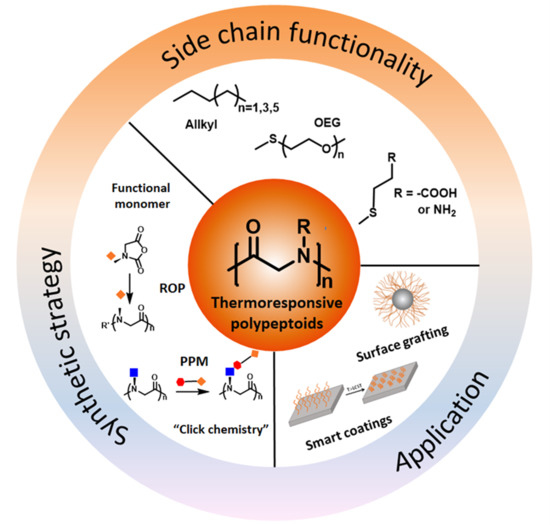
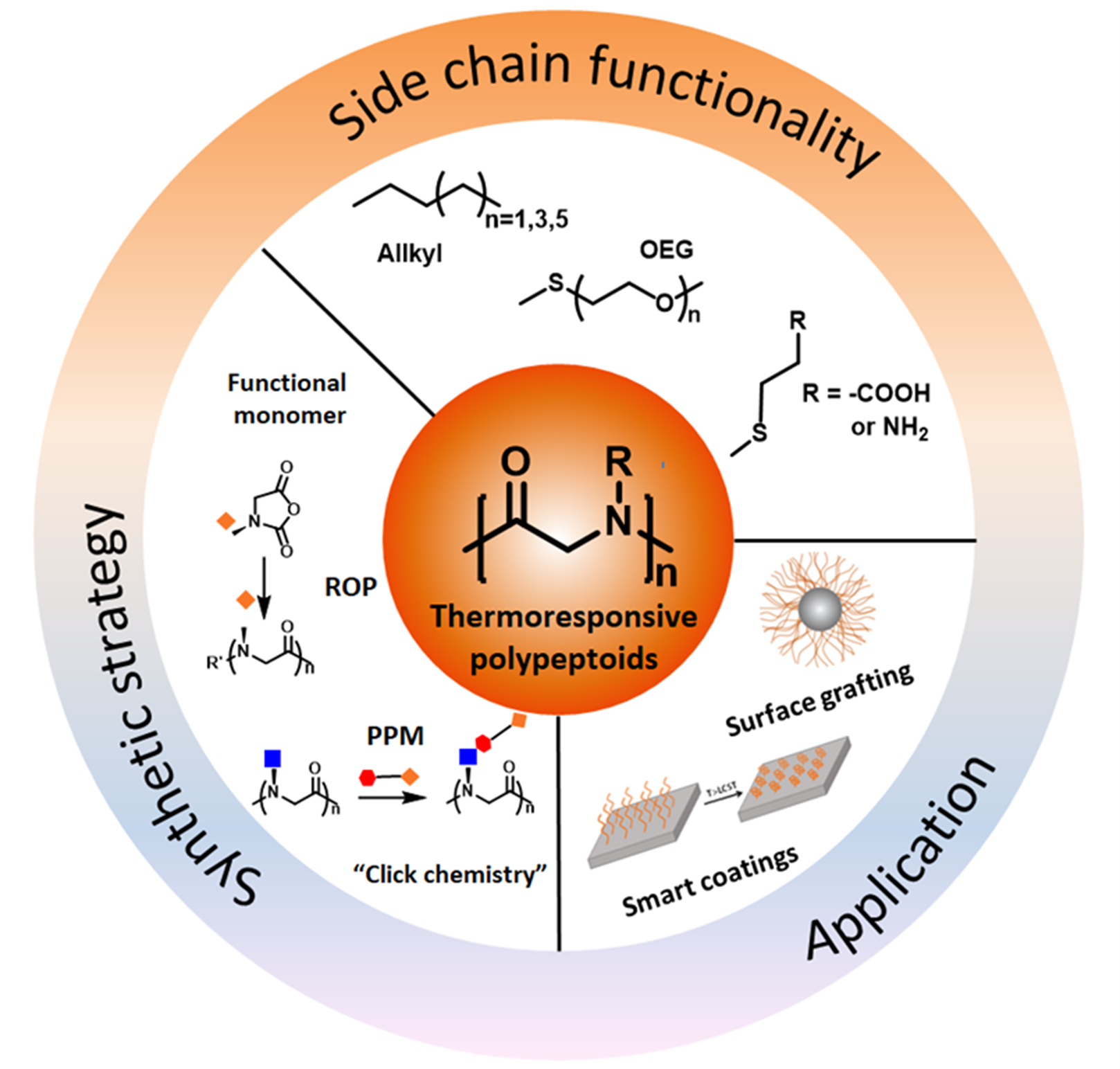
Thermoresponsive Polypeptoids


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Synthetic Strategies of Thermoresponsive Polypeptoids
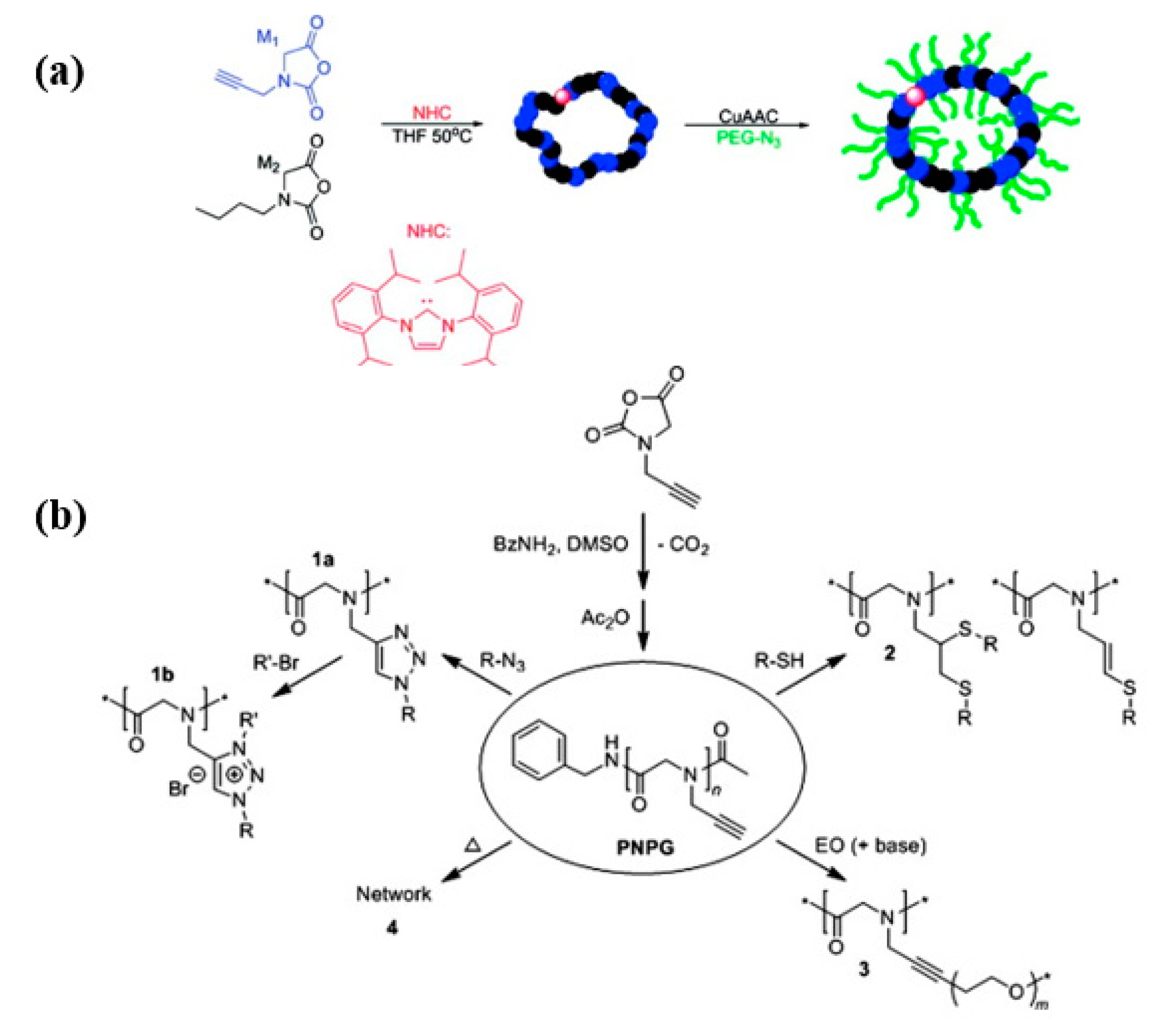
2.1. The Controlled Polymerization of Functional Monomer Strategy
2.2. Post-Polymerization Modification (PPM) Strategy
3. Thermoresponsive Polypeptoids
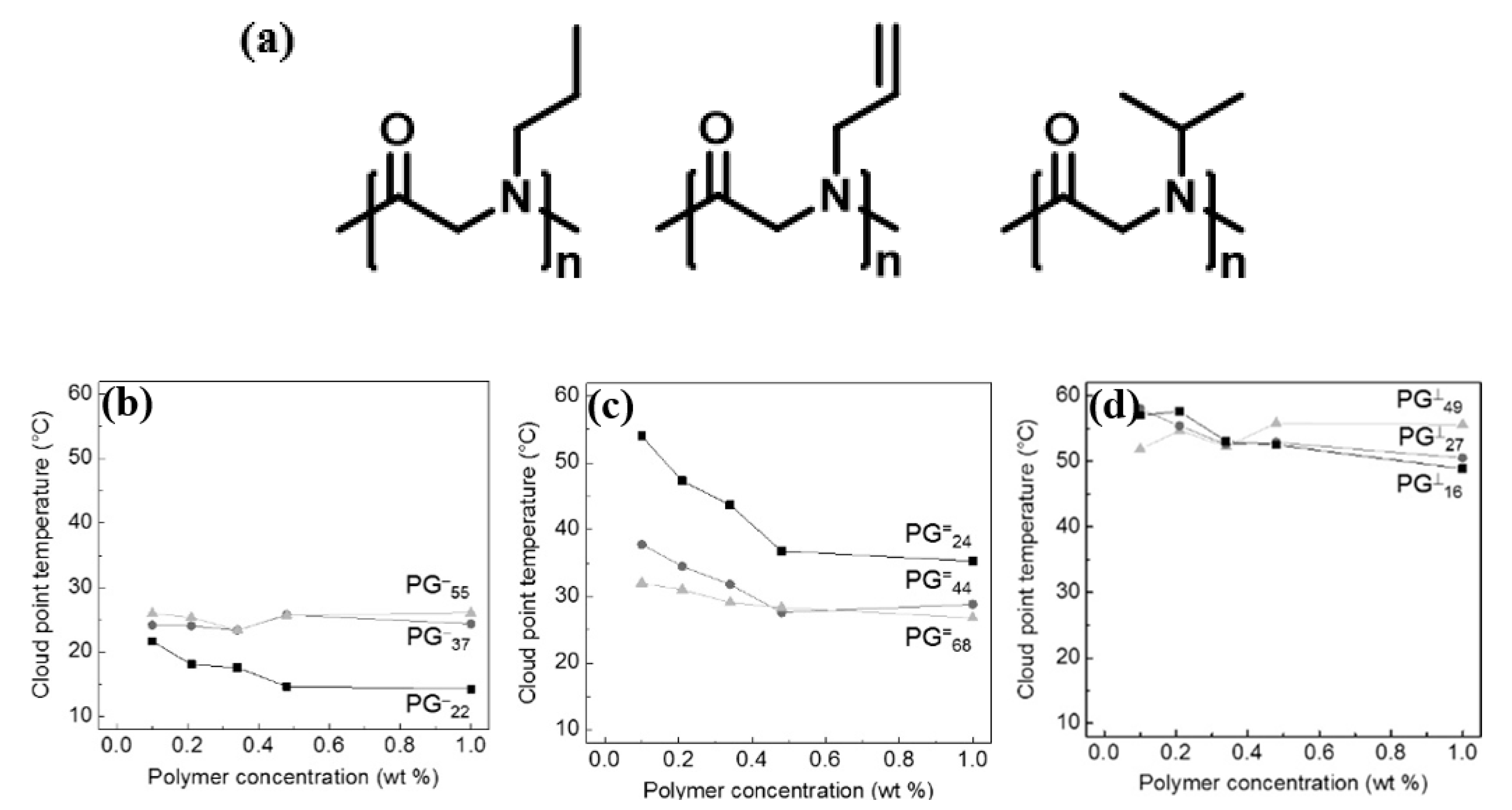
3.1. Thermoresponsive Polypeptoids Containing Alkyl Side Chains
3.2. OEGlyated Thermoresponsive Polypeptoids
3.3. Charge-Determined Thermoresponsive Polypeptoids
4. Applications
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Hoffman, A.S. Stimuli-responsive polymers: Biomedical applications and challenges for clinical translation. Adv. Drug Deliv. Rev. 2013, 65, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Vanparijs, N.; Nuhn, L.; De Geest, B.G. Transiently thermoresponsive polymers and their applications in biomedicine. Chem. Soc. Rev. 2017, 46, 1193–1239. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, G.; Tsitsilianis, C. LCST polymers: Thermoresponsive nanostructured assemblies towards bioapplications. Polymer 2020, 211, 123146. [Google Scholar] [CrossRef]
- Hogan, K.J.; Mikos, A.G. Biodegradable thermoresponsive polymers: Applications in drug delivery and tissue engineering. Polymer 2020, 211, 123063. [Google Scholar] [CrossRef]
- Zhu, Y.; Batchelor, R.; Lowe, A.B.; Roth, P.J. Design of Thermoresponsive Polymers with Aqueous LCST, UCST, or Both: Modification of a Reactive Poly(2-vinyl-4,4-dimethylazlactone) Scaffold. Macromolecules 2016, 49, 672–680. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Thijs, H.M.L.; Jochems, M.J.H.C.; van Lankvelt, B.M.; Fijten, M.W.M.; Schubert, U.S. Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: Alternatives to poly(N-isopropylacrylamide)? Chem. Commun. 2008, 44, 5758–5760. [Google Scholar] [CrossRef]
- Ko, C.-H.; Claude, K.-L.; Niebuur, B.-J.; Jung, F.A.; Kang, J.-J.; Schanzenbach, D.; Frielinghaus, H.; Barnsley, L.C.; Wu, B.; Pipich, V.; et al. Temperature-Dependent Phase Behavior of the Thermoresponsive Polymer Poly(N-isopropylmethacrylamide) in an Aqueous Solution. Macromolecules 2020, 53, 6816–6827. [Google Scholar] [CrossRef]
- Fischer, T.; Demco, D.E.; Fechete, R.; Möller, M.; Singh, S. Poly(vinylamine-co-N-isopropylacrylamide) linear polymer and hydrogels with tuned thermoresponsivity. Soft Matter 2020, 16, 6549–6562. [Google Scholar] [CrossRef]
