation and deletion mutants | ↑26S PS association with Ecm | [162,163] | |
| α7 (Ser243, Ser250) | FPS, myelin basic protein | CKII | NRK cells, purified 20PS {rat liver} Recombinant pTHC8, C8.1, C8.2, C8.3, C8 S243A, and C8 S243,250A proteins | No changes in proteolysis of FPS or protein substrate | [141] | |
| α7, α3 | FPS | Plk, PP, PP inhibitors | Purified 20S PS, 20S precipitated from CA46 and HEK cells, cells expressing HA-K82R mutant | ↑ChTL | [143] | |
| α1, α2, α3, β2, β3, β7 (Ser) α1, α3, β3, β7 (Thr) | FPS | PKA/PP2A | Purified murine cardiac 20PS | ↑ChTL ↑TL ↑CL | [145] | |
| 28-30-kDa subunit | FPS, β-amyloid precursor protein | PKA, forskolin, 8-bromo cAMP | HEK293 cells | ↑ChTL, ↑APPα secretion | [146] | |
| Rpt6 (Ser120) | GFP-CL1 {in vivo}, FPS {in vitro} | PKA, Forskolin, rolipram | HEK293, NRK cells | ↑ChTL ↑TL ↑CL ↑degradation GFP-CL1 | [148] | |
| Rpt6 | paGFP-CL1, -ODC | CMPK II, bicuculline | Rat hippocampal neurons | ↑degradation GFP-CL1 and GFP-ODC | [149] | |
| p45/Sug1/Rpt6 | - | p45 kinase | Purified cardiac 26S PS | ↑interaction with α2 | [150] | |
| Rpt3 (Thr25) | FPS, 3H-Phe-proteins, polyUb GFP-titinV15P-cyclin-PY, GFP-CL1, UBL-YFP-ODC, -PEST, casein, p27Kip1, p21Cip1 | DYRK2 | HEK293T, MDA-MB-468, HaCaT cells, MEFs, T25A knock-in and DYRK2 knockout cells, mouse brain, purified 26S PS from cells | ↑ChTL ↑TL ↑CL ↑Degradation of proteins, degron-reporters and ATP; ↑S → M phase in cell cycle | [151] | |
| Rpn1 | FPS, NLS-GFP-CL1 and NLS-UbG76V-GFP | UBLCP1 | ZR751 breast cancer and HEK293Tcells, HaCaT double-stable lines | ↑nuclear ChTL ↑nuclear 26S PS assembly ↑degradation NLS-GFP-CL1 and NLS-UbG76V-GFP | [136] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| PA28α,β | FPS | Alkaline phosphatase | Rabbit reticulocytes, human erythrocytes | ↑ChTL | [142] | |
| 3.1.4. Phosphorylation (continued) | Rpn6 (Ser14) | FPS, GFP-CL1, Ub-R-GFP, UbG76V-GFP, 32P-Ub5-DHFR, Ubn-Sic1, [3H]Phe and [3Tyr]-proteins, mutant FUS, SOD1, TDP43, and tau | PKA, forskolin, rolipram, dibutyryl cAMP, glucagon, epinephrin, exercise, food deprivation | HEK293, C2C12, SH-SY5Y, H9C2, renal cells, hepatocytes, Rpn6 mutants, purified PS from rTg4510 mice, human and rat muscles and liver | ↑ChTL ↑TL ↑CL ↑degradation short lived but not long lived proteins, polyUb- and aggregation-prone proteins, ATP | [154,155,156] |
| α4 (Tyr153) | FPS, 35S-labeled proteins | c-Abl, Arg | 293T, MCF-7, MCF-7/c-Abl(K290R), c-abl−/−arg−/−−/− MEFs, cells expressing α4 Y153F or c-Abl (K290R) mutant, purified 26S PS | ↓ChTL, ↓32S-labeled proteins degradation, ↑destabilisation of p53, p27, cyclin A, and cyclin E and G1/S cell cycle arrest in 293T α4 Y153F mutants | [158] | |
| Rpt5 | FPS, polyUb lysosyme, UbG76V-GFP, GFP-ODC, GFP-CL1, IκBα, ATP | ASK1 | HeLa, HEK293, neuroblastoma N2a, C6 glioma cells, cells expressing truncation and catalytically inactive ASK1 mutants | ↑apoptosis ↓ChTL ↓TL ↓CL ↓Degradation of total proteins, IκBα, reporters and ATP hydrolysis | [160] | |
| Rpn2 (Thr273) | UbG76V-GFP, Ub-R-GFP, GFP-CL1, GFP-ODC, endogenous IκBα | Osmotic stress p38 MAPK | HeLa expressing constitutively active form of p38 activator MKK6, T273A mutants, purif. 26S PS | ↓ChTL ↓TL ↓CL ↑stabilisation of UbG76V-GFP, Ub-R-GFP, GFP-CL1, GFP-ODC, polyUb proteins and IκBα | [164] | |
