Surfactant-Modulation of the Cationic-Polymer-Induced Aggregation of Anionic Particulate Dispersions


Abstract
:
1. Introduction
2. Materials and Methods
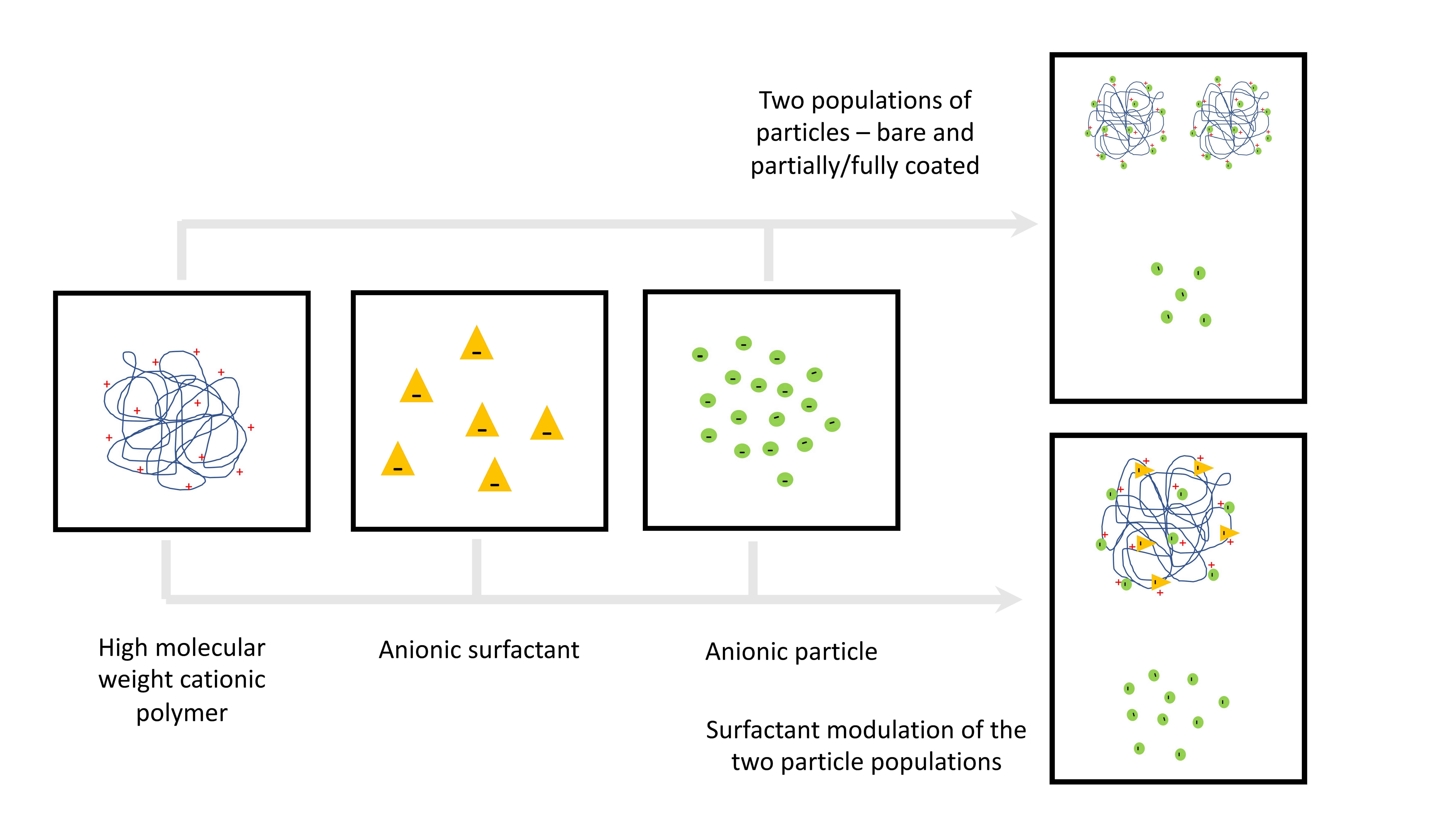
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grządka, E.; Matusiak, J.; Paszkiewicz, M. Factors influencing the stability of the 2-hydroxyethyl cellulose/alumina system. Cellulose 2018, 25, 2839–2847. [Google Scholar] [CrossRef]
- Mabire, F.; Audebert, R.; Quivoron, C. Flocculation properties of some water-soluble cationic copolymers toward silica suspensions: A semiquantitative interpretation of the role of molecular weight and cationicity through a “patchwork” model. J. Colloid Interface Sci. 1984, 97, 120–136. [Google Scholar] [CrossRef]
- Lee, C.S.; Robinson, J.; Chong, M.F. A review of application of flocculants in waterwater treatment. Process Safety Environ. Prot. 2014, 92, 489–508. [Google Scholar] [CrossRef]
- Mészáros, R.; Varga, I.; Gilányi, T. Adsorption of poly(ethyleneimine) on silica surfaces: Effect of pH on the reversibility of adsorption. Langmuir 2004, 20, 5026–5029. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Bratskaya, S.; Jaeger, W.; Paulke, B.R. Effect of charge density, molecular weight, and hydrophobicity on polycations adsorption and flocculation of polystyrene latices and silica. J. Appl. Polym. Sci. 2006, 101, 3422–3429. [Google Scholar] [CrossRef]
- Netz, R.R.; Andelman, D. Neutral and charged polymers at interfaces. Phys. Rep. 2003, 380, 1–95. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, E.; Biesheuvel, P.M.; De Vos, W.M. Adsorption of charged and neutral polymer chains on silica surfaces: The role of electrostatics, volume exclusion, and hydrogen bonding. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2015, 91, 012601. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, X.; Thormann, E. Surface Forces between Highly Charged Cationic Polyelectrolytes Adsorbed to Silica: How Control of pH and the Adsorbed Amount Determines the Net Surface Charge. Langmuir 2018, 34, 7264–7271. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gan, Y.; Wanless, E.J.; Jameson, G.J.; Franks, G.V. Interaction forces between silica surfaces in aqueous solutions of cationic polymeric flocculants: Effect of polymer charge. Langmuir 2008, 24, 10920–10928. [Google Scholar] [CrossRef]
- Szilagyi, I.; Trefalt, G.; Tiraferri, A.; Maroni, P.; Borkovec, M. Polyelectrolyte adsorption, interparticle forces, and colloidal aggregation. Soft Matter 2014, 10, 2479–2502. [Google Scholar] [CrossRef] [Green Version]