- Sharker, K.K.; Takeshima, S.; Toyama, Y.; Ida, S.; Kanaoka, S.; Yusa, S.-I. pH- and thermo-responsive behavior of PNIPAM star containing terminal carboxy groups in aqueous solutions. Polymer 2020, 203, 122735. [Google Scholar] [CrossRef]
- Tang, L.; Wang, L.; Yang, X.; Feng, Y.; Li, Y.; Feng, W. Poly(N-isopropylacrylamide)-based smart hydrogels: Design, properties and applications. Prog. Mater. Sci. 2021, 115, 100702. [Google Scholar] [CrossRef]
- Wischerhoff, E.; Uhlig, K.; Lankenau, A.; Börner, H.G.; Laschewsky, A.; Duschl, C.; Lutz, J.-F. Controlled Cell Adhesion on PEG-Based Switchable Surfaces. Angew. Chem. Int. Ed. 2008, 47, 5666–5668. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kataoka, K. Precise Control of Lower Critical Solution Temperature of Thermosensitive Poly(2-isopropyl-2-oxazoline) via Gradient Copolymerization with 2-Ethyl-2-oxazoline as a Hydrophilic Comonomer. Macromolecules 2006, 39, 6622–6630. [Google Scholar] [CrossRef]
- Diehl, C.; Schlaad, H. Thermo-Responsive Polyoxazolines with Widely Tuneable LCST. Macromol. Biosci. 2009, 9, 157–161. [Google Scholar] [CrossRef]
- Shen, Y.; Fu, X.; Fu, W.; Li, Z. Biodegradable stimuli-responsive polypeptide materials prepared by ring opening polymerization. Chem. Soc. Rev. 2015, 44, 612–622. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Akdemir, Ö.; Hoth, A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of Poly(NIPAM) Over? J. Am. Chem. Soc. 2006, 128, 13046–13047. [Google Scholar] [CrossRef]
- Mäkinen, L.; Varadharajan, D.; Tenhu, H.; Hietala, S. Triple Hydrophilic UCST–LCST Block Copolymers. Macromolecules 2016, 49, 986–993. [Google Scholar] [CrossRef]
- Seuring, J.; Bayer, F.M.; Huber, K.; Agarwal, S. Upper Critical Solution Temperature of Poly(N-acryloyl glycinamide) in Water: A Concealed Property. Macromolecules 2012, 45, 374–384. [Google Scholar] [CrossRef]
- Woodfield, P.A.; Zhu, Y.; Pei, Y.; Roth, P.J. Hydrophobically Modified Sulfobetaine Copolymers with Tunable Aqueous UCST through Postpolymerization Modification of Poly(pentafluorophenyl acrylate). Macromolecules 2014, 47, 750–762. [Google Scholar] [CrossRef]
- Seuring, J.; Agarwal, S. Non-Ionic Homo- and Copolymers with H-Donor and H-Acceptor Units with an UCST in Water. Macromol. Chem. Phys. 2010, 211, 2109–2117. [Google Scholar] [CrossRef]