| Rpn3 (Ser6) | FPS, ubiquitin-AMC, 35S-ODC, 35S-Ub-cIAP1 | CKIIα′ | MEFs, HeLa, HEK293T cells Rpn11-Flag/EGFP MEFs infected with Cre-retrovirus | ↑deubiquitinaton and ↓nuclear localisation of old proteasomes, no change in proteolysis of FPS and 35S-ODC, 35S-Ub-cIAP1 | [157] | |
| 3.1.5. O-GlcNAcylation | Rpt2 | FPS, transcription factor SP1, GFP-degron, ATP | OGT/O-GlcNAcase | Nuclear extract from glucose-starved/forskolin-treated NRK cells, purified 20S and 26S PS, OGT siRNA cells | ↓generation of SpX from SP1, ↓ChTL {suc-LLVY-AMC}, unchanged TL and ChTL {Z-GGL-AMC} ↓ATPase activity ↓GFP-degron in OGT siRNA cells | [165] |
| α1 (Ser5), α5 (Ser198), α4 (Ser130), β6 (Ser208 and Ser57) | FPS | 10mM glucose | Murine fibroblast cells, 20S PS purified from spleen, liver, and brain tissues | ↓ChTL | [166] | |
| α5 | - | diabetes | Human erythrocytes | ↑O-GlcNAc site occupancy | [169] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| 3.1.6. N-acetylation | α6 (Lys30, Lys115), β3 (Lys77), β6 (Lys203), β7 (Lys201) | FPS | Acute I/R injury, end-stage heart failure, HDAC inhibitors | Human and mice cardiac tissue; purified cardiac 20S PS | ↑TL ↓TL after I/R restored by HDAC inhibitors | [175] |
| PA28γ (Lys195) | p21, HCV core protein | CBP/SIRT | HEK293/293T, H1299, HeLa, A549 cells HEK293 cells expressing PA28γ-K195R mutant | ↑degradation p21 and HCV core-173; ↑ PA28γ heptamerization and cell cycle progression | [176] | |
| PA28α, β | FPS {Cell-based bioluminescent assays and standard in vitro assay} | Naa10p protein independent on its activity | RKO, H1299, PG cells, shNaa10p, siPA28α cells, cells expressing mutant Naa10p (R82A) or overexpressing Naa10p; purified PS complexes | ↓ChTL | [177] | |
| β1, β2, β5 | FPS | N-acetyltransferase ARD1-NAT1 | Yeast, β-subunit propeptide and NAT1 deletion mutants | Corresponding ↓ChTL ↓TL ↓CL; growth defects | [178] | |
| 3.1.7. Ubiquitination | α2 {mono-ubiquitination} | FPS | Binding to δ-ALAD, HDAC inhibitor (SAHA) | Human prostate cancer DU145, LNCaP and PC-3 cells, ALAD overexpressing cells | ↓ChTL Interaction with δ-ALAD; ↑Interaction with δ-ALAD, acetylation and nuclear translocation in response to SAHA | [183] |
| Rpn10 (Lys84, Lys268, Lys71, Lys99) {mono-ubiquitination} | cyclin B | Rsp5/Ubp2; Rpn10-conjugated Ub clashes with Rpn9 leading to disassembly of Rpn10:Rpn9 complex | Yeast; rsp5, ubp2, Rpn10, Dsk2 mutants; Purified 26S PS | ↓affinity to polyUb substrates; ↓cyclin B degradation; Dissoc. of Rpn10/interaction with Dsk2 26S PS “Rpn10 low/Dsk2 high” state; ↑processivity of PS through cytosolic Rpn10 | [184,185,186] | |
| Rpn10 (multiple sites; Lys74, Lys122, Lys262, Lys365 ubiquitinated by both MuRF1 and E6AP){poly-ubiquitination} | - | Ring finger E3s (MuRF1, Siah2, Parkin, APC, SCFβTRCP1), U-box E3 (CHIP), HECT domain E3s (E6AP and Nedd4)/UbcH5 | Purified WT and mutant proteins | Polyubiquitination, release and degradation of Rpn10 | [187] | |