- Kizilay, E.; Kayitmazer, A.B.; Dubin, P.L. Complexation and coacervation of polyelectrolytes with oppositely charged colloids. Adv. Colloid Interface Sci. 2011, 167, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, R.; Prévost, S.; Schweins, R.; Appavou, M.S.; Gradzielski, M. Interactions of silica nanoparticles with poly(ethylene oxide) and poly(acrylic acid): Effect of the polymer molecular weight and of the surface charge. J. Colloid Interface Sci. 2013, 394, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Roberts, J.E.; Santore, M.M. The relationship between polymer/substrate charge density and charge overcompensation by adsorbed polyelectrolyte layers. J. Colloid Interface Sci. 2002, 247, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrynin, A.V.; Rubinstein, M. Theory of polyelectrolytes in solutions and at surfaces. Prog. Polym. Sci. 2005, 30, 1049–1118. [Google Scholar] [CrossRef]
- Hasan, A.; Fatehi, P. Flocculation of kaolin particles with cationic lignin polymers. Sci. Rep. 2019, 9, 2672. [Google Scholar] [CrossRef]
- Zhou, Y.; Franks, G.V. Flocculation mechanism induced by cationic polymers investigated by light scattering. Langmuir 2006, 22, 6775–6786. [Google Scholar] [CrossRef]
- Matusiak, J.; Grządka, E.; Bastrzyk, A. Stability, adsorption and electrokinetic properties of the chitosan/silica system. Colloids Surfaces A Physicochem. Eng. Asp. 2018, 554, 245–252. [Google Scholar] [CrossRef]
- Renault, F.; Sancey, B.; Badot, P.M.; Crini, G. Chitosan for coagulation/flocculation processes—An eco-friendly approach. Eur. Polym. J. 2009, 45, 1337–1348. [Google Scholar] [CrossRef]
- Bolto, B.; Gregory, J. Organic polyelectrolytes in water treatment. Water Res. 2007, 41, 2301–2324. [Google Scholar] [CrossRef]
- Lee, K.E.; Morad, N.; Teng, T.T.; Poh, B.T. Development, characterization and the application of hybrid materials in coagulation/flocculation of wastewater: A review. Chem. Eng. J. 2012, 203, 370–386. [Google Scholar] [CrossRef]
- Biggs, S.; Habgood, M.; Jameson, G.J.; Yan, Y.D. Aggregate structures formed via a bridging flocculation mechanism. Chem. Eng. J. 2000, 80, 13–22. [Google Scholar] [CrossRef]
- Barany, S.; Skvarla, J. Effect of Polyelectrolytes and Polyelectrolyte Mixtures on the Electrokinetic Potential of Dispersed Particles. 1. Electrokinetic Potential of Polystyrene Particles in Solutions of Surfactants, Polyelectrolytes and their Mixtures. Colloid J. 2013, 75, 129–136. [Google Scholar] [CrossRef]