- Plamper, F.A.; Ballauff, M.; Müller, A.H.E. Tuning the Thermoresponsiveness of Weak Polyelectrolytes by pH and Light: Lower and Upper Critical-Solution Temperature of Poly(N,N-dimethylaminoethyl methacrylate). J. Am. Chem. Soc. 2007, 129, 14538–14539. [Google Scholar] [CrossRef] [PubMed]
- Seuring, J.; Agarwal, S. First Example of a Universal and Cost-Effective Approach: Polymers with Tunable Upper Critical Solution Temperature in Water and Electrolyte Solution. Macromolecules 2012, 45, 3910–3918. [Google Scholar] [CrossRef]
- Roth, P.J. Composing Well-Defined Stimulus-Responsive Materials through Postpolymerization Modification Reactions. Macromol. Chem. Phys. 2014, 215, 825–838. [Google Scholar] [CrossRef]
- Chockalingam, K. Design and application of stimulus-responsive peptide systems. Protein Eng. Des. Sel. 2007, 20, 155–161. [Google Scholar] [CrossRef]
- Chopko, C.M.; Lowden, E.L.; Engler, A.C.; Griffith, L.G.; Hammond, P.T. Dual Responsiveness of a Tunable Thermosensitive Polypeptide. ACS Macro Lett. 2012, 1, 727–731. [Google Scholar] [CrossRef]
- Sun, J.; Zuckermann, R.N. Peptoid Polymers: A Highly Designable Bioinspired Material. ACS Nano 2013, 7, 4715–4732. [Google Scholar] [CrossRef]
- Gangloff, N.; Ulbricht, J.; Lorson, T.; Schlaad, H.; Luxenhofer, R. Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering. Chem. Rev. 2016, 116, 1753–1802. [Google Scholar] [CrossRef]
- Sun, J.; Li, Z. 7—Peptoid applications in biomedicine and nanotechnology. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Cambridge, UK, 2018; pp. 183–213. [Google Scholar]
- Figliozzi, G.; Goldsmith, R.; Ng, S.; Banville, S.; Zuckermann, R. Synthesis of N-Substituted Glycine Peptoid Libraries. Methods Enzymol. 1996, 267, 437–447. [Google Scholar]
- Richter, L.; Spellmeyer, D.; Martin, E.; Figliozzi, G.; Zuckermann, R. ChemInform Abstract: Automated Synthesis of Nonnatural Oligomer Libraries: The Peptoid Concept. Cheminform 2010, 28. [Google Scholar] [CrossRef]
- Zuckermann, R.; Goff, D. Synthesis of (N-Substituted)Glycine Polymers of Defined Length and Sequence. Polym. Prepr. 1994, 35, 975–976. [Google Scholar]
- Zhang, D.; Lahasky, S.H.; Guo, L.; Lee, C.-U.; Lavan, M. Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules 2012, 45, 5833–5841. [Google Scholar] [CrossRef]