| Rpn13 (Lys21, Lys34) {poly-ubiquitination} | FPS, 32P-Ub5-DHFR, 32P-Ubn-Sic1, 32P-casein | Ube3c PS inhibitors Heat shock Arsenite | HEK293F cells, cells stably expressing Dss1/Sem1, Ube3c siRNA transfected cells, cells re-expressing Ube3c; Purified 26S PS | ↓ Ub5-DHFR, Ubn-Sic1 degradation; Unchanged ChTL and casein degradation; ↓26S PS binding to Ub5-DHFR | [188] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| α5, β4 | - | Heat shock | U2OS cells | - | [189] | |
| 3.1.8. SUMOylation | Rpn2 (Lys932) | Cyclin B | PIASy/xSENP1 | Xenopus egg extract Purified WT and mutant proteins | Decrease in association of Ub receptor Rpn13 with Rpn2, ↓cyclin B degradation, delayed anaphase progression and mitotic arrest | [190] |
| 41 modified Lys residues | - | MG132 | HEK293 stably expressing SUMO3m, SUMO interacting motif (SIM) mutants | Nuclear translocation and colocalization with PML NB | [191] | |
| 3.1.8. N-myristoylation | Rpt2 (Gly2) | FPS, CFP-Ubi4, Gcn4 | N-myristoyltransferase Canavanine, elevated temperature | Yeast, Cells expressing rpt2-G2A, and rpt2-G2Δ mutants | Nuclear translocation ↑degradation nuclear polyUb proteins and Gcn4; Unchanged peptidase activity or 26S assembly; Cytoplasmic PS aggregates, and ↓growth in mutant cells | [192,193] |
| 3.1.8. N-methylation | Rpt1 (Pro3) | - | N-methyltransferase Canavanine, H2O2 | Yeast Cells expressing PK-deleted Rpt1 mutants | ↓ growth, and ↑ sensitivity to stress in mutant cells | [195] |
| Lys in 25kDa subunit | FPS, CYP2E1 | N-methyltransferases EtOH SAM:SAH, IFN-γ, Tubercidin | Huh7CYP cells, liver cytosolic and nuclear extracts, HCV+ mice and hepatocytes, purified 20S PS | ↓ChTL larger reduction in nuclear PS ↑CYP2E1 stabilization ↑LMP7 (β5i) (HCV+, HCV−) ↓PA28α (HCV+, HCV−) ↓LMP2 (β1i) (HCV+) ↓Ecm29, UCHL5 (HCV+, HCV−) ↓SAM:SAH ↓SIINFEKL presentation (HCV+>HCV−) | [197,198] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| 3.2.1. Carbonylation | α2, α4, α6, β3, β7 (Thr94) | - | Paraquat | Purified murine cardiac 20S PS | - | [127] |
| Rpt3 | Flag-Ub ATP | 15d-PGJ2 Cu2+/H2O2 | SH-SY5Y cells Purified 26S PS siRNA Rpt3 transfected cells | ↓ATPase activity ↑Ub-proteins | [202] | |
| Rpt3, Rpt5 | FPS polyUb proteins Bax | Ischemia/reperfusion Ischemic preconditioning | I/R murin model Isolated murine hearts | ↓ChTL {ATP-dependent and –independent} ↑Ub-proteins and Bax | [205] | |
| Rpt5 | FPS {fluorescent probe, MV151} polyUb proteins Bax, p53, Akt | Human heart failure and hypertrophic cardiomyopathy | Human heart tissue | ↓ChTL ↓CL ↑oxidized and Ub-proteins, p53, Akt | [206] | |
| 3.2.2. 4-hydroxy-nonenalyation | α7 | - | HNE | FF95 cells Purified 20S PS | unstable and reversible HNE adduct | [211] |
| α1, α2, α7 | FPS | Coronary occlusion/reperfusion | Rat cardiac cytosolic fraction; purified 20S PS | ↓ChTL ↓TL ↓CL in cytosolic fraction ↓TL in purified PS; ↑oxidized and Ub-proteins; slight ↓26S association | [215] | |
| α, β | FPS | Cerebral ischemia/reperfusion | Brain homogenates of GPX1(+) and GPX deficient mice (GPX−/−) | ↓ChTL ↓TL ↓CL; ↓ChTL attenuated in GPX1(+) mice | [216] | |
| 20S PS | FPS HNE-proteins and HNE-GAPDH | Ferric nitrilotriacetate PS inhibitor {in vitro} | Renal tissue homogenates | Transient ↓TL ↓CL Transient ↑oxidative stress markers, HNE- and Ub-proteins; ↑clearance of HNE-proteins in vitro | [217] | |