- Taubaeva, R.; Meszaros, R.; Musabekov, K.; Barany, S. Electrokinetic Potential and Flocculation of Bentonite Suspensions in Solutions of Surfactants, Polyelectrolytes and Their Mixtures. Colloid J. 2015, 77, 91–98. [Google Scholar] [CrossRef]
- Besra, L.; Sengupta, D.K.; Roy, S.K.; Ay, P. Influence of polymer adsorption and conformation on flocculation and dewatering of kaolin suspension. Sep. Purif. Technol. 2004, 37, 231–246. [Google Scholar] [CrossRef]
- Karlson, L.; Olsson, M.; Bostrom, G.; Piculell, L. Influence of added surfactant on particle flocculation in waterborne polymer–particle systems. J. Coat. Technol. Res. 2008, 5, 447–454. [Google Scholar] [CrossRef]
- Lele, B.J.; Tilton, R.D. Colloidal Depletion and Structural Force Synergism or Antagonism in Solutions of Mutually Repelling Polyelectrolytes and Ionic Surfactants. Langmuir 2019, 35, 15937–15947. [Google Scholar] [CrossRef]
- Matusiak, J.; Grządka, E. Cationic starch as the effective flocculant of silica in the presence of different surfactants. Sep. Purif. Technol. 2020, 234, 116132. [Google Scholar] [CrossRef]
- Wang, K.; Ye, L. Structure and property of cationic hydroxyethyl cellulose. Polym. Plast. Technol. Eng. 2010, 49, 807–811. [Google Scholar] [CrossRef]
- Shubin, V. Adsorption of Cationic Polymer onto Negatively Charged Surfaces in the Presence of Anionic Surfactant. Langmuir 1994, 10, 1093–1100. [Google Scholar] [CrossRef]
- Terada, E.; Samoshina, Y.; Nylander, T. Adsorption of cationic cellulose derivatives/anionic surfactant complexes onto solid surfaces. Langmuir 2004, 20, 1753–1762. [Google Scholar] [CrossRef]
- Roos, P.; Westling, Å.; Chronakis, I.S. Hydrophilic Monolayer Formation of Adsorbed Cationic Starch and Cationic Hydroxyethyl Cellulose Derivatives on Polyester Surfaces. Biosci. Biotechnol. Biochem. 2004, 68, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Mansour, O.; Crossman, M.; Griffiths, P. Electrophoretic NMR Characterization of Charged Side Chain Cationic Polyelectrolytes and Their Interaction with the Anionic Surfactant, Sodium Dodecyl Sulfate. Langmuir 2019, 35, 9233–9238. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.L.; van Duijneveldt, J.S.; Cosgrove, T.; Prescott, S.W.; Murray, M. Competition between Polymers for Adsorption on Silica: A Solvent Relaxation NMR and Small-Angle Neutron Scattering Study. Langmuir 2013, 29, 12670–12678. [Google Scholar] [CrossRef] [PubMed]
- Van der Beek, G.P.; Cohen Stuart, M.A.; Cosgrove, T. Polymer Adsorption and Desorption Studies via 1H NMR Relaxation of the Solvent. Langmuir 1991, 7, 327–334. [Google Scholar] [CrossRef]