- Birke, A.; Huesmann, D.; Kelsch, A.; Weilbächer, M.; Xie, J.; Bros, M.; Bopp, T.; Becker, C.; Landfester, K.; Barz, M. Polypeptoid-block-polypeptide Copolymers: Synthesis, Characterization, and Application of Amphiphilic Block Copolypept(o)ides in Drug Formulations and Miniemulsion Techniques. Biomacromolecules 2014, 15, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Deming, T.J. Synthesis of Side-Chain Modified Polypeptides. Chem. Rev. 2016, 116, 786–808. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N. Peptoid origins. Pept. Sci. 2011, 96, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Dill, K.; Zuckermann, R. Synthesis of Long Non-natural Sequence-Specific Heteropolymers. Polym. Prepr. 2005, 46, 174–175. [Google Scholar]
- Guo, L.; Zhang, D. Cyclic Poly(α-peptoid)s and Their Block Copolymers from N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxylanhydrides. J. Am. Chem. Soc. 2009, 131, 18072–18074. [Google Scholar] [CrossRef]
- Tao, X.; Deng, C.; Ling, J. PEG-Amine-Initiated Polymerization of Sarcosine N-Thiocarboxyanhydrides Toward Novel Double-Hydrophilic PEG-b-Polysarcosine Diblock Copolymers. Macromol. Rapid Commun. 2014, 35, 875–881. [Google Scholar] [CrossRef]
- Guo, L.; Lahasky, S.H.; Ghale, K.; Zhang, D. N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhydrides toward Poly(α-peptoid)s: Kinetic, Mechanism, and Architectural Control. J. Am. Chem. Soc. 2012, 134, 9163–9171. [Google Scholar] [CrossRef]
- Lahasky, S.H.; Serem, W.K.; Guo, L.; Garno, J.C.; Zhang, D. Synthesis and Characterization of Cyclic Brush-Like Polymers by N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Propargyl N-Carboxyanhydride and the Grafting-to Approach. Macromolecules 2011, 44, 9063–9074. [Google Scholar] [CrossRef]
- Tao, X.; Zheng, B.; Bai, T.; Zhu, B.; Ling, J. Hydroxyl Group Tolerated Polymerization of N-Substituted Glycine N-Thiocarboxyanhydride Mediated by Aminoalcohols: A Simple Way to α-Hydroxyl-ω-aminotelechelic Polypeptoids. Macromolecules 2017, 50, 3066–3077. [Google Scholar] [CrossRef]
- Tao, X.; Zheng, B.; Bai, T.; Li, M.-H.; Ling, J. Polymerization of N-Substituted Glycine N-Thiocarboxyanhydride through Regioselective Initiation of Cysteamine: A Direct Way toward Thiol-Capped Polypeptoids. Macromolecules 2018, 51, 4494–4501. [Google Scholar] [CrossRef]