| α2, α5, β1, β5i (α1, α7, β7 in young and middle age) | FPS | Human ageing | Leukocyte-enriched fractions of healthy human blood (from 20- to 63-year-old donors) | no dissociation into 20S ↓ChTL | [218] | |
| α, β | FPS | Rat ageing HNE, FeSO4 {in vitro} | Spinal cord of aged Fisher 344 rats, NSC-19 cells | ↓ChTL ↓TL ↓CL ↓α, β expression | [219] | |
| α6 | FPS | HNE | Purified 20S PS from rat liver | ↓ChTL, ↓TL ↓CL ↓ChTL at time of α6 modification | [220] |
| PTM | Subunit | Substrate | Stimuli/Mechanism | Model | Effect | Ref. |
|---|---|---|---|---|---|---|
| 3.2.3. Glycation | β2 {in vivo} β2, β4, β5 {in vitro} | FPS | AGE-BSA, MGO, GA {purified PS} HG, MGO, GO, GA {cells} | C57BL/6 WT mice, diabetic Ins2Akita, GLO1-knockdown (GLO1-KD), and STZ diabetic and GLO1-knockdown diabetic (GLO1-KD-STZ) mice; EA-hy.926 and bovine retinal endothelial cells, purified 20S PS from cells | ↓ChTL in cells ↓ChTL in purified PS {no effect of AGE-BSA} ↓ChTL ↓TL↓CL {kidney diab. Ins2Akita mice} ↓ChTL {aorta diab Ins2Akita mice} ↓ChTL {kidney GLO1-KD, STZ diab. and GLO1-KD-STZ} ↑Ub-proteins in vivo and in vitro ↓19S Rpn10 {in vivo} ↓19S RP {in vitro} | [224] |
| - | FPS | HG, MGO, GO, GA, AGE-BSA {BSA pre-glycated with glucose, MGO, GO, GA} | J774A.1 mouse macrophage cell extracts | ↑ChTL ↑TL ↑CL {MGO, GO, GA} ↓ChTL ↓CL ↑TL {AGE-BSA} | [227] | |
| 3.2.4. Tyrosine nitration | α1, α2, α7, β1, β3, β5, β7 | - | - | Purified murine cardiac 20S PS | - | [127] |
| Rpt4 | FPS Endogenous BH4, GTPCH and Trx | STZ, Angiotensin II, HFD, MG132 {in vivo}; ONOO−, uric acid, L-NAME, MG132 {in vitro}, Tempol, MG132, SNP, Ach {ex vivo} | C57BL/6J WT, LDLr−/− knockout and PA700/S10B knockdown mice; HUVEC WT and p67-DN Nox cells, purified 26S PS | ↑ChTL {ATP-dependent} ↑26S PS assembly ↓Levels of BH4, GTPCH I and Trx ↓Ach-induced vessel relaxation Restorative effect of antoxidants and PS, Nox, NOS inhibition | [232,233] | |
| 20S PS | FPS, β-casein | ONOO− | Bovine brain 20S PS and thymus 20S iPS | ↑ChTL↑CL ↑BrAAP ↓TL:20S PS ↓ChTL↓CL ↓BrAAP: 20S iPS ↓β-casein degradation by iPS | [221] | |
| 20S PS {high level ONOO−} | FPS | Molsidomine {in vivo} ONOO− SIN-1 SNAP | Mouse liver, Hep G2 cells, liver soluble fractions, crude and purified PS preparations | ↑ChTL {low level ONOO−} ↓ChTL {high level ONOO−} ChTL stimulated by ATP and PA28 {low level ONOO−} | [235] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Račková, L.; Csekes, E. Proteasome Biology: Chemistry and Bioengineering Insights. Polymers 2020, 12, 2909. https://doi.org/10.3390/polym12122909
Račková L, Csekes E. Proteasome Biology: Chemistry and Bioengineering Insights. Polymers. 2020; 12(12):2909. https://doi.org/10.3390/polym12122909
Chicago/Turabian StyleRačková, Lucia, and Erika Csekes. 2020. "Proteasome Biology: Chemistry and Bioengineering Insights" Polymers 12, no. 12: 2909. https://doi.org/10.3390/polym12122909
APA StyleRačková, L., & Csekes, E. (2020). Proteasome Biology: Chemistry and Bioengineering Insights. Polymers, 12(12), 2909. https://doi.org/10.3390/polym12122909