- Cosgrove, T.; Griffiths, P.C.; Lloyd, P.M. Polymer Adsorption. The Effect of the Relative Sizes of Polymer and Particle. Langmuir 1995, 11, 1457–1463. [Google Scholar] [CrossRef]
- Nelson, A.; Jack, K.S.; Cosgrove, T.; Kozak, D. NMR solvent relaxation in studies of multicomponent polymer adsorption. Langmuir 2002, 18, 2750–2755. [Google Scholar] [CrossRef]
- Cooper, C.L.; Cosgrove, T.; Van Duijneveldt, J.S.; Murray, M.; Prescott, S.W. The use of solvent relaxation NMR to study colloidal suspensions. Soft Matter 2013, 9, 7211–7228. [Google Scholar] [CrossRef]
- Cosgrove, T.; Obey, T.M.; Taylor, M. Solvent relaxation NMR: Bound fraction determination for sodium Poly(styrene sulphonate) at the solid/solution interface. Colloids Surfaces 1992, 64, 311–316. [Google Scholar] [CrossRef]
- Cattoz, B.; Cosgrove, T.; Crossman, M.; Prescott, S.W. Surfactant-mediated desorption of polymer from the nanoparticle interface. Langmuir 2012, 28, 2485–2492. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis of the Equilibrium Phase Extracted from the Following Samples | Zeta Potential/mV |
|---|---|
| Stock, as-supplied Ludox, 1-in-10 dilution | −40.4 (±1.8) |
| Dialysed Ludox | −35.5 (±1.5) |
| Dialysed Ludox plus polymer N = 0.5% | −30.9 (±1.3) |
| Dialysed Ludox plus polymer N = 0.95% | −30.8 (±1.7) |
| Dialysed Ludox plus polymer N = 1.8% | −31.4 (±2.4) |
| Dialysed Ludox plus polymer N = 0.5% and SDS | −31.4 (±1.6) |
| Dialysed Ludox plus polymer N = 0.95% and SDS | −29.9 (±2.2) |
| Dialysed Ludox plus polymer N = 1.8% and SDS | −32.6 (±2.8) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullahi, W.; Crossman, M.; Griffiths, P.C. Surfactant-Modulation of the Cationic-Polymer-Induced Aggregation of Anionic Particulate Dispersions. Polymers 2020, 12, 287. https://doi.org/10.3390/polym12020287
Abdullahi W, Crossman M, Griffiths PC. Surfactant-Modulation of the Cationic-Polymer-Induced Aggregation of Anionic Particulate Dispersions. Polymers. 2020; 12(2):287. https://doi.org/10.3390/polym12020287
Chicago/Turabian StyleAbdullahi, Wasiu, Martin Crossman, and Peter Charles Griffiths. 2020. "Surfactant-Modulation of the Cationic-Polymer-Induced Aggregation of Anionic Particulate Dispersions" Polymers 12, no. 2: 287. https://doi.org/10.3390/polym12020287
APA StyleAbdullahi, W., Crossman, M., & Griffiths, P. C. (2020). Surfactant-Modulation of the Cationic-Polymer-Induced Aggregation of Anionic Particulate Dispersions. Polymers, 12(2), 287. https://doi.org/10.3390/polym12020287