- Robinson, J.W.; Schlaad, H. A versatile polypeptoid platform based on N-allyl glycine. Chem. Commun. 2012, 48, 7835–7837. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Xuan, S.; Li, A.; Simpson, J.M.; Sternhagen, G.L.; Yu, T.; Darvish, O.A.; Jiang, N.; Zhang, D. Polypeptoid polymers: Synthesis, characterization, and properties. Biopolymers 2018, 109, e23070. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Deng, Y.; Shen, Z.; Ling, J. Controlled Polymerization of N-Substituted Glycine N-Thiocarboxyanhydrides Initiated by Rare Earth Borohydrides toward Hydrophilic and Hydrophobic Polypeptoids. Macromolecules 2014, 47, 6173–6180. [Google Scholar] [CrossRef]
- Secker, C.; Robinson, J.W.; Schlaad, H. Alkyne-X modification of polypeptoids. Eur. Polym. J. 2015, 62, 394–399. [Google Scholar] [CrossRef]
- Tian, J.; Sun, J.; Li, Z. Biomimetic pegylated polypeptoids with thermoresponsive properties. Polymer 2018, 138, 132–138. [Google Scholar] [CrossRef]
- Fu, X.; Tian, J.; Li, Z.; Sun, J.; Li, Z. Dual-responsive pegylated polypeptoids with tunable cloud point temperatures. Biopolymers 2019, 110, e23243. [Google Scholar] [CrossRef]
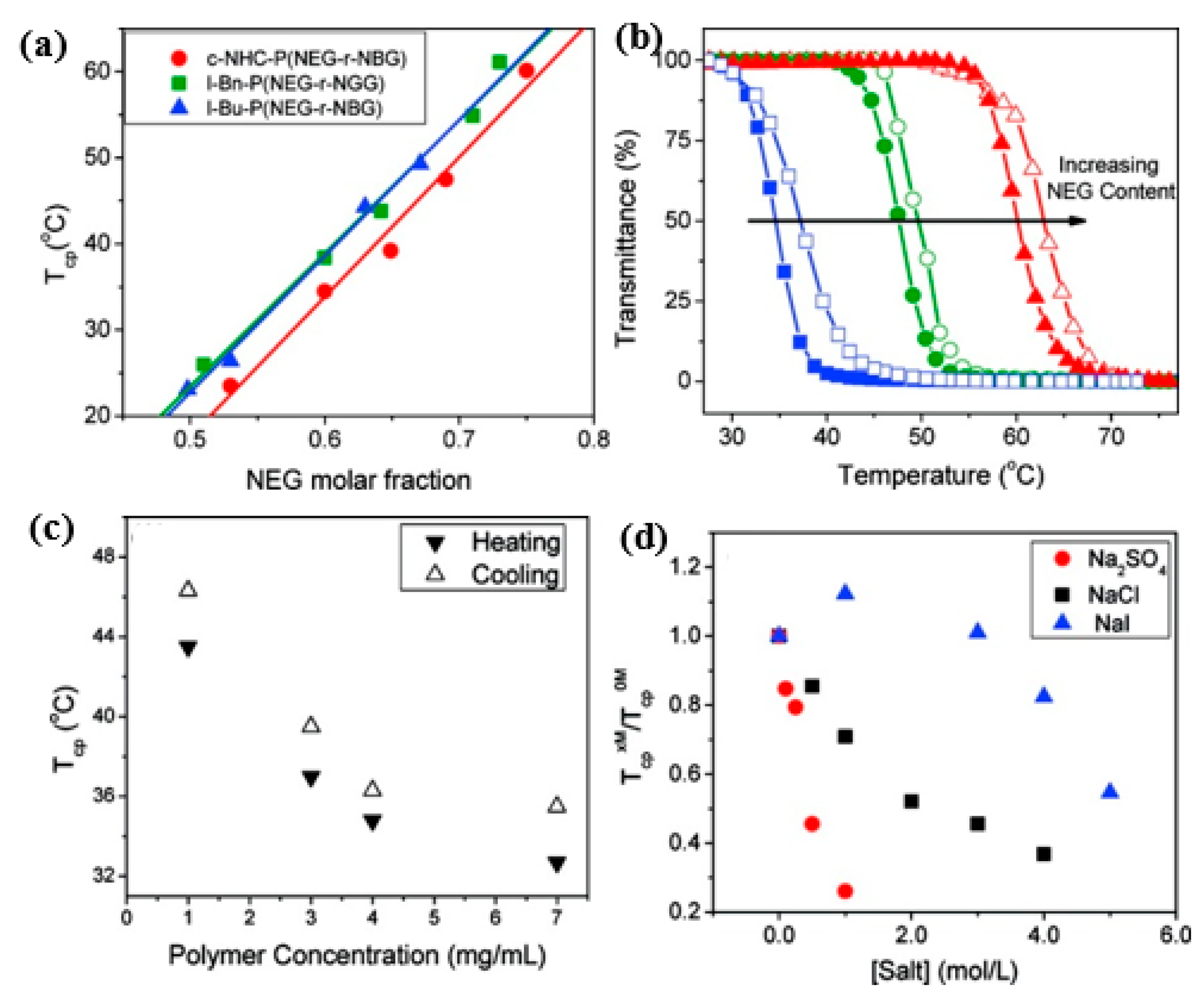
- Xing, C.; Shi, Z.; Tian, J.; Sun, J.; Li, Z. Charge-Determined LCST/UCST Behavior in Ionic Polypeptoids. Biomacromolecules 2018, 19, 2109–2116. [Google Scholar] [CrossRef]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from N-Substituted Glycine N-Carboxyanhydrides: Hydrophilic, Hydrophobic, and Amphiphilic Polymers with Poisson Distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Robinson, J.W.; Secker, C.; Weidner, S.; Schlaad, H. Thermoresponsive Poly(N-C3 glycine)s. Macromolecules 2013, 46, 580–587. [Google Scholar] [CrossRef]
- Secker, C.; Völkel, A.; Tiersch, B.; Koetz, J.; Schlaad, H. Thermo-Induced Aggregation and Crystallization of Block Copolypeptoids in Water. Macromolecules 2016, 49, 979–985. [Google Scholar] [CrossRef]
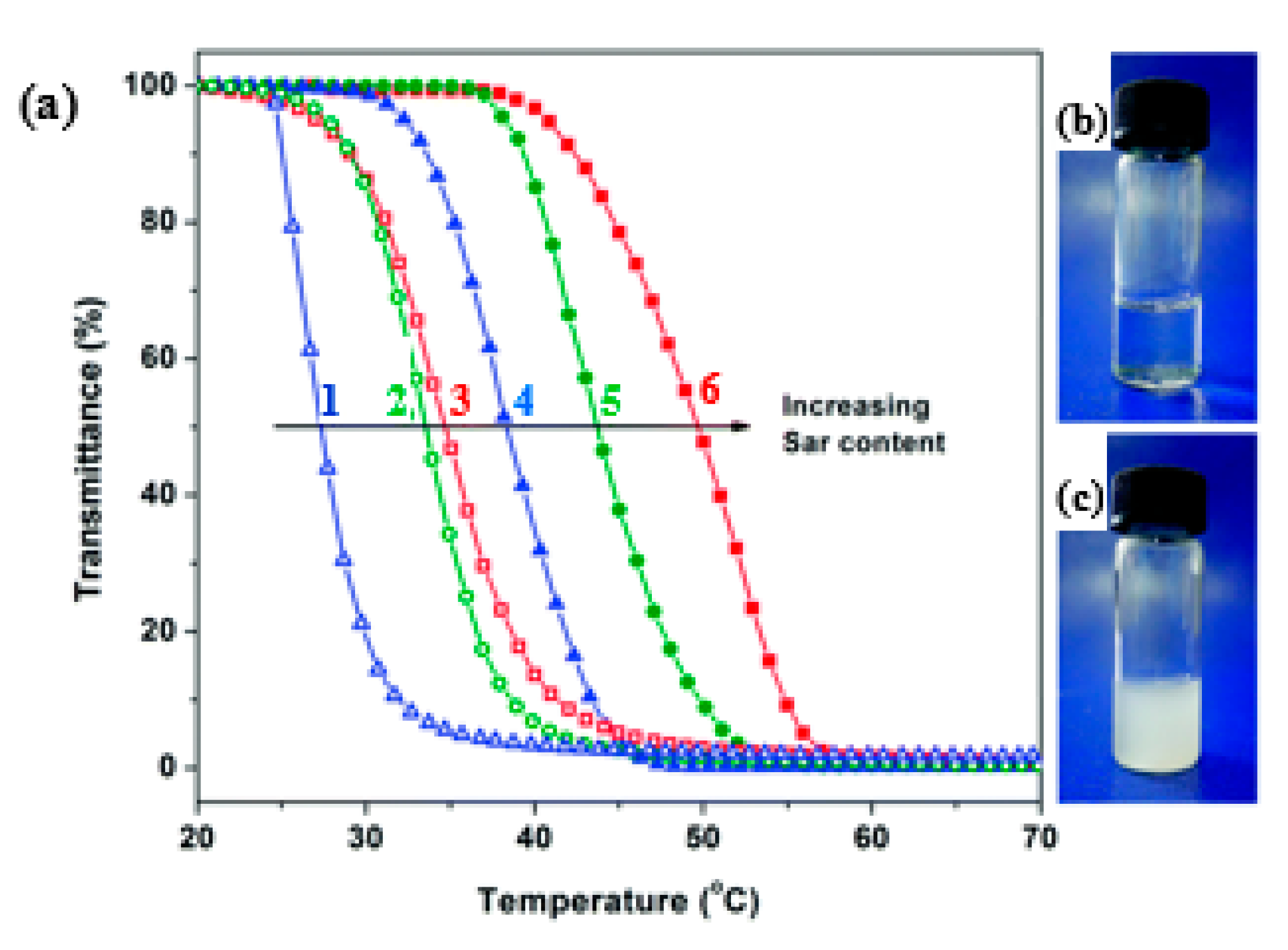
- Lahasky, S.H.; Hu, X.; Zhang, D. Thermoresponsive Poly(α-peptoid)s: Tuning the Cloud Point Temperatures by Composition and Architecture. ACS Macro Lett. 2012, 1, 580–584. [Google Scholar] [CrossRef]
- Lee, C.-U.; Lu, L.; Chen, J.; Garno, J.C.; Zhang, D. Crystallization-Driven Thermoreversible Gelation of Coil-Crystalline Cyclic and Linear Diblock Copolypeptoids. ACS Macro Lett. 2013, 2, 436–440. [Google Scholar] [CrossRef]
- Xuan, S.; Lee, C.-U.; Chen, C.; Doyle, A.B.; Zhang, Y.; Guo, L.; John, V.T.; Hayes, D.; Zhang, D. Thermoreversible and Injectable ABC Polypeptoid Hydrogels: Controlling the Hydrogel Properties through Molecular Design. Chem. Mater. 2016, 28, 727–737. [Google Scholar] [CrossRef]
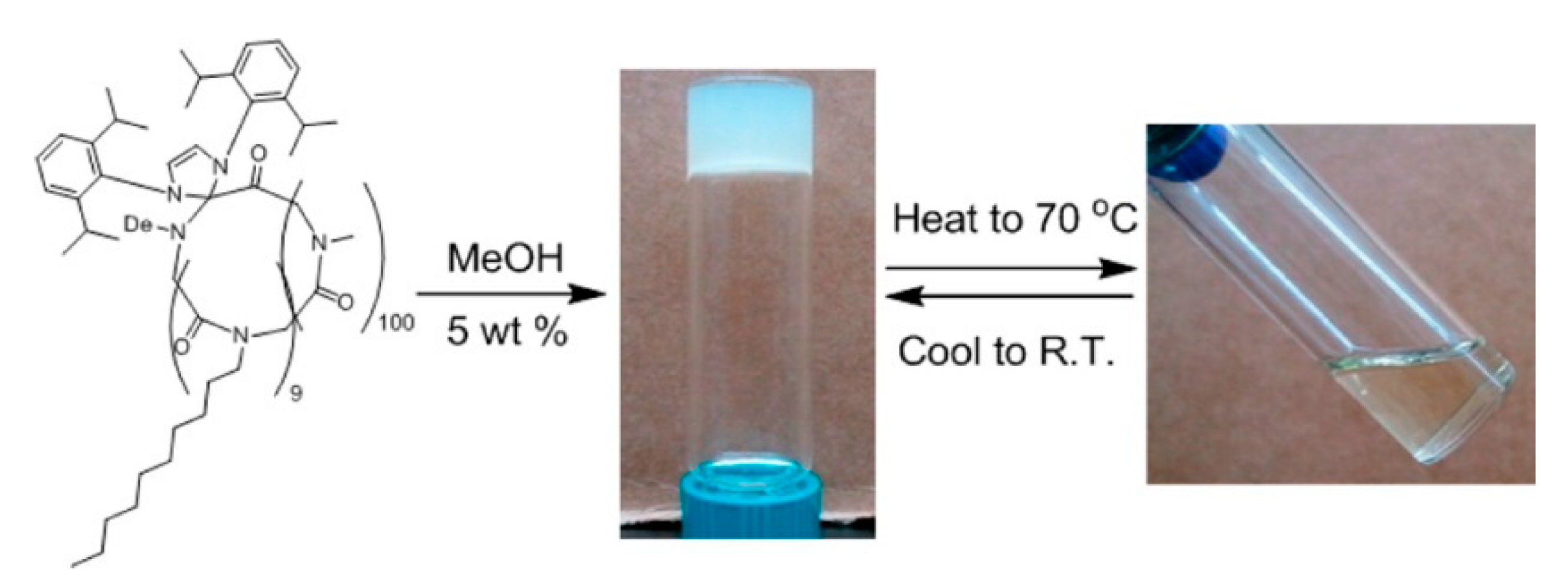
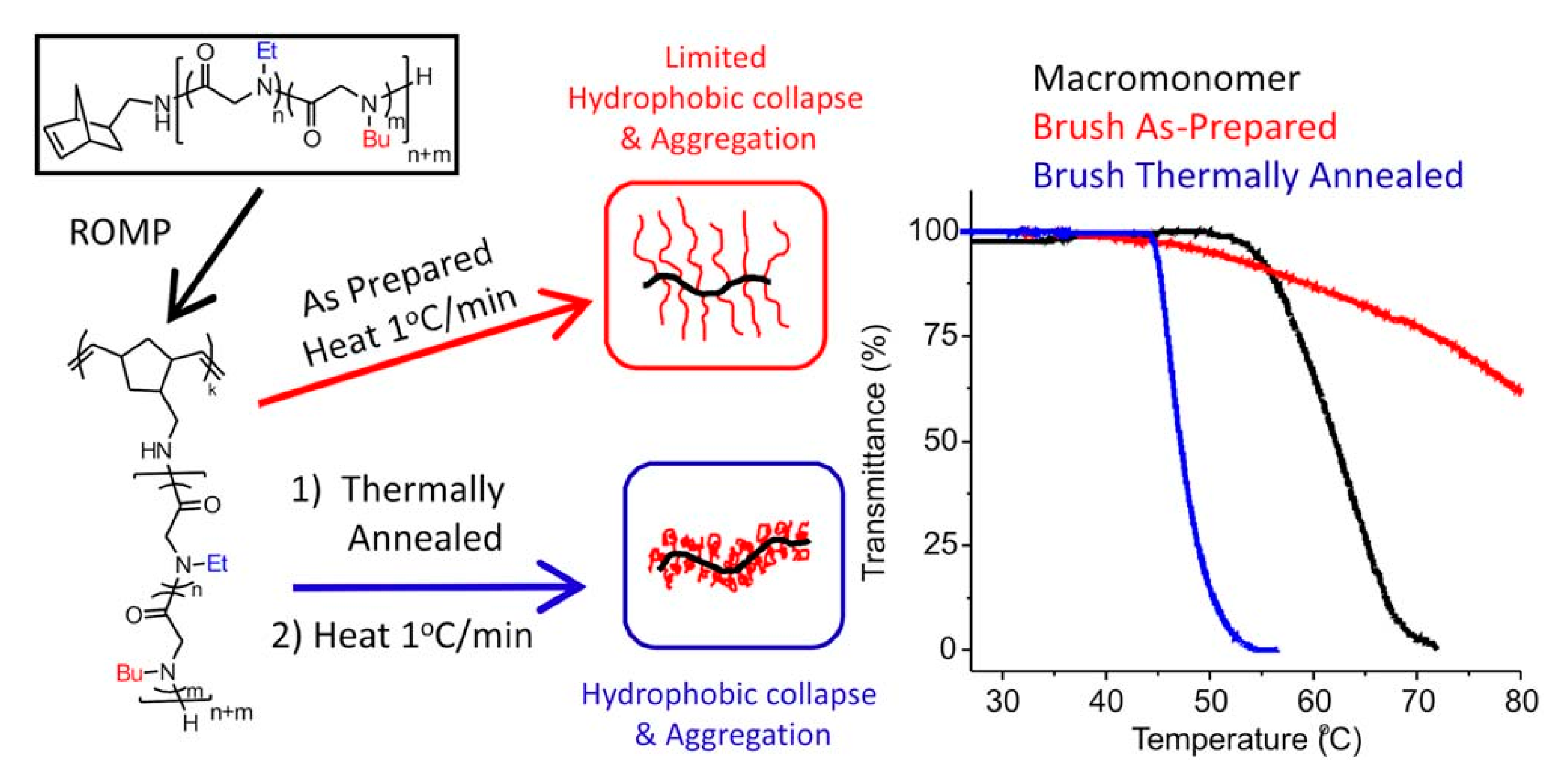
- Lahasky, S.H.; Lu, L.; Huberty, W.A.; Cao, J.; Guo, L.; Garno, J.C.; Zhang, D. Synthesis and characterization of thermo-responsive polypeptoid bottlebrushes. Polym. Chem. 2014, 5, 1418–1426. [Google Scholar] [CrossRef]
- Ma, J.; Xuan, S.; Guerin, A.C.; Yu, T.; Zhang, D.; Kuroda, D.G. Unusual molecular mechanism behind the thermal response of polypeptoids in aqueous solutions. Phys. Chem. Chem. Phys. 2017, 19, 10878–10888. [Google Scholar] [CrossRef]
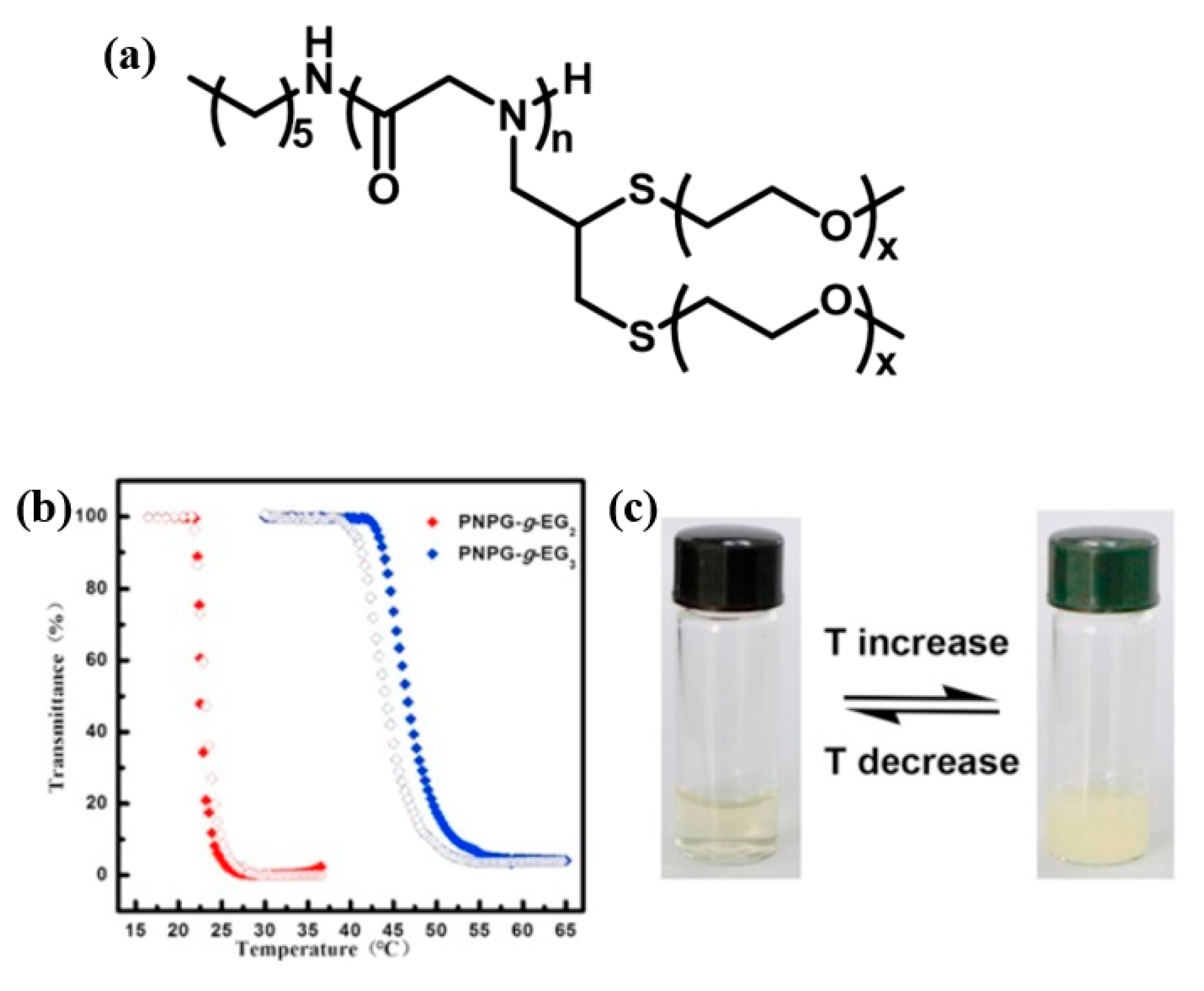
- Tao, X.; Du, J.; Wang, Y.; Ling, J. Polypeptoids with tunable cloud point temperatures synthesized from N-substituted glycine N-thiocarboxyanhydrides. Polym. Chem. 2015, 6, 3164–3174. [Google Scholar] [CrossRef]
- Salzinger, S.; Seemann, U.B.; Plikhta, A.; Rieger, B. Poly(vinylphosphonate)s Synthesized by Trivalent Cyclopentadienyl Lanthanide-Induced Group Transfer Polymerization. Macromolecules 2011, 44, 5920–5927. [Google Scholar] [CrossRef]
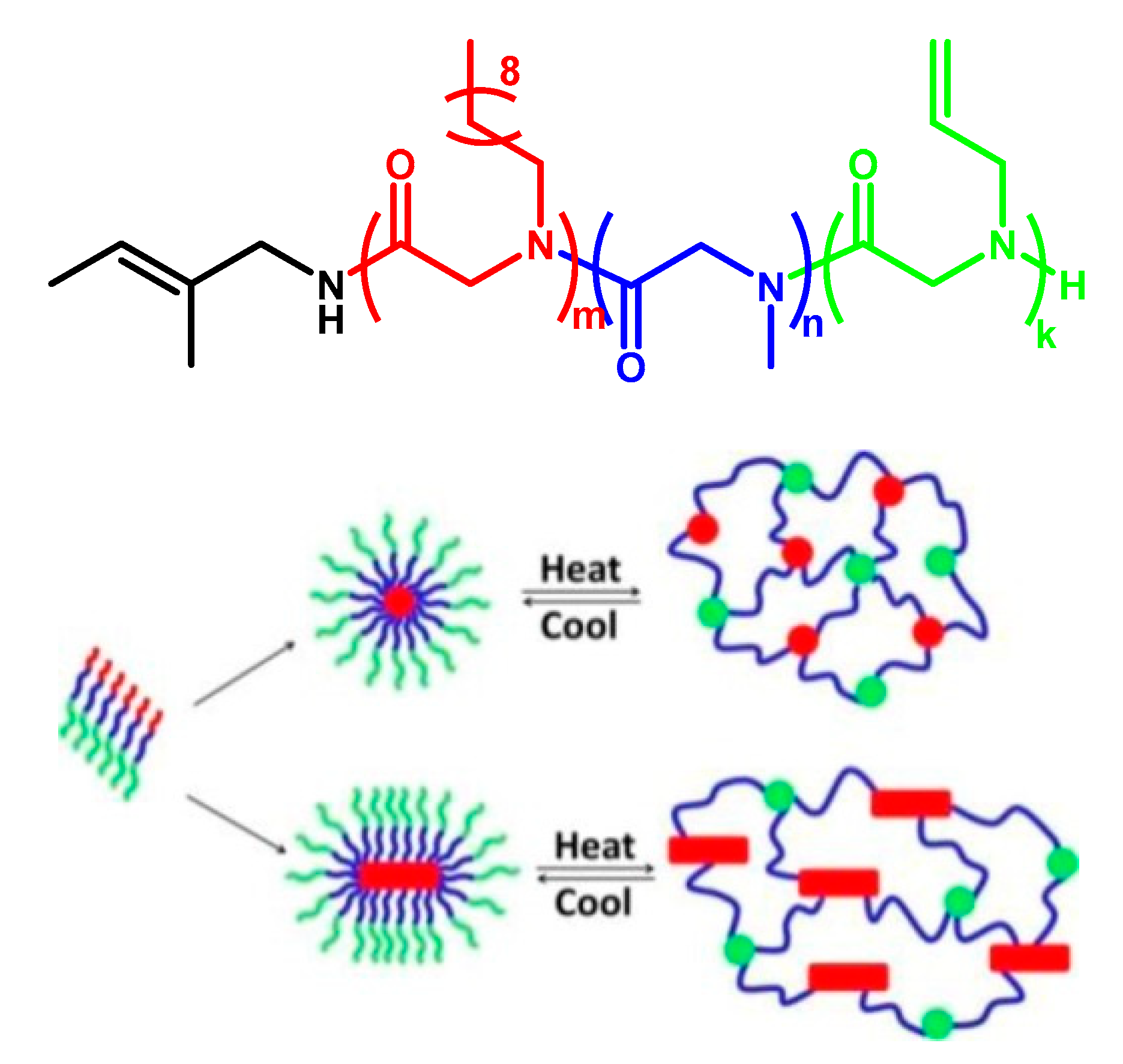
- Li, Y.; Tom, J.C.; Biehl, P.; Ling, J.; Schacher, F.H. Block Polypeptoids: Synthesis, Characterization, and Response toward Irradiation with UV Light and Temperature. Macromolecules 2020, 53, 5218–5226. [Google Scholar] [CrossRef]
- Hedir, G.G.; Arno, M.C.; Langlais, M.; Husband, J.T.; O’Reilly, R.K.; Dove, A.P. Poly(oligo(ethylene glycol) vinyl acetate)s: A Versatile Class of Thermoresponsive and Biocompatible Polymers. Angew. Chem. Int. Ed. 2017, 56, 9178–9182. [Google Scholar] [CrossRef] [PubMed]
- Aoki, D.; Ajiro, H. Thermoresponsive Polyurethane Bearing Oligo(Ethylene Glycol) as Side Chain Without Polyol at Polymer Backbone Achieved Excellent Hydrophilic and Hydrophobic Switching. Macromol. Rapid. Commun. 2018, 39, e1800239. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.-F.; Hoth, A. Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules 2006, 39, 893–896. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, M.; Li, M.; Ling, Y.; Tang, H. Synthesis and LCST-type phase behavior of water-soluble polypeptide with Y-shaped and charged side-chains. Polym. Chem. 2016, 7, 1922–1930. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Xiao, C.; Ding, J.; Zhuang, X.; Chen, X. Versatile synthesis of temperature-sensitive polypeptides by click grafting of oligo(ethylene glycol). Polym. Chem. 2011, 2, 2627–2634. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Li, Z. Thermoresponsive polypeptides from pegylated poly-L-glutamates. Biomacromolecules 2011, 12, 2859–2863. [Google Scholar] [CrossRef]
- Fu, X.; Shen, Y.; Fu, W.; Li, Z. Thermoresponsive Oligo(ethylene glycol) Functionalized Poly-l-cysteine. Macromolecules 2013, 46, 3753–3760. [Google Scholar] [CrossRef]
- Kramer, J.R.; Deming, T.J. Multimodal Switching of Conformation and Solubility in Homocysteine Derived Polypeptides. J. Am. Chem. Soc. 2014, 136, 5547–5550. [Google Scholar] [CrossRef]
- Xuan, S.; Gupta, S.; Li, X.; Bleuel, M.; Schneider, G.J.; Zhang, D. Synthesis and Characterization of Well-Defined PEGylated Polypeptoids as Protein-Resistant Polymers. Biomacromolecules 2017, 18, 951–964. [Google Scholar] [CrossRef]
- Sun, J.; Stone, G.M.; Balsara, N.P.; Zuckermann, R.N. Structure-Conductivity Relationship for Peptoid-Based PEO-Mimetic Polymer Electrolytes. Macromolecules 2012, 45, 5151–5156. [Google Scholar] [CrossRef]
- Statz, A.R.; Meagher, R.J.; Barron, A.E.; Messersmith, P.B. New Peptidomimetic Polymers for Antifouling Surfaces. J. Am. Chem. Soc. 2005, 127, 7972–7973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, C.; Li, Z. Effects of molecular weight on thermal responsive property of pegylated poly-l-glutamates. Chin. J. Polym. Sci. 2013, 31, 201–210. [Google Scholar] [CrossRef]
- Xia, Y.; Yin, X.; Burke, N.; Stöver, H. Thermal Response of Narrow-Disperse Poly(N-isopropylacrylamide) Prepared by Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 5937–5943. [Google Scholar] [CrossRef]
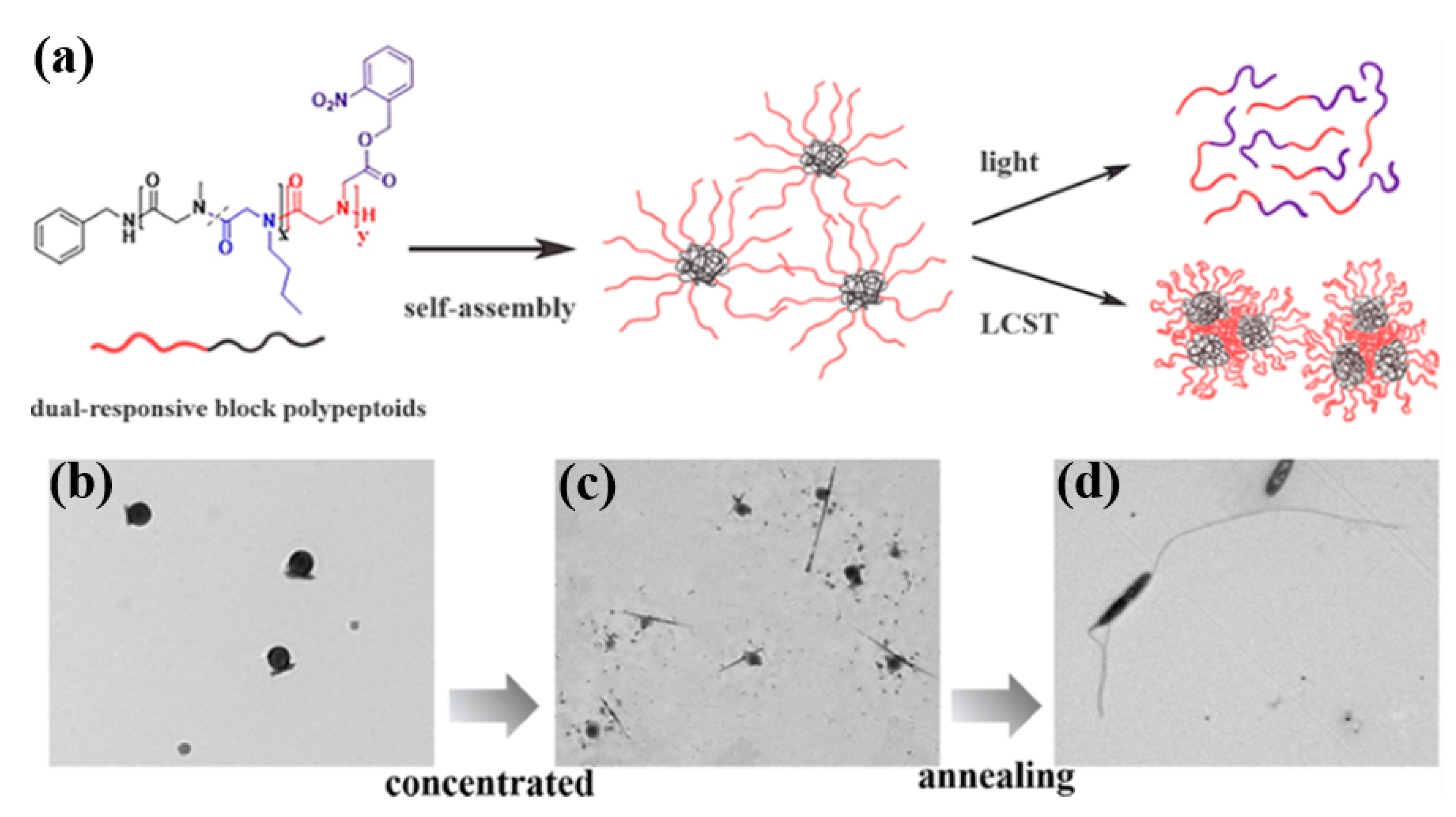
- Wang, Z.; Lin, M.; Bonduelle, C.; Li, R.; Shi, Z.; Zhu, C.; Lecommandoux, S.; Li, Z.; Sun, J. Thermoinduced Crystallization-Driven Self-Assembly of Bioinspired Block Copolymers in Aqueous Solution. Biomacromolecules 2020, 21, 3411–3419. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; An, Z.; Wu, P. UCST or LCST? Composition-Dependent Thermoresponsive Behavior of Poly(N-acryloylglycinamide-co-diacetone acrylamide). Macromolecules 2017, 50, 2175–2182. [Google Scholar] [CrossRef]
- Wu, G.; Chen, S.-C.; Zhan, Q.; Wang, Y.-Z. Well-Defined Amphiphilic Biodegradable Comb-Like Graft Copolymers: Their Unique Architecture-Determined LCST and UCST Thermoresponsivity. Macromolecules 2011, 44, 999–1008. [Google Scholar] [CrossRef]
- Fu, X.; Xing, C.; Sun, J. Tunable LCST/UCST-Type Polypeptoids and Their Structure-Property Relationship. Biomacromolecules 2020. [Google Scholar] [CrossRef]
- Hu, J.; Liu, S. Responsive Polymers for Detection and Sensing Applications: Current Status and Future Developments. Macromolecules 2010, 43, 8315–8330. [Google Scholar] [CrossRef]
- Liu, L.; Li, W.; Liu, K.; Yan, J.; Hu, G.; Zhang, A. Comblike Thermoresponsive Polymers with Sharp Transitions: Synthesis, Characterization, and Their Use as Sensitive Colorimetric Sensors. Macromolecules 2011, 44, 8614–8621. [Google Scholar] [CrossRef]
- Lau, K.H.A. Peptoids for biomaterials science. Biomater. Sci. 2014, 2, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Okano, T.; Sakai, H.; Karikusa, F.; Sawasaki, Y.; Sakurai, Y. Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Die Makromol. Chem. Rapid Commun. 1990, 11, 571–576. [Google Scholar] [CrossRef]
- Lu, D.; Wang, H.; Li, T.e.; Li, Y.; Wang, X.; Niu, P.; Guo, H.; Sun, S.; Wang, X.; Guan, X.; et al. Versatile Surgical Adhesive and Hemostatic Materials: Synthesis, Properties, and Application of Thermoresponsive Polypeptides. Chem. Mater. 2017, 29, 5493–5503. [Google Scholar] [CrossRef]
- Kurzhals, S.; Pretzner, B.; Reimhult, E.; Zirbs, R. Thermoresponsive Polypeptoid-Coated Superparamagnetic Iron Oxide Nanoparticles by Surface-Initiated Polymerization. Macromol. Chem. Phys. 2017, 218, 1700116. [Google Scholar] [CrossRef]
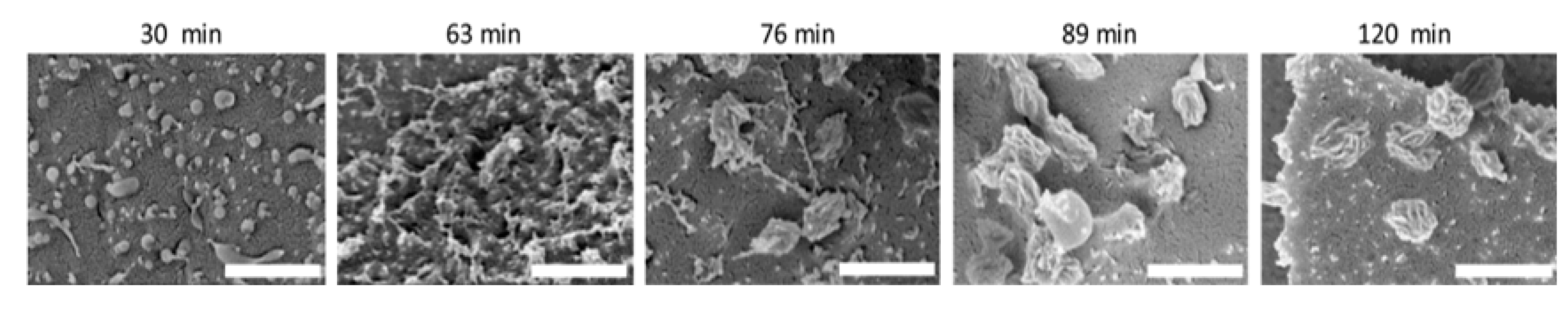
- Lu, L.; Lahasky, S.H.; McCandless, G.T.; Zhang, D.; Garno, J.C. Thermoresponsive Behavior of Polypeptoid Nanostructures Investigated with Heated Atomic Force Microscopy: Implications toward the Development of Smart Coatings for Surface-Based Sensors. ACS Appl. Nano Mater. 2019, 2, 7617–7625. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Sun, J. Thermoresponsive Polypeptoids. Polymers 2020, 12, 2973. https://doi.org/10.3390/polym12122973
Liu D, Sun J. Thermoresponsive Polypeptoids. Polymers. 2020; 12(12):2973. https://doi.org/10.3390/polym12122973
Chicago/Turabian StyleLiu, Dandan, and Jing Sun. 2020. "Thermoresponsive Polypeptoids" Polymers 12, no. 12: 2973. https://doi.org/10.3390/polym12122973
APA StyleLiu, D., & Sun, J. (2020). Thermoresponsive Polypeptoids. Polymers, 12(12), 2973. https://doi.org/10.3390/polym12